The Influence of Physical Activity on the Bioactive Lipids Metabolism in Obesity-Induced Muscle Insulin Resistance


{kind=link}
{kind=link}
Abstract
:1. Introduction
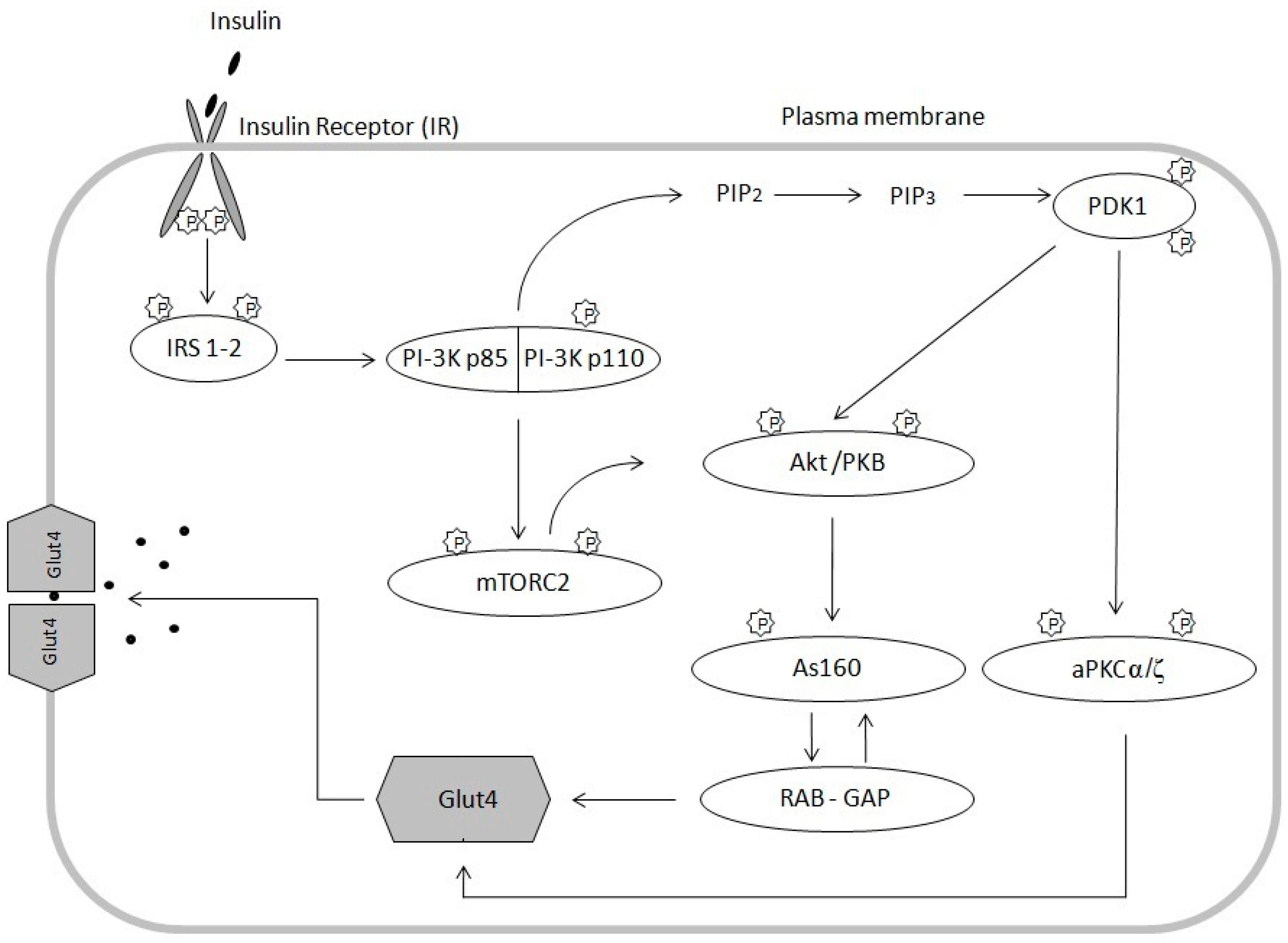
2. Insulin Signal Transduction Pathway and Mechanisms of Insulin Resistance
2.1. Insulin Signaling Pathway
2.2. Obesity as a Factor of IRes
2.3. Role of Ceramides in IRes
2.4. Role of Diacylglycerols in IR
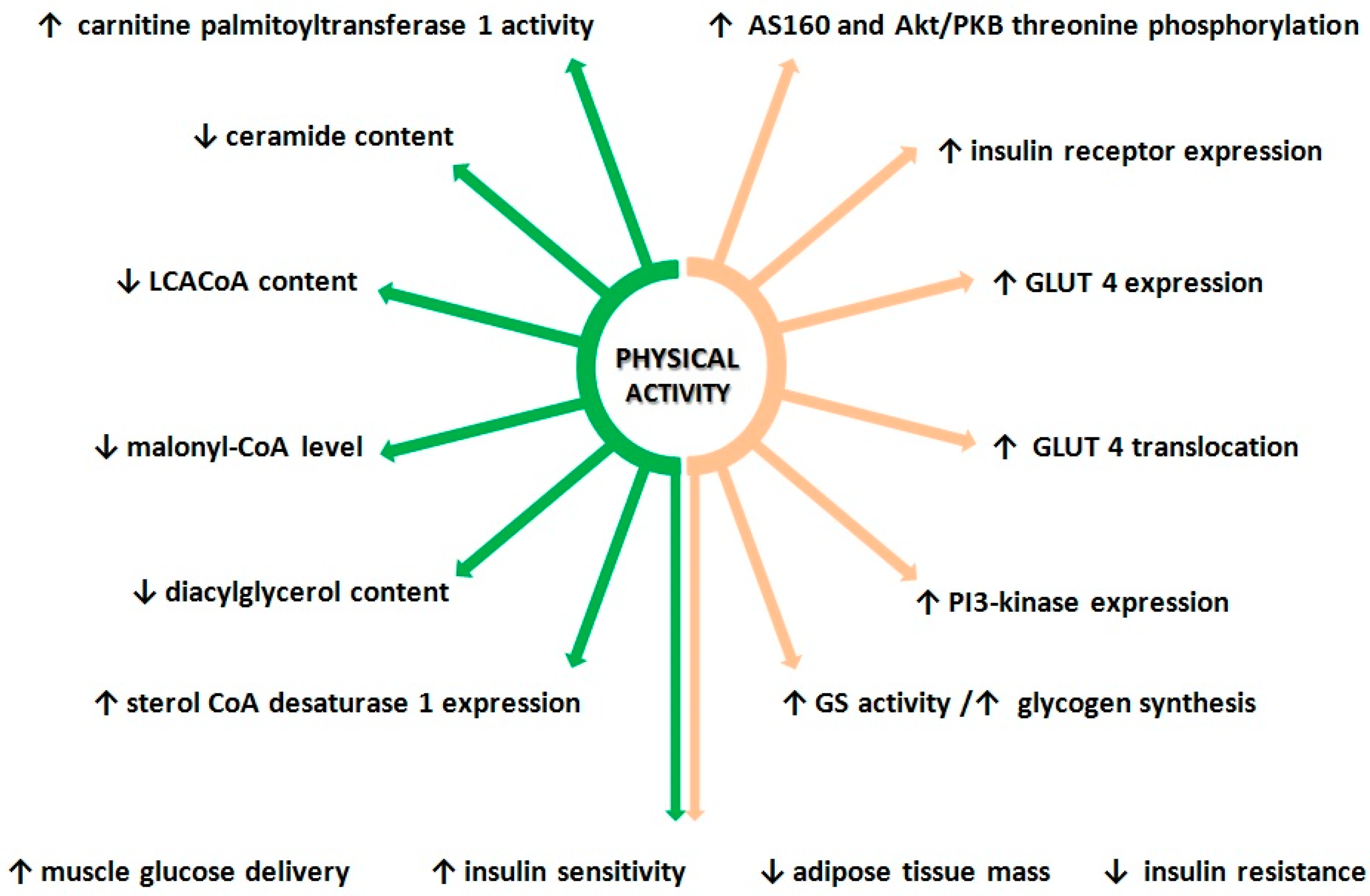
3. Effect of Exercise on Lipid Metabolism and Insulin Sensitivity
3.1. Effect of Physical Activity on Insulin Signaling Pathway
3.2. Physical Activity and Lipid Metabolism
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Garrow, J.S. Obesity and Related Diseases; Churchill Livingstone: Edimburgh, UK, 1988; pp. 1–16. [Google Scholar]
- Ofei, F. Obesity—A preventable disease. Ghana Med. J. 2005, 39, 98–101. [Google Scholar] [PubMed]
- Kelly, T.; Yang, W.; Chen, C.S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. 2008, 32, 1431–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Marin-Penalver, J.J.; Martin-Timon, I.; Sevillano-Collantes, C.; Del Canizo-Gomez, F.J. Update on the treatment of type 2 diabetes mellitus. World J. Diabetes 2016, 7, 354–395. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, Z.S.; Guo, C.S. New hope for type 2 diabetics: Targeting insulin resistance through the immune modulation of stem cells. Autoimmun. Rev. 2011, 11, 137–142. [Google Scholar] [CrossRef]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, Z.J. Current views on type 2 diabetes. J. Endocrinol. 2010, 204, 1–11. [Google Scholar] [CrossRef]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17. [Google Scholar] [CrossRef]
- Taylor, R. Insulin Resistance and Type 2 Diabetes. Diabetes 2012, 61, 778–779. [Google Scholar] [CrossRef] [Green Version]
- Batty, G.D.; Shipley, M.J.; Marmot, M.; Smith, G.D. Physical activity and cause-specific mortality in men with Type 2 diabetes/impaired glucose tolerance: Evidence from the Whitehall study. Diabet. Med. 2002, 19, 580–588. [Google Scholar] [CrossRef]
- Colberg, S.R.; Sigal, R.J.; Fernhall, B.; Regensteiner, J.G.; Blissmer, B.J.; Rubin, R.R.; Chasan-Taber, L.; Albright, A.L.; Braun, B. Exercise and Type 2 Diabetes The American College of Sports Medicine and the American Diabetes Association joint position statement executive summary. Diabetes Care 2010, 33, 2692–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or Hyperplasia: Dynamics of Adipose Tissue Growth. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorntorp, P.; Bergman, H.; Varnauskas, E. Plasma Free Fatty Acid Turnover Rate in Obesity. Acta Med. Scand. 1969, 185. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Newgard, C.B. Obesity-related derangements in metabolic regulation. Annu. Rev. Biochem. 2006, 75, 367–401. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J. Mechanisms of insulin resistance in obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmukh, A.S. Proteomics of Skeletal Muscle: Focus on Insulin Resistance and Exercise Biology. Proteomes 2016, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Defronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The Effect of Insulin on the Disposal of Intravenous Glucose—Results from Indirect Calorimetry and Hepatic and Femoral Venous Catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef]
- Furler, S.M.; Poynten, A.M.; Kriketos, A.D.; Lowy, A.J.; Ellis, B.A.; Maclean, E.L.; Courtenay, B.G.; Kraegen, E.W.; Campbell, L.V.; Chisholm, D.J. Independent influences of central fat and skeletal muscle lipids on insulin sensitivity. Obes. Res. 2001, 9, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.C.; Shillabeer, G.; Bar-Tana, J.; Lau, D.C.W.; Richardson, M.; Wenzel, L.M.; Graham, S.E.; Dolphin, P.J. Development of insulin resistance in the JCR: LA-cp rat—Role of triacylglycerols and effects of MEDICA 16. Diabetes 1998, 47, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Newsholme, E.A.; Hales, C.N. Glucose Fatty-Acid Cycle—Its Role in Insulin Sensitivity and Metabolic Disturbances of Diabetes Mellitus. Lancet 1963, 1, 785. [Google Scholar] [CrossRef]
- Boden, G.; Chen, X.H.; Ruiz, J.; White, J.V.; Rossetti, L. Mechanisms of Fatty Acid-Induced Inhibition of Glucose-Uptake. J. Clin. Investig. 1994, 93, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and I kappa B-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, G.; Jadali, F.; White, J.; Liang, Y.; Mozzoli, M.; Chen, X.; Coleman, E.; Smith, C. Effects of Fat on Insulin-Stimulated Carbohydrate-Metabolism in Normal Men. J. Clin. Investig. 1991, 88, 960–966. [Google Scholar] [CrossRef]
- Yu, C.L.; Chen, Y.; Cline, G.W.; Zhang, D.Y.; Zong, H.H.; Wang, Y.L.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [Green Version]
- Zeigerer, A.; Lampson, M.A.; Karylowski, O.; Sabatini, D.D.; Adesnik, M.; Ren, M.D.; McGraw, T.E. GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol. Biol. Cell 2002, 13, 2421–2435. [Google Scholar] [CrossRef] [Green Version]
- Kahn, C.R.; White, M.F. The Insulin-Receptor and the Molecular Mechanism of Insulin Action. J. Clin. Investig. 1988, 82, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, H.K.R.; Zierath, J.R. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem. Biophys. 2007, 48, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.M.; Hou, W.M.; Johnson, J.; Graham, L.K.; Lee, M.H.; Chen, C.S.; Newton, A.C.; Schaffhausen, B.S.; Toker, A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr. Biol. 1998, 8, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.H.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J. Biol. Chem. 2004, 279, 41189–41196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farese, R.V.; Sajan, M.P.; Standaert, M.L. Insulin-sensitive protein kinases (atypical protein kinase C and protein kinase B/Akt): Actions and defects in obesity and type II diabetes. Exp. Biol. Med. 2005, 230, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.Q.; Liu, F. PDK2: The missing piece in the receptor tyrosine kinase signaling pathway puzzle. Am. J. Physiol.-Endocrinol. Metab. 2005, 289, E187–E196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miinea, C.P.; Sano, H.; Kane, S.; Sano, E.; Fukuda, M.; Peranen, J.; Lane, W.S.; Lienhard, G.E. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem. J. 2005, 391, 87–93. [Google Scholar] [CrossRef]
- Cartee, G.D.; Wojtaszewski, J.F.P. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl. Physiol. Nutr. Metab. 2007, 32, 557–566. [Google Scholar] [CrossRef]
- Funaki, M.; Randhawa, P.; Janmey, P.A. Separation of insulin signaling into distinct GLUT4 translocation and activation steps. Mol. Cell. Biol. 2004, 24, 7567–7577. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D. Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M.; Newgard, C.B. Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Beattie, J.H. Physiological role of adipose tissue: White adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc. 2001, 60, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, J.B.; ORahilly, S. Regulation of adipose cell number in man. Clin. Sci. 1997, 92, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Kissebah, A.H.; Krakower, G.R. Regional adiposity and morbidity. Physiol. Rev. 1994, 74, 761–811. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, F.X. NHLBI Obesity Education Initiative Expert Panel on the identification, evaluation, and treatment of overweight and obesity in adults—The evidence report. Obes. Res. 1998, 51s–209s. [Google Scholar]
- Boden, G. Obesity and Free Fatty Acids. Endocrinol. Metab. Clin. 2008, 37, 635. [Google Scholar] [CrossRef] [Green Version]
- Petta, S.; Gastaldelli, A.; Rebelos, E.; Bugianesi, E.; Messa, P.; Miele, L.; Svegliati-Baroni, G.; Valenti, L.; Bonino, F. Pathophysiology of Non Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 2082. [Google Scholar] [CrossRef]
- Byrne, C.D. Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proc. Nutr. Soc. 2013, 72, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.T.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.H.; Brickey, W.J.; Ting, J.P.Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Alwahsh, S.M.; Dwyer, B.J.; Forbes, S.; van Thiel, D.H.; Lewis, P.J.S.; Ramadori, G. Insulin Production and Resistance in Different Models of Diet-Induced Obesity and Metabolic Syndrome. Int. J. Mol. Sci. 2017, 18, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, M.J.; Holmes, A.G.; Pinnamaneni, S.K.; Garnham, A.P.; Steinberg, G.R.; Kemp, B.E.; Febbraio, M.A. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am. J. Physiol.-Endocrinol. Metab. 2006, 290, E500–E508. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Stingl, H.; Chandramouli, V.; Schumann, W.C.; Hofer, A.; Landau, B.R.; Nowotny, P.; Waldhausl, W.; Shulman, G.I. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes 2000, 49, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samra, J.S.; Clark, M.L.; Humphreys, S.M.; MacDonald, I.A.; Bannister, P.A.; Frayn, K.N. Effects of physiological hypercortisolemia on the regulation of lipolysis in subcutaneous adipose tissue. J. Clin. Endocrinol. Metab. 1998, 83, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Engfeldt, P.; Hellmer, J.; Wahrenberg, H.; Arner, P. Effects of Insulin on Adrenoceptor Binding and the Rate of Catecholamine-Induced Lipolysis in Isolated Human Fat-Cells. J. Biol. Chem. 1988, 263, 15553–15560. [Google Scholar]
- Lan, Y.L.; Lou, J.C.; Lyu, W.; Zhang, B. Update on the synergistic effect of HSL and insulin in the treatment of metabolic disorders. Ther. Adv. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Wang, S.P.; Laurin, N.; Himms-Hagen, J.; Rudnicki, M.A.; Levy, E.; Robert, M.F.; Pan, L.; Oligny, L.; Mitchell, G.A. The adipose tissue phenotype of hormone-sensitive lipase deficiency in mice. Obes. Res. 2001, 9, 119–128. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Hamm, J.K.; Verhagen, L.A.; Peroni, O.; Katic, M.; Flier, J.S. Adipose triglyceride lipase: Function, regulation by insulin, and comparison with adiponutrin. Diabetes 2006, 55, 148–157. [Google Scholar] [CrossRef]
- Bray, G.A.; Champagne, C.M. Obesity and the Metabolic Syndrome: Implications for dietetics practitioners. J. Am. Dietet. Assoc. 2004, 104, 86–89. [Google Scholar] [CrossRef]
- Trujillo, M.E.; Scherer, P.E. Adipose tissue-derived factors: Impact on health and disease. Endocrinol. Rev. 2006, 27, 762–778. [Google Scholar] [CrossRef] [Green Version]
- Clement, K. Leptin and the genetics of obesity. Acta Paediatr. 1999, 88, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Wasim, M.; Awan, F.R.; Najam, S.S.; Khan, A.R.; Khan, H.N. Role of Leptin Deficiency, Inefficiency, and Leptin Receptors in Obesity. Biochem. Genet. 2016, 54, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef]
- Kanasaki, K.; Koya, D. Biology of Obesity: Lessons from Animal Models of Obesity. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Lobo, A.M.; Donato, J., Jr. The role of leptin in health and disease. Temperature 2017, 4, 258–291. [Google Scholar] [CrossRef] [Green Version]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Combs, T.P.; Berg, A.H.; Obici, S.; Scherer, P.E.; Rossetti, L. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J. Clin. Investig. 2001, 108, 1875–1881. [Google Scholar] [CrossRef]
- Sethi, J.K.; Vidal-Puig, A.J. Thematic review series: Adipocyte biology—Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 2007, 48, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Stefan, N.; Vozarova, B.; Funahashi, T.; Matsuzawa, Y.; Weyer, C.; Lindsay, R.S.; Youngren, J.F.; Havel, P.J.; Pratley, R.E.; Bogardus, C.; et al. Plasma Adiponectin Concentration Is Associated With Skeletal Muscle Insulin Receptor Tyrosine Phosphorylation, and Low Plasma Concentration Precedes a Decrease in Whole-Body Insulin Sensitivity in Humans. Diabetes 2002, 51, 1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppan, C.M.; Lazar, M.A. Resistin and obesity-associated insulin resistance. Trends Endocrinol. Met. 2002, 13, 18–23. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Zhou, L.; Sell, H.; Eckardt, K.; Yang, Z.Q.; Eckel, Y. Conditioned medium obtained from in vitro differentiated adipocytes and resistin induce insulin resistance in human hepatocytes. FEBS Lett. 2007, 581, 4303–4308. [Google Scholar] [CrossRef] [Green Version]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Sethi, J.K.; Hotamisligil, G.S. The role of TNF alpha in adipocyte metabolism. Semin. Cell Dev. Biol. 1999, 10, 19–29. [Google Scholar] [CrossRef]
- Feinstein, R.; Kanety, H.; Papa, M.Z.; Lunenfeld, B.; Karasik, A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J. Biol. Chem. 1993, 268, 26055–26058. [Google Scholar]
- Hotamisligil, G.S.; Budavari, A.; Murray, D.; Spiegelman, B.M. Reduced Tyrosine Kinase-Activity of the Insulin-Receptor in Obesity-Diabetes—Central Role of Tumor-Necrosis-Factor-Alpha. J. Clin. Investig. 1994, 94, 1543–1549. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, D.E. Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol. Metab. 2000, 11, 212–217. [Google Scholar] [CrossRef]
- Hostamisligil, G.S. Mechanisms of TNF-alpha-induced insulin resistance. Exp. Clin. Endocrinol. Diabetes 1999, 107, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Boden, G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997, 46, 3–10. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: Depot difference and regulation by glucocorticoid. J. Clin. Endocrinol. Metab. 1998, 83, 847–850. [Google Scholar] [CrossRef]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar]
- Fruhbeck, G.; Gomez-Ambrosi, J.; Muruzabal, F.J.; Burrell, M.A. The adipocyte: A model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am. J. Physiol.-Endocrinol. Metab. 2001, 280, E827–E847. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine 1-phosphate, a key cell signaling molecule. J. Biol. Chem. 2002, 277, 25851–25854. [Google Scholar] [CrossRef] [Green Version]
- Mencarelli, C.; Martinez-Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaurasia, B.; Summers, S.A. Ceramides—Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati, F. Revisiting the diacylglycerol-induced insulin resistance hypothesis. Obes. Rev. 2012, 13, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Progr. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef]
- Summers, S.A.; Garza, L.A.; Zhou, H.; Birnbaum, M.J. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol. Cell. Biol. 1998, 18, 5457–5464. [Google Scholar] [CrossRef] [Green Version]
- Stratford, S.; DeWald, D.B.; Summers, S.A. Ceramide dissociates 3 ’-phosphoinositide production from pleckstrin homology domain translocation. Biochem. J. 2001, 354, 359–368. [Google Scholar] [CrossRef]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of insulin action by ceramide—Dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef] [Green Version]
- Hajduch, E.; Balendran, A.; Batty, I.H.; Litherland, G.J.; Blair, A.S.; Downes, C.P.; Hundal, H.S. Ceramide impairs the insulin-dependent membrane recruitment of Protein Kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia 2001, 44, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Resjo, S.; Goransson, O.; Harndahl, L.; Zolnierowicz, S.; Manganiello, V.; Degerman, E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal 2002, 14, 231–238. [Google Scholar] [CrossRef]
- Cortright, R.N.; Azevedo, J.L.; Zhou, Q.; Sinha, M.; Pories, W.J.; Itani, S.I.; Dohm, G.L. Protein kinase C modulates insulin action in human skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2000, 278, E553–E562. [Google Scholar] [CrossRef] [PubMed]
- Bourbon, N.A.; Sandirasegarane, L.; Kester, M. Ceramide-induced inhibition of Akt is mediated through protein kinase C zeta—Implications for growth arrest. J. Biol. Chem. 2002, 277, 3286–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farese, R.V. Function and dysfunction of aPKC isoforms for glucose transport in insulin-sensitive and insulin-resistant states. Am. J. Physiol.-Endocrinol. Metab. 2002, 283, E1–E11. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.; Hajduch, E.; Kular, G.; Hundal, H.S. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKC zeta-dependent mechanism. Mol. Cell. Biol. 2003, 23, 7794–7808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doornbos, R.P.; Theelen, M.; van der Hoeven, P.C.J.; van Blitterswijk, W.J.; Verkleij, A.J.; Henegouwen, P.M.P.V.E. Protein kinase C zeta is a negative regulator of protein kinase B activity. J. Biol. Chem. 1999, 274, 8589–8596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajduch, E.; Turban, S.; Le Liepvre, X.; Le Lay, S.; Lipina, C.; Dimopoulos, N.; Dugail, I.; Hundal, H.S. Targeting of PKC zeta and PKB to caveolin-enriched microdomains represents a crucial step underpinning the disruption in PKB-directed signalling by ceramide. Biochem. J. 2008, 410, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, T.C.A.; Graciano, M.F.R.; Anhe, G.F.; Curi, R.; Bordin, S.; Carpinelli, A.R. Short-Term Modulation of Extracellular Signal-Regulated Kinase 1/2 and Stress-Activated Protein Kinase/c-Jun NH2-Terminal Kinase in Pancreatic Islets by Glucose and Palmitate Possible Involvement of Ceramide. Pancreas 2009, 38, 585–592. [Google Scholar] [CrossRef]
- Hassan, R.H.; de Sousa, A.C.P.; Mahfouz, R.; Hainault, I.; Blachnio-Zabielska, A.; Bourron, O.; Koskas, F.; Gorski, J.; Ferre, P.; Foufelle, F.; et al. Sustained Action of Ceramide on the Insulin Signaling Pathway in Muscle Cells IMPLICATION OF THE DOUBLE-STRANDED RNA-ACTIVATED PROTEIN KINASE. J. Biol. Chem. 2016, 291, 3019–3029. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, P.P. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol. Res. 2003, 47, 383–392. [Google Scholar] [CrossRef]
- Daniluk, U.; Alifier, M.; Kaczmarski, M.; Stasiak-Barmuta, A.; Lebensztejn, D. Longitudinal observation of children with enhanced total serum IgE. Ann. Allergy Asthma Immunol. 2015, 114, 404–410. [Google Scholar] [CrossRef]
- Gual, P.; Le Marchand-Brustel, Y.; Tanti, J.F. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 2005, 87, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kitessa, S.M.; Abeywardena, M.Y. Lipid-Induced Insulin Resistance in Skeletal Muscle: The Chase for the Culprit Goes from Total Intramuscular Fat to Lipid Intermediates, and Finally to Species of Lipid Intermediates. Nutrients 2016, 8, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, R.H. Lipotoxicity in the Pathogenesis of Obesity-Dependent Niddm—Genetic and Clinical Implications. Diabetes 1995, 44, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Csh. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Erion, D.M.; Shulman, G.I. Diacylglycerol-mediated insulin resistance. Nat. Med. 2010, 16, 400–402. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.Y.; Jelenik, T.; Muller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKC theta in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [Green Version]
- Morino, K.; Petersen, K.F.; Shulman, G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006, 55 (Suppl. 2), S9–S15. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Alkhawam, H.; Madanieh, R.; Shah, N.; Kosmas, C.E.; Vittorio, T.J. Aerobic vs. anaerobic exercise training effects on the cardiovascular system. World J. Cardiol. 2017, 9, 134–138. [Google Scholar] [CrossRef]
- Wahid, A.; Manek, N.; Nichols, M.; Kelly, P.; Foster, C.; Webster, P.; Kaur, A.; Smith, C.F.; Wilkins, E.; Rayner, M.; et al. Quantifying the Association Between Physical Activity and Cardiovascular Disease and Diabetes: A Systematic Review and Meta-Analysis. J. Am. Heart. Assoc. 2016, 5, e002495. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Atkin, S.L.; Simental-Mendia, L.E.; Sahebkar, A. Molecular mechanisms by which aerobic exercise induces insulin sensitivity. J. Cell Physiol. 2019, 234, 12385–12392. [Google Scholar] [CrossRef]
- Hall, K.E.; McDonald, M.W.; Grise, K.N.; Campos, O.A.; Noble, E.G.; Melling, C.W.J. The role of resistance and aerobic exercise training on insulin sensitivity measures in STZ-induced Type 1 diabetic rodents. Metabolism 2013, 62, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, C.; Layne, J.E.; Munoz-Orians, L.; Gordon, P.L.; Walsmith, J.; Foldvari, M.; Roubenoff, R.; Tucker, K.L.; Nelson, M.E. A Randomized controlled trial of resistance exercise training to improve glycemic control in older adults with type 2 diabetes. Diabetes Care 2002, 25, 2335–2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaspelkis, B.B.; Singh, M.K.; Trevino, B.; Krisan, A.D.; Collins, D.E. Resistance training increases glucose uptake and transport in rat skeletal muscle. Acta Physiol. Scand. 2002, 175, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Gibala, M.J.; Little, J.P.; Macdonald, M.J.; Hawley, J.A. Physiological adaptations to low-volume, high-intensity interval training in health and disease. J. Physiol. 2012, 590, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Wisløff, U.; Ellingsen, Ø.; Kemi, O.J. High-Intensity Interval Training to Maximize Cardiac Benefits of Exercise Training? Exerc. Sport Sci. Rev. 2009, 37, 139–146. [Google Scholar]
- Kessler, H.S.; Sisson, S.B.; Short, K.R. The potential for high-intensity interval training to reduce cardiometabolic disease risk. Sports Med. 2012, 42, 489–509. [Google Scholar] [CrossRef]
- Gallo-Villegas, J.; Aristizabal, J.C.; Estrada, M.; Valbuena, L.H.; Narvaez-Sanchez, R.; Osorio, J.; Aguirre-Acevedo, D.C.; Calderón, J.C. Efficacy of high-intensity, low-volume interval training compared to continuous aerobic training on insulin resistance, skeletal muscle structure and function in adults with metabolic syndrome: Study protocol for a randomized controlled clinical trial (Intraining-MET). Trials 2018, 19, 144. [Google Scholar]
- Jelleyman, C.; Yates, T.; O’Donovan, G.; Gray, L.J.; King, J.A.; Khunti, K.; Davies, M.J. The effects of high-intensity interval training on glucose regulation and insulin resistance: A meta-analysis. Obes. Rev. 2015, 16, 942–961. [Google Scholar] [CrossRef] [Green Version]
- de Matos, M.A.; Vieira, D.V.; Pinhal, K.C.; Lopes, J.F.; Dias-Peixoto, M.F.; Pauli, J.R.; de Castro Magalhães, F.; Little, J.P.; Rocha-Vieira, E.; Amorim, F.T. High-Intensity Interval Training Improves Markers of Oxidative Metabolism in Skeletal Muscle of Individuals With Obesity and Insulin Resistance. Front. Physiol. 2018, 9, 1451. [Google Scholar] [CrossRef] [Green Version]
- Little, J.P.; Gillen, J.B.; Percival, M.E.; Safdar, A.; Tarnopolsky, M.A.; Punthakee, Z.; Jung, M.E.; Gibala, M.J. Low-volume high-intensity interval training reduces hyperglycemia and increases muscle mitochondrial capacity in patients with type 2 diabetes. J. Appl. Physiol. 2011, 111, 1554–1560. [Google Scholar] [CrossRef] [Green Version]
- Little, J.P.; Safdar, A.; Bishop, D.; Tarnopolsky, M.A.; Gibala, M.J. An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1α and activates mitochondrial biogenesis in human skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2011, 300, R1303–R1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgomaster, K.A.; Howarth, K.R.; Phillips, S.M.; Rakobowchuk, M.; Macdonald, M.J.; McGee, S.L.; Gibala, M.J. Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J. Physiol. 2008, 586, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.J.; Richter, E.A. Skeletal muscle glucose uptake during exercise: How is it regulated? Physiology 2005, 20, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtaszewski, J.F.P.; Richter, E.A. Effects of acute exercise and training on insulin action and sensitivity: Focus on molecular mechanisms in muscle. Essays Biochem. 2006, 42, 31–46. [Google Scholar]
- Blachnio-Zabielska, A.; Zabielski, P.; Baranowski, M.; Gorski, J. Aerobic Training in Rats Increases Skeletal Muscle Sphingomyelinase and Serine Palmitoyltransferase Activity, While Decreasing Ceramidase Activity. Lipids 2011, 46, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Caponi, P.W.; Lehnen, A.M.; Pinto, G.H.; Borges, J.; Markoski, M.; Machado, U.F.; Schaan, B.D. Aerobic exercise training induces metabolic benefits in rats with metabolic syndrome independent of dietary changes. Clinics 2013, 68, 1010–1017. [Google Scholar] [CrossRef]
- Nassis, G.P.; Papantakou, K.; Skenderi, K.; Triandafillopoulou, M.; Kavouras, S.A.; Yannakoulia, M.; Chrousos, G.P.; Sidossis, L.S. Aerobic exercise training improves insulin sensitivity without changes in body weight, body fat, adiponectin, and inflammatory markers in overweight and obese girls. Metabolism 2005, 54, 1472–1479. [Google Scholar] [CrossRef]
- Goulet, E.D.; Melancon, M.O.; Aubertin-Leheudre, M.; Dionne, I.J. Aerobic training improves insulin sensitivity 72-120 h after the last exercise session in younger but not in older women. Eur. J. Appl. Physiol. 2005, 95, 146–152. [Google Scholar] [CrossRef]
- Ivey, F.M.; Ryan, A.S.; Hafer-Macko, C.E.; Goldberg, A.P.; Macko, R.F. Treadmill aerobic training improves glucose tolerance and indices of insulin sensitivity in disabled stroke survivors: A preliminary report. Stroke 2007, 38, 2752–2758. [Google Scholar] [CrossRef] [Green Version]
- Winnick, J.J.; Sherman, W.M.; Habash, D.L.; Stout, M.B.; Failla, M.L.; Belury, M.A.; Schuster, D.P. Short-term aerobic exercise training in obese humans with type 2 diabetes mellitus improves whole-body insulin sensitivity through gains in peripheral, not hepatic insulin sensitivity. J. Clin. Endocrinol. Metab. 2008, 93, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Chibalin, A.V.; Yu, M.; Ryder, J.W.; Song, X.M.; Galuska, D.; Krook, A.; Wallberg-Henriksson, H.; Zierath, J.R. Exercise-induced changes in expression and activity of proteins involved in insulin signal transduction in skeletal muscle: Differential effects on insulin-receptor substrates 1 and 2. Proc. Natl. Acad. Sci. USA 2000, 97, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, A.S.R.; Pauli, J.R.; Ropelle, E.R.; Oliveira, A.G.; Cintra, D.E.; De Souza, C.T.; Velloso, L.A.; Carvalheira, J.B.C.; Saad, M.J.A. Exercise Intensity, Inflammatory Signaling, and Insulin Resistance in Obese Rats. Med. Sci. Sport Exerc. 2010, 42, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Inoue, T.; Nakajima, R.; Shirai-Morishita, Y.; Tokuyama, K.; Suzuki, M. Effect of long-term exercise on gene expression of insulin signaling pathway intermediates in skeletal muscle. Biochem. Biophys. Res. Commun. 1999, 254, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.E.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of Glycogen-Synthase Kinase-3 by Insulin-Mediated by Protein-Kinase-B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Lauritzen, H.P.M.M.; Galbo, H.; Toyoda, T.; Goodyear, L.J. Kinetics of Contraction-Induced GLUT4 Translocation in Skeletal Muscle Fibers From Living Mice. Diabetes 2010, 59, 2134–2144. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.W.; Hirshman, M.F.; Gervino, E.V.; Ocel, J.V.; Forse, R.A.; Hoenig, S.J.; Aronson, D.; Goodyear, L.J.; Horton, E.S. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes 1999, 48, 1192–1197. [Google Scholar] [CrossRef]
- Richter, E.A.; Hargreaves, M. Exercise, Glut4, and Skeletal Muscle Glucose Uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [Green Version]
- Sylow, L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Exercise-stimulated glucose uptake—Regulation and implications for glycaemic control. Nat. Rev. Endocrinol. 2017, 13, 133–148. [Google Scholar] [CrossRef]
- Daniluk, U.; Alifier, M.; Kaczmarski, M. Probiotic-induced apoptosis and its potential relevance to mucosal inflammation of gastrointestinal tract. Adv. Med. Sci 2012, 57, 175–182. [Google Scholar] [CrossRef]
- Friedman, J.E.; Sherman, W.M.; Reed, M.J.; Elton, C.W.; Dohm, G.L. Exercise training increases glucose transporter protein GLUT-4 in skeletal muscle of obese Zucker (fa/fa) rats. FEBS Lett. 1990, 268, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Hughes, V.A.; Fiatarone, M.A.; Fielding, R.A.; Kahn, B.B.; Ferrara, C.M.; Shepherd, P.; Fisher, E.C.; Wolfe, R.R.; Elahi, D.; Evans, W.J. Exercise Increases Muscle Glut-4 Levels and Insulin Action in Subjects with Impaired Glucose-Tolerance. Am J. Physiol. 1993, 264, E855–E862. [Google Scholar] [CrossRef] [PubMed]
- Filimoniuk, A.; Daniluk, U.; Samczuk, P.; Wasilewska, N.; Jakimiec, P.; Kucharska, M.; Lebensztejn, D.M.; Ciborowski, M. Metabolomic profiling in children with inflammatory bowel disease. Adv. Med. Sci 2020, 65, 65–70. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; Hargreaves, M. Exercise and skeletal muscle glucose transporter 4 expression: Molecular mechanisms. Clin. Exp. Pharmacol. Physiol. 2006, 33, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Funai, K.; Schweitzer, G.G.; Sharma, N.; Kanzaki, M.; Cartee, G.D. Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E242–E251. [Google Scholar] [CrossRef] [Green Version]
- Frosig, C.; Rose, A.J.; Treebak, J.T.; Kiens, B.; Richter, E.A.; Wojtaszewski, J.F.P. Effects of endurance exercise training on insulin signaling in human skeletal muscle—Interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes 2007, 56, 2093–2102. [Google Scholar] [CrossRef]
- Christ-Roberts, C.Y.; Pratipanawatr, T.; Pratipanawatr, W.; Berria, R.; Belfort, R.; Kashyap, S.; Mandarino, L.J. Exercise training increases glycogen synthase activity and GLUT4 expression but not insulin signaling in overweight nondiabetic and type 2 diabetic subjects. Metabolism 2004, 53, 1233–1242. [Google Scholar] [CrossRef]
- Nielsen, J.N.; Wojtaszewski, J.F. Regulation of glycogen synthase activity and phosphorylation by exercise. Proc. Nutr. Soc. 2004, 63, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Tabata, I.; Suzuki, Y.; Fukunaga, T.; Yokozeki, T.; Akima, H.; Funato, K. Resistance training affects GLUT-4 content in skeletal muscle of humans after 19 days of head-down bed rest. J. Appl. Physiol. 1999, 86, 909–914. [Google Scholar] [CrossRef]
- Ahmadizad, S.; Haghighi, A.H.; Hamedinia, M.R. Effects of resistance versus endurance training on serum adiponectin and insulin resistance index. Eur. J. Endocrinol. 2007, 157, 625–631. [Google Scholar] [CrossRef]
- Ross, R.; Bradshaw, A.J. The future of obesity reduction: Beyond weight loss. Nat. Rev. Endocrinol. 2009, 5, 319–326. [Google Scholar] [CrossRef]
- Mourier, A.; Gautier, J.F.; DeKerviler, E.; Bigard, A.X.; Villette, J.M.; Garnier, J.P.; Duvallet, A.; Guezennec, C.Y.; Cathelineau, G. Mobilization of visceral adipose tissue related to the improvement in insulin sensitivity in response to physical training in NIDDM—Effects of branched-chain amino acid supplements. Diabetes Care 1997, 20, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, E.; Negri, C.; Zanolin, M.E.; Milanese, C.; Faccioli, N.; Trombetta, M.; Zoppini, G.; Cevese, A.; Bonadonna, R.C.; Schena, F.; et al. Metabolic Effects of Aerobic Training and Resistance Training in Type 2 Diabetic Subjects A randomized controlled trial (the RAED2 study). Diabetes Care 2012, 35, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karstoft, K.; Pedersen, B.K. Exercise and type 2 diabetes: Focus on metabolism and inflammation. Immunol. Cell Biol. 2016, 94, 146–150. [Google Scholar] [CrossRef]
- Ziccardi, P.; Nappo, F.; Giugliano, G.; Esposito, K.; Marfella, R.; Cioffi, M.; D’Andrea, F.; Molinari, A.M.; Giugliano, D. Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation 2002, 105, 804–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandona, P.; Weinstock, R.; Thusu, K.; Abdel-Rahman, E.; Aljada, A.; Wadden, T. Tumor necrosis factor-alpha in sera of obese patients: Fall with weight loss. J. Clin. Endocrinol. Metab. 1998, 83, 2907–2910. [Google Scholar] [PubMed] [Green Version]
- Ryan, A.S.; Ge, S.; Blumenthal, J.B.; Serra, M.C.; Prior, S.J.; Goldberg, A.P. Aerobic Exercise and Weight Loss Reduce Vascular Markers of Inflammation and Improve Insulin Sensitivity in Obese Women. J. Am. Geriatr. Soc. 2014, 62, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, A.S.; Nicklas, B.J. Reductions in plasma cytokine levels with weight loss improve insulin sensitivity in overweight and obese postmenopausal women. Diabetes Care 2004, 27, 1699–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milan, G.; Granzotto, M.; Scarda, A.; Calcagno, A.; Pagano, C.; Federspil, G.; Vettor, R. Resistin and adiponectin expression in visceral fat of obese rats: Effect of weight loss. Obes. Res. 2002, 10, 1095–1103. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.; Park, K.S. Responses of inflammatory cytokines following moderate intensity walking exercise in overweight or obese individuals. J. Exerc. Rehabil. 2017, 13, 472–476. [Google Scholar] [CrossRef]
- Dube, J.J.; Amati, F.; Stefanovic-Racic, M.; Toledo, F.G.S.; Sauers, S.E.; Goodpaster, B.H. Exercise-induced alterations in intramyocellular lipids and insulin resistance: The athlete’s paradox revisited. Am. J. Physiol.-Endocrinol. Metab. 2008, 294, E882–E888. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Horowitz, J.F. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J. Clin. Investig. 2007, 117, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- van Loon, L.J.C.; Goodpaster, B.H. Increased intramuscular lipid storage in the insulin-resistant and endurance-trained state. Pflug. Arch. Eur. J. Physiol. 2006, 451, 606–616. [Google Scholar] [CrossRef]
- Oakes, N.D.; Bell, K.S.; Furler, S.M.; Camilleri, S.; Saha, A.K.; Ruderman, N.B.; Chisholm, D.J.; Kraegen, E.W. Diet-induced muscle insulin resistance in rats is ameliorated by acute dietary lipid withdrawal or a single bout of exercise—Parallel relationship between insulin stimulation of glucose uptake and suppression of long-chain fatty acyl-CoA. Diabetes 1997, 46, 2022–2028. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.K.; Ruderman, N.B. Malonyl-CoA and AMP-activated protein kinase: An expanding partnership. Mol. Cell. Biochem. 2003, 253, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Blachnio-Zabielska, A.; Baranowski, M.; Zabielski, P.; Gorski, J. Effect of exercise duration on the key pathways of ceramide metabolism in rat skeletal muscles. J. Cell Biochem. 2008, 105, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Bergman, B.C.; Brozinick, J.T.; Strauss, A.; Bacon, S.; Kerege, A.; Bui, H.H.; Sanders, P.; Siddall, P.; Wei, T.; Thomas, M.K.; et al. Muscle sphingolipids during rest and exercise: A C18:0 signature for insulin resistance in humans. Diabetologia 2016, 59, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Bruce, C.R.; Thrush, A.B.; Mertz, V.A.; Bezaire, V.; Chabowski, A.; Heigenhauser, G.J.F.; Dyck, D.J. Endurance training in obese humans improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid content. Am. J. Physiol.-Endocrinol. Metab. 2006, 291, E99–E107. [Google Scholar] [CrossRef] [Green Version]
- Stephens, T.J.; Chen, Z.P.; Canny, B.J.; Michell, B.J.; Kemp, B.E.; McConell, G.K. Progressive increase in human skeletal muscle AMPK alpha 2 activity and ACC phosphorylation during exercise. Am. J. Physiol.-Endocrinol. Metab. 2002, 282, E688–E694. [Google Scholar]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Hagberg, J.M.; Hurley, B.F.; Ehsani, A.A.; Holloszy, J.O. Effects of endurance training on glucose tolerance and plasma lipid levels in older men and women. JAMA 1984, 252, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.S. Insulin Resistance with Aging. Sports Med. 2000, 30, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Keshel, T.E.; Coker, R.H. Exercise Training and Insulin Resistance: A Current Review. J. Obes. Weight Loss Ther. 2015, 5, S5. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Rizza, R.A.; Coenen-Schimke, J.M.; Nair, K.S. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 2003, 52, 1888–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.H.; Cortright, R.N.; Dohm, G.L.; Houmard, J.A. Effect of aging on response to exercise training in humans: Skeletal muscle GLUT-4 and insulin sensitivity. J. Appl. Physiol. 1999, 86, 2019–2025. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imierska, M.; Kurianiuk, A.; Błachnio-Zabielska, A. The Influence of Physical Activity on the Bioactive Lipids Metabolism in Obesity-Induced Muscle Insulin Resistance. Biomolecules 2020, 10, 1665. https://doi.org/10.3390/biom10121665
Imierska M, Kurianiuk A, Błachnio-Zabielska A. The Influence of Physical Activity on the Bioactive Lipids Metabolism in Obesity-Induced Muscle Insulin Resistance. Biomolecules. 2020; 10(12):1665. https://doi.org/10.3390/biom10121665
Chicago/Turabian StyleImierska, Monika, Adam Kurianiuk, and Agnieszka Błachnio-Zabielska. 2020. "The Influence of Physical Activity on the Bioactive Lipids Metabolism in Obesity-Induced Muscle Insulin Resistance" Biomolecules 10, no. 12: 1665. https://doi.org/10.3390/biom10121665
APA StyleImierska, M., Kurianiuk, A., & Błachnio-Zabielska, A. (2020). The Influence of Physical Activity on the Bioactive Lipids Metabolism in Obesity-Induced Muscle Insulin Resistance. Biomolecules, 10(12), 1665. https://doi.org/10.3390/biom10121665