Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease

 , , and
, , and
Abstract
:1. Introduction
2. Ferroptosis: Three Hallmarks for Specific Dysfunctions
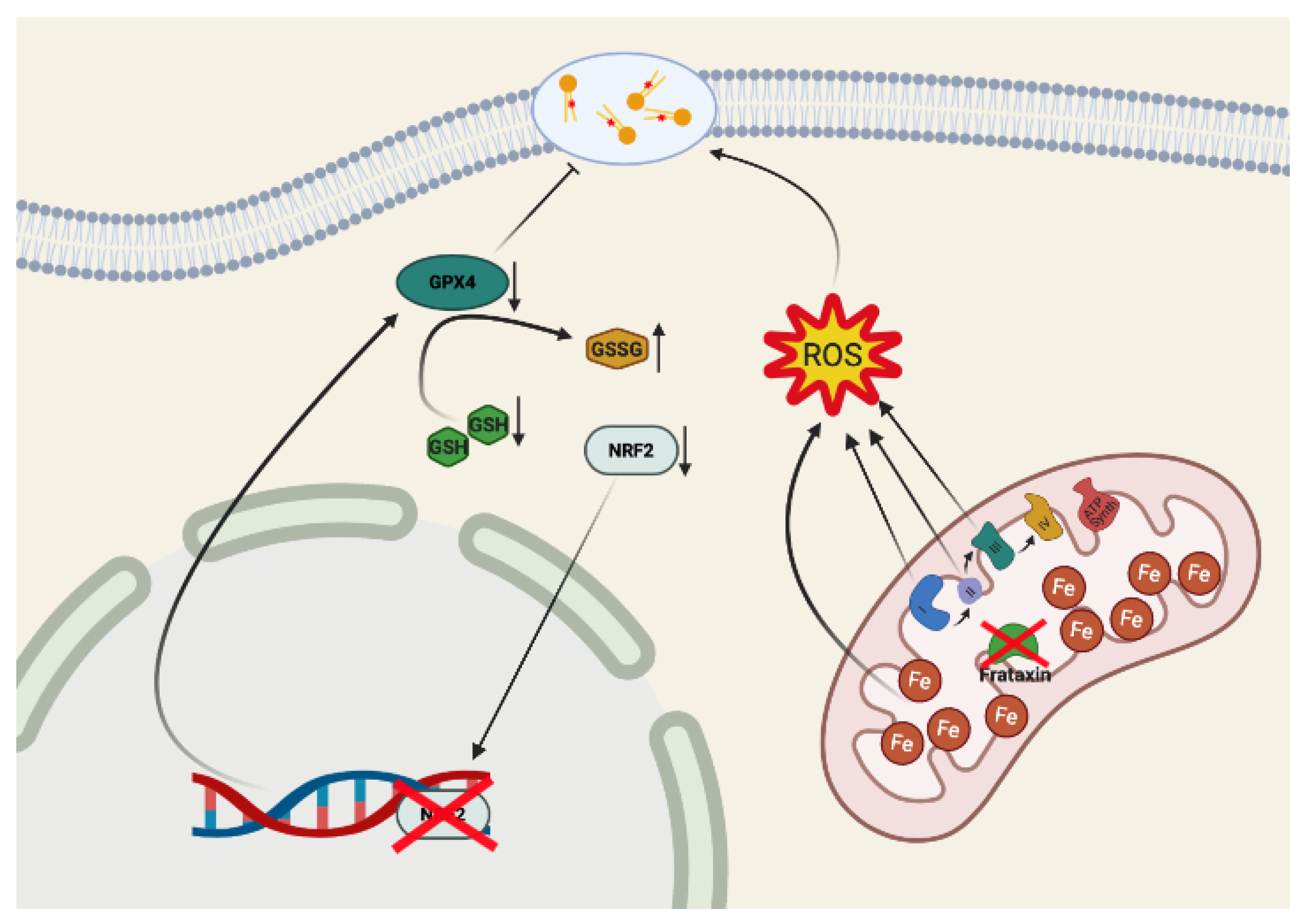
3. Ferroptosis in Neurodegeneration and in Friedreich’s Ataxia
4. Conclusions

{kind=link}
{kind=link}
| Biochemical Features | Affected Tissues | References |
|---|---|---|
| Iron accumulation | Mouse models | [105,116,117,118] |
| Cellular models | [84,85,119] | |
| Human heart | [111,112] | |
| Human nervous system | [113,114,115] | |
| Human fibroblasts | [83,120] | |
| Morphological mitochondrial changes | Mouse models | [127] |
| Cellular models | [84,93] | |
| ROS and lipid peroxidation | Animal models | [86,88,121,122,123,124,125,126,127] |
| Cellular models | [86,92,93,130,131,132,133] | |
| Human blood | [87,128,129] | |
| Human fibroblasts | [87,92,120] | |
| Glutathione imbalance | Human blood | [136] |
| Human fibroblasts | [87,135,137] | |
| Negative regulation of NRF2 | Animal models | [91,140] |
| Cellular models | [47,90,91] | |
| Human fibroblasts | [90,135] |
Author Contributions
Funding
Conflicts of Interest
References
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Büttner, S.; Eisenberg, T.; Herker, E.; Carmona-Gutierrez, D.; Kroemer, G.; Madeo, F. Why yeast cells can undergo apoptosis: Death in times of peace, love, and war. J. Cell Biol. 2006, 175, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Cornillon, S.; Foa, C.; Davoust, J.; Buonavista, N.; Gross, J.D.; Golstein, P. Programmed cell death in Dictyostelium. J. Cell Sci. 1994, 107, 2691. [Google Scholar]
- Bayles, K.W. Bacterial programmed cell death: Making sense of a paradox. Nat. Rev. Microbiol. 2014, 12, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef]
- Conradt, B. Genetic control of programmed cell death during animal development. Annu. Rev. Genet. 2009, 43, 493–523. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-l.; Huang, Z.-j.; Lin, Z.-t.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Fearnhead, H.O.; Vandenabeele, P.; Vanden Berghe, T. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017, 24, 1991–1998. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weïwer, M.; Bittker, J.A.; Lewis, T.A.; Shimada, K.; Yang, W.S.; MacPherson, L.; Dandapani, S.; Palmer, M.; Stockwell, B.R.; Schreiber, S.L.; et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 2012, 22, 1822–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Shimada, K.; Delva, D.; Patel, M.; Ode, E.; Skouta, R.; Stockwell, B.R. Identification of Simple Compounds with Microtubule-Binding Activity That Inhibit Cancer Cell Growth with High Potency. ACS Med. Chem. Lett. 2012, 3, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodríguez Martínez, M.; López, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Abrams, R.P.; Carroll, W.L.; Woerpel, K.A. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem Biol 2016, 11, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.R.; Lane, D.J.; Becker, E.M.; Huang, M.L.; Whitnall, M.; Suryo Rahmanto, Y.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Comm. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Minotti, G.; Aust, S.D. The role of iron in oxygen radical mediated lipid peroxidation. Chem.-Biol. Interact. 1989, 71, 1–19. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Gregolin, C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Corenblum, M.J.; Ray, S.; Remley, Q.W.; Long, M.; Harder, B.; Zhang, D.D.; Barnes, C.A.; Madhavan, L. Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle-age period. Aging Cell 2016, 15, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Antón, N.; Rojo, A.I.; Ferreiro, E.; Núñez, Á.; Krause, K.H.; Jaquet, V.; Cuadrado, A. Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol. 2017, 13, 393–401. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.; Chattopadhyay, A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Gladwell, W.; Wang, X.; Chorley, B.; Bell, D.; Reddy, S.P.; Kleeberger, S.R. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2010, 182, 170–182. [Google Scholar] [CrossRef]
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.-J.; Gao, Y.; Bennett, M.V.L.; et al. Peroxisome proliferator-activated receptor γ (PPARγ): A master gatekeeper in CNS injury and repair. Prog. Neurobiol. 2018, 163-164, 27–58. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell 2017, 68, 224–232.e224. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X. A Physiological Function for Ferroptosis in Tumor Suppression by the Immune System. Cell Met. 2019, 30, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J. Alzheimers Dis. 2013, 37, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Lei, P. Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. BioMed Res. Int. 2014, 2014, 581256. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef]
- Gerlach, M.; Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Altered brain metabolism of iron as a cause of neurodegenerative diseases? J. Neurochem. 1994, 63, 793–807. [Google Scholar] [CrossRef]
- Connor, J.R.; Snyder, B.S.; Beard, J.L.; Fine, R.E.; Mufson, E.J. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J. Neurosci. Res. 1992, 31, 327–335. [Google Scholar] [CrossRef]
- Zecca, L.; Gallorini, M.; Schünemann, V.; Trautwein, A.X.; Gerlach, M.; Riederer, P.; Vezzoni, P.; Tampellini, D. Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: Consequences for iron storage and neurodegenerative processes. J. Neurochem. 2001, 76, 1766–1773. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Veyrat-Durebex, C.; Corcia, P.; Mucha, A.; Benzimra, S.; Mallet, C.; Gendrot, C.; Moreau, C.; Devos, D.; Piver, E.; Pagès, J.-C.; et al. Iron Metabolism Disturbance in a French Cohort of ALS Patients. BioMed Res. Int. 2014, 2014, 485723. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H.; Moore, D.H.; Miller, R.G.; Florence, J.M.; Verheijde, J.L.; Doorish, C.; Hilton, J.F.; Spitalny, G.M.; MacArthur, R.B.; Mitsumoto, H.; et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: A phase III randomised trial. Lancet Neurol. 2007, 6, 1045–1053. [Google Scholar] [CrossRef]
- Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 2007, 69, 1480–1490. [CrossRef] [PubMed]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2020, 101890. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef]
- Petillon, C.; Hergesheimer, R.; Puy, H.; Corcia, P.; Vourc’h, P.; Andres, C.; Karim, Z.; Blasco, H. The Relevancy of Data Regarding the Metabolism of Iron to Our Understanding of Deregulated Mechanisms in ALS; Hypotheses and Pitfalls. Front. Neurosci. 2018, 12, 1031. [Google Scholar] [CrossRef]
- Devos, D.; Cabantchik, Z.I.; Moreau, C.; Danel, V.; Mahoney-Sanchez, L.; Bouchaoui, H.; Gouel, F.; Rolland, A.S.; Duce, J.A.; Devedjian, J.C. Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J. Neural. Transm. 2020, 127, 189–203. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Kyheng, M.; Garçon, G.; Rolland, A.S.; Blasco, H.; Gelé, P.; Timothée Lenglet, T.; Veyrat-Durebex, C.; Corcia, P.; et al. A ferroptosis-based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 2918. [Google Scholar] [CrossRef] [Green Version]
- Delatycki, M.B.; Williamson, R.; Forrest, S.M. Friedreich ataxia: An overview. J. Med. Genet. 2000, 37, 1–8. [Google Scholar] [CrossRef]
- Bürk, K. Friedreich Ataxia: Current status and future prospects. Cerebellum Ataxias 2017, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Delatycki, M.B.; Camakaris, J.; Brooks, H.; Evans-Whipp, T.; Thorburn, D.R.; Williamson, R.; Forrest, S.M. Direct evidence that mitochondrial iron accumulation occurs in Friedreich ataxia. Ann. Neurol. 1999, 45, 673–675. [Google Scholar] [CrossRef]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [Green Version]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef]
- Bradley, J.L.; Homayoun, S.; Hart, P.E.; Schapira, A.H.; Cooper, J.M. Role of oxidative damage in Friedreich’s ataxia. Neurochem. Res. 2004, 29, 561–567. [Google Scholar] [CrossRef]
- Navarro, J.A.; Ohmann, E.; Sanchez, D.; Botella, J.A.; Liebisch, G.; Moltó, M.D.; Ganfornina, M.D.; Schmitz, G.; Schneuwly, S. Altered lipid metabolism in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 2010, 19, 2828–2840. [Google Scholar] [CrossRef] [Green Version]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchère, F.; Lönn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef] [Green Version]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J. Pharmacol Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef] [PubMed]
- Dürr, A.; Cossee, M.; Agid, Y.; Campuzano, V.; Mignard, C.; Penet, C.; Mandel, J.L.; Brice, A.; Koenig, M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef]
- Nair, M.; Adinolfi, S.; Pastore, C.; Kelly, G.; Temussi, P.; Pastore, A. Solution structure of the bacterial frataxin ortholog, CyaY: Mapping the iron binding sites. Structure 2004, 12, 2037–2048. [Google Scholar] [CrossRef] [Green Version]
- Pastore, C.; Franzese, M.; Sica, F.; Temussi, P.; Pastore, A. Understanding the binding properties of an unusual metal-binding protein--a study of bacterial frataxin. Febs J. 2007, 274, 4199–4210. [Google Scholar] [CrossRef] [Green Version]
- Bou-Abdallah, F.; Adinolfi, S.; Pastore, A.; Laue, T.M.; Dennis Chasteen, N. Iron binding and oxidation kinetics in frataxin CyaY of Escherichia coli. J. Mol. Biol. 2004, 341, 605–615. [Google Scholar] [CrossRef]
- Gentry, L.E.; Thacker, M.A.; Doughty, R.; Timkovich, R.; Busenlehner, L.S. His86 from the N-terminus of frataxin coordinates iron and is required for Fe-S cluster synthesis. Biochemistry 2013, 52, 6085–6096. [Google Scholar] [CrossRef]
- Adamec, J.; Rusnak, F.; Owen, W.G.; Naylor, S.; Benson, L.M.; Gacy, A.M.; Isaya, G. Iron-dependent self-assembly of recombinant yeast frataxin: Implications for Friedreich ataxia. Am. J. Hum. Genet. 2000, 67, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Cavadini, P.; O’Neill, H.A.; Benada, O.; Isaya, G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Schagerlöf, U.; Elmlund, H.; Gakh, O.; Nordlund, G.; Hebert, H.; Lindahl, M.; Isaya, G.; Al-Karadaghi, S. Structural basis of the iron storage function of frataxin from single-particle reconstruction of the iron-loaded oligomer. Biochemistry 2008, 47, 4948–4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Alam, S.L.; Proteasa, S.V.; Zhang, Y.; Lesuisse, E.; Dancis, A.; Stemmler, T.L. Yeast frataxin solution structure, iron binding, and ferrochelatase interaction. Biochemistry 2004, 43, 16254–16262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söderberg, C.; Gillam, M.E.; Ahlgren, E.C.; Hunter, G.A.; Gakh, O.; Isaya, G.; Ferreira, G.C.; Al-Karadaghi, S. The Structure of the Complex between Yeast Frataxin and Ferrochelatase: CHARACTERIZATION AND PRE-STEADY STATE REACTION OF FERROUS IRON DELIVERY AND HEME SYNTHESIS. J. Biol Chem 2016, 291, 11887–11898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesuisse, E.; Santos, R.; Matzanke, B.F.; Knight, S.A.; Camadro, J.M.; Dancis, A. Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum. Mol. Genet. 2003, 12, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Becker, E.M.; Whitnall, M.; Suryo Rahmanto, Y.; Ponka, P.; Richardson, D.R. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. USA 2009, 106, 16381–16386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinkellner, H.; Singh, H.N.; Muckenthaler, M.U.; Goldenberg, H.; Moganty, R.R.; Scheiber-Mojdehkar, B.; Sturm, B. No changes in heme synthesis in human Friedreich’s ataxia erythroid progenitor cells. Gene 2017, 621, 5–11. [Google Scholar] [CrossRef]
- Alsina, D.; Purroy, R.; Ros, J.; Tamarit, J. Iron in Friedreich Ataxia: A Central Role in the Pathophysiology or an Epiphenomenon? Pharmaceuticals 2018, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Prischi, F.; Pastore, A. Hybrid Methods in Iron-Sulfur Cluster Biogenesis. Front. Mol. Biosci. 2017, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Gerber, J.; Mühlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, S.; Iannuzzi, C.; Prischi, F.; Pastore, C.; Iametti, S.; Martin, S.R.; Bonomi, F.; Pastore, A. Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009, 16, 390–396. [Google Scholar] [CrossRef]
- Sanchez-Casis, G.; Cote, M.; Barbeau, A. Pathology of the heart in Friedreich’s ataxia: Review of the literature and report of one case. Can. J. Neurol Sci 1976, 3, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamarche, J.B.; Côté, M.; Lemieux, B. The cardiomyopathy of Friedreich’s ataxia morphological observations in 3 cases. Can. J. Neurol. Sci. 1980, 7, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilha da Silva, C.; Bergo, F.P.G.; D’Abreu, A.; Cendes, F.; Lopes-Cendes, I.; França, M.C., Jr. Dentate nuclei T2 relaxometry is a reliable neuroimaging marker in Friedreich’s ataxia. Eur. J. Neurol. 2014, 21, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue atrophy and elevated iron concentration in the extrapyramidal motor system in Friedreich ataxia: The IMAGE-FRDA study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, A.H.; Morral, J.A.; Davis, A.N.; Qian, J.; Petrocine, S.V.; Knutson, M.D.; Gibson, W.M.; Cusack, M.J.; Li, D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009, 118, 763–776. [Google Scholar] [CrossRef]
- Llorens, J.V.; Soriano, S.; Calap-Quintana, P.; Gonzalez-Cabo, P.; Moltó, M.D. The Role of Iron in Friedreich’s Ataxia: Insights From Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci. 2019, 13, 75. [Google Scholar] [CrossRef] [Green Version]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Whitnall, M.; Suryo Rahmanto, Y.; Huang, M.L.; Saletta, F.; Lok, H.C.; Gutiérrez, L.; Lázaro, F.J.; Fleming, A.J.; St Pierre, T.G.; Mikhael, M.R.; et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. USA 2012, 109, 20590–20595. [Google Scholar] [CrossRef] [Green Version]
- Calmels, N.; Schmucker, S.; Wattenhofer-Donzé, M.; Martelli, A.; Vaucamps, N.; Reutenauer, L.; Messaddeq, N.; Bouton, C.; Koenig, M.; Puccio, H. The first cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia. PLoS ONE 2009, 4, e6379. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Irazusta, V.; Moreno-Cermeño, A.; Cabiscol, E.; Ros, J.; Tamarit, J. Major targets of iron-induced protein oxidative damage in frataxin-deficient yeasts are magnesium-binding proteins. Free Radic. Biol. Med. 2008, 44, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Dancis, A.; Gareil, M.; Montagne, J.J.; Camadro, J.M.; Lesuisse, E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med. 2007, 42, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.R.; Kirby, K.; Orr, W.C.; Hilliker, A.J.; Phillips, J.P. Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich’s ataxia. Proc. Natl. Acad. Sci. USA 2008, 105, 611–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, S.; Llorens, J.V.; Blanco-Sobero, L.; Gutiérrez, L.; Calap-Quintana, P.; Morales, M.P.; Moltó, M.D.; Martínez-Sebastián, M.J. Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich’s ataxia. Gene 2013, 521, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Llorens, J.V.; Navarro, J.A.; Martínez-Sebastián, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.A.; Moltó, M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. Faseb J. 2007, 21, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.S.; Khdour, O.; Hecht, S.M. Does oxidative stress contribute to the pathology of Friedreich’s ataxia? A radical question. Faseb J. 2010, 24, 2152–2163. [Google Scholar] [CrossRef]
- Schulz, J.B.; Dehmer, T.; Schöls, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar] [CrossRef]
- Cotticelli, M.G.; Crabbe, A.M.; Wilson, R.B.; Shchepinov, M.S. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 2013, 1, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. ‘Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia’. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Uzun, E.; Renganathan, I.; Honda, T.; Pook, M.A.; Giunti, P. Targeting lipid peroxidation and mitochondrial imbalance in Friedreich’s ataxia. Pharmacol. Res. 2015, 99, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, P.; Petrillo, S.; Bertini, E.S.; Piemonte, F. Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement. Biomolecules 2020, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef] [Green Version]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Investig. 2001, 31, 1007–1011. [Google Scholar] [CrossRef]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [Green Version]
- Miranda, C.J.; Santos, M.M.; Ohshima, K.; Smith, J.; Li, L.; Bunting, M.; Cossée, M.; Koenig, M.; Sequeiros, J.; Kaplan, J.; et al. Frataxin knockin mouse. FEBS Lett. 2002, 512, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.; Seznec, H.; Gansmuller, A.; Carelle, N.; Weber, P.; Metzger, D.; Rustin, P.; Koenig, M.; Puccio, H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 2004, 24, 1987–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, R.M.; De Biase, I.; Malykhina, A.P.; Al-Mahdawi, S.; Pook, M.; Bidichandani, S.I. The GAA triplet-repeat is unstable in the context of the human FXN locus and displays age-dependent expansions in cerebellum and DRG in a transgenic mouse model. Hum. Genet. 2007, 120, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandolfo, M. Friedreich ataxia: New pathways. J. Child. Neurol. 2012, 27, 1204–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Rosa, P.; Petrillo, S.; Fiorenza, M.T.; Bertini, E.S.; Piemonte, F. Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease. Biomolecules 2020, 10, 1551. https://doi.org/10.3390/biom10111551
La Rosa P, Petrillo S, Fiorenza MT, Bertini ES, Piemonte F. Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease. Biomolecules. 2020; 10(11):1551. https://doi.org/10.3390/biom10111551
Chicago/Turabian StyleLa Rosa, Piergiorgio, Sara Petrillo, Maria Teresa Fiorenza, Enrico Silvio Bertini, and Fiorella Piemonte. 2020. "Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease" Biomolecules 10, no. 11: 1551. https://doi.org/10.3390/biom10111551
APA StyleLa Rosa, P., Petrillo, S., Fiorenza, M. T., Bertini, E. S., & Piemonte, F. (2020). Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease. Biomolecules, 10(11), 1551. https://doi.org/10.3390/biom10111551