The Mitochondrial Lon Protease: Novel Functions off the Beaten Track?

{kind=link}
{kind=link}
Abstract
:1. Introduction
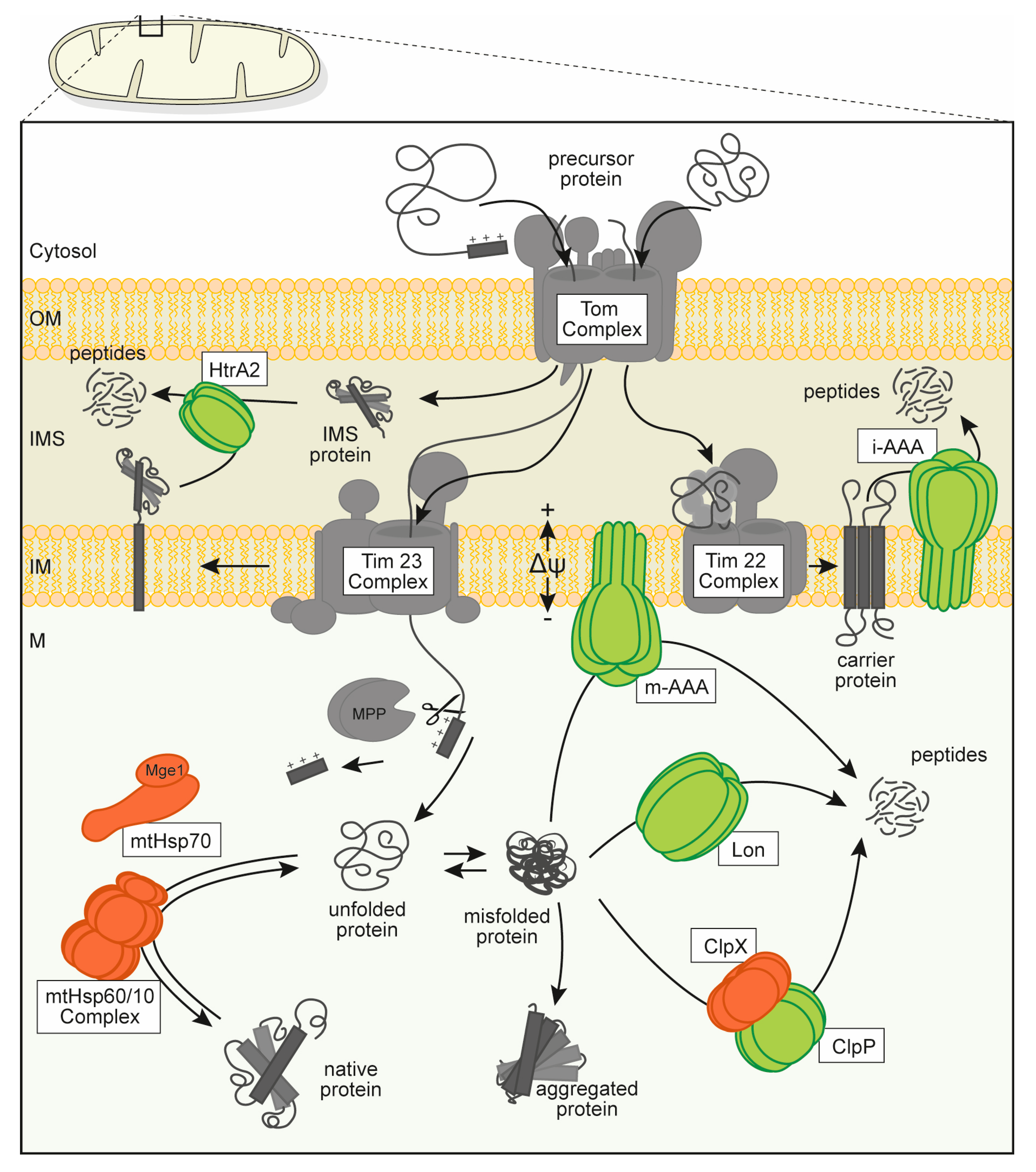
2. Lon Function in Mitochondrial PQC
2.1. Proteolysis Properties
2.2. Protection against Oxidative Stress
2.3. Prevention of Aggregation
2.4. Role in Gene Expression
3. Involvement in Metazoan Cellular Stress Protection
3.1. Stress Induced Upregulation of Lon
3.2. Role during ER Stress
3.3. Role in Aging
3.4. Role during Cancer
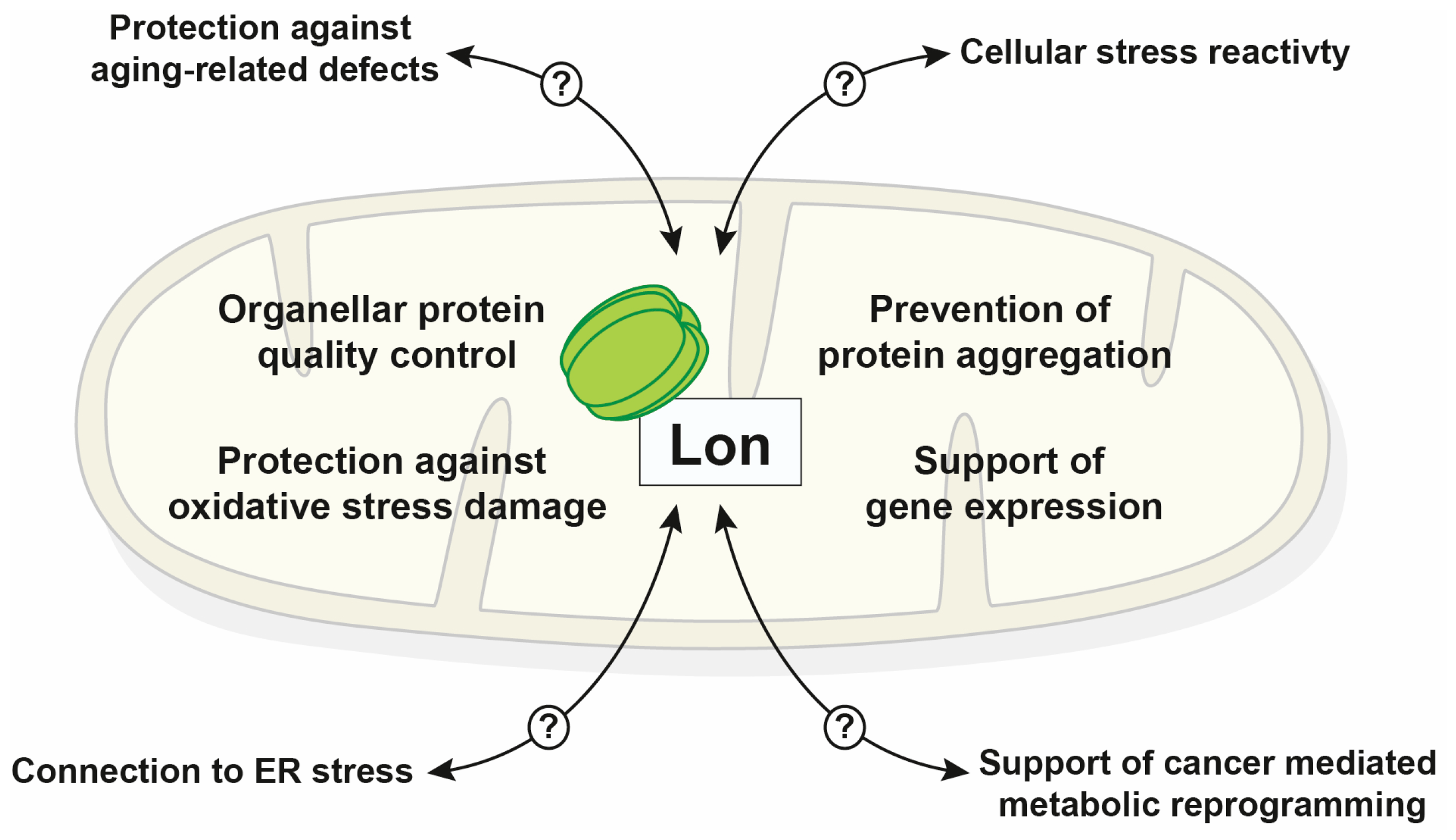
4. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Hanson, P.I.; Whiteheart, S.W. AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef]
- Pickart, C.M.; Cohen, R.E. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 2004, 5, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Langer, T.; Lopez-Otin, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Voos, W.; Röttgers, K. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim. Biophys. Acta 2002, 1592, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [Green Version]
- Voos, W.; Jaworek, W.; Wilkening, A.; Bruderek, M. Protein quality control at the mitochondrion. Essays Biochem. 2016, 60, 213–225. [Google Scholar]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria retrograde signaling and the UPR mt: Where are we in mammals? Int. J. Mol. Sci. 2015, 16, 18224–188251. [Google Scholar] [CrossRef] [Green Version]
- Morishita, H.; Mizushima, N. Diverse cellular roles of autophagy. Annu. Rev. Cell. Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Olson, M.; Kornbluth, S. Mitochondria in apoptosis and human disease. Curr. Mol. Med. 2001, 1, 91–122. [Google Scholar] [CrossRef]
- Desautels, M.; Goldberg, A.L. Liver mitochondria contain an ATP-dependent, vanadate-sensitive pathway for the degradation of proteins. Proc. Natl. Acad. Sci. USA 1982, 79, 1869–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Gottesman, S.; Willingham, M.C.; Gottesman, M.M.; Maurizi, M.R. A human mitochondrial ATP-dependent protease that is highly homologous to bacterial Lon protease. Proc. Natl. Acad. Sci. USA 1993, 90, 11247–11251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, C.K.; Suda, K.; Wang, N.; Schatz, G. Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science 1994, 264, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, L.; Pearce, D.A.; Sherman, F. PIM1 encodes a mitochondrial ATP-dependent protease that is required for mitochondrial function in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1994, 269, 238–242. [Google Scholar]
- Gottesman, S. Proteases and their targets in Escherichia coli. Annu. Rev. Genet. 1996, 30, 465–506. [Google Scholar] [CrossRef]
- Kikuchi, M.; Hatano, N.; Yokota, S.; Shimozawa, N.; Imanaka, T.; Taniguchi, H. Proteomic analysis of rat liver peroxisome: Presence of peroxisome-specific isozyme of lon protease. J. Biol. Chem. 2004, 279, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Fox, T.D. Mitochondrial protein synthesis, import, and assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, S.; Lee, J.; Singh, K.; Lee, I.; Suzuki, C.K. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta 2012, 1823, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Stahlberg, H.; Kutejova, E.; Suda, K.; Wolpensinger, B.; Lustig, A.; Schatz, G.; Engel, A.; Suzuki, C.K. Mitochondrial Lon of Saccharomyces cerevisiae is a ring-shaped protease with seven flexible subunits. Proc. Natl. Acad. Sci. USA 1999, 96, 6787–6790. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Wagner, I.; Arlt, H.; van Dyck, L.; Langer, T.; Neupert, W. Molecular chaperones cooperate with PIM1 protease in the degradation of misfolded proteins in mitochondria. EMBO J. 1994, 13, 5135–5145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Janowsky, B.; Knapp, K.; Major, T.; Krayl, M.; Guiard, B.; Voos, W. Structural properties of substrate proteins determine their proteolysis by the mitochondrial AAA+ protease Pim1. Biol. Chem. 2005, 386, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Ondrovicova, G.; Liu, T.; Singh, K.; Tian, B.; Li, H.; Gakh, O.; Perecko, D.; Janata, J.; Granot, Z.; Orly, J.; et al. Cleavage site selection within a folded substrate by the ATP-dependent lon protease. J. Biol. Chem. 2005, 280, 25103–25110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Major, T.; von Janowsky, B.; Ruppert, T.; Mogk, A.; Voos, W. Proteomic analysis of mitochondrial protein turnover: Identification of novel substrate proteins of the matrix protease Pim1. Mol. Cell. Biol. 2006, 26, 762–776. [Google Scholar] [CrossRef] [Green Version]
- Song, J.Y.; Marszalek, J.; Craig, E.A. Cysteine desulfurase Nfs1 and Pim1 protease control levels of Isu, the Fe-S cluster biogenesis scaffold. Proc. Natl. Acad. Sci. USA 2012, 109, 10370–10375. [Google Scholar] [CrossRef] [Green Version]
- Ciesielski, S.J.; Schilke, B.; Marszalek, J.; Craig, E.A. Protection of scaffold protein Isu from degradation by the Lon protease Pim1 as a component of Fe-S cluster biogenesis regulation. Mol. Biol. Cell 2016, 27, 1060–1068. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef] [Green Version]
- Quiros, P.M.; Espanol, Y.; Acin-Perez, R.; Rodriguez, F.; Barcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernandez-Garcia, M.S.; Fueyo, A.; et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, L.; Pinti, M.; Boraldi, F.; Giorgio, V.; Bernardi, P.; Bartolomeo, R.; Nasi, M.; De Biasi, S.; Missiroli, S.; Carnevale, G.; et al. Silencing of mitochondrial Lon protease deeply impairs mitochondrial proteome and function in colon cancer cells. FASEB J. 2014, 28, 5122–5135. [Google Scholar] [CrossRef] [Green Version]
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines disturb mitochondrial function across eukaryotic models: A call for caution in biomedical research. Cell Rep. 2015, 10, 1681–1691. [Google Scholar] [CrossRef] [Green Version]
- Key, J.; Kohli, A.; Barcena, C.; Lopez-Otin, C.; Heidler, J.; Wittig, I.; Auburger, G. Global proteome of LonP1(+/-) mouse embryonal fibroblasts reveals impact on respiratory chain, but no interdependence between Eral1 and mitoribosomes. Int. J. Mol. Sci. 2019, 20, 4523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, T.Y.; Chiu, Y.C.; Fang, W.C.; Cheng, C.W.; Kuo, C.Y.; Juan, H.F.; Wu, S.H.; Lee, A.Y. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015, 6, e1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bota, D.A.; Davies, K.J. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002, 4, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Bayot, A.; Gareil, M.; Rogowska-Wrzesinska, A.; Roepstorff, P.; Friguet, B.; Bulteau, A.L. Identification of novel oxidized protein substrates and physiological partners of the mitochondrial ATP-dependent Lon-like protease Pim1. J. Biol. Chem. 2010, 285, 11445–114457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, T.; Leidhold, C.; Ruppert, T.; Franken, S.; Voos, W. The role of protein quality control in mitochondrial protein homeostasis under oxidative stress. Proteomics 2010, 10, 1426–1443. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Taanman, J.W.; Schapira, A.H. A LON-ClpP proteolytic axis degrades complex I to extinguish ROS production in depolarized mitochondria. Cell Rep. 2016, 17, 2522–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, E.; Dehay, B.; Ben-Haiem, L.; Trottier, Y.; Morange, M.; Bertolotti, A. Targeting expression of expanded polyglutamine proteins to the endoplasmic reticulum or mitochondria prevents their aggregation. Proc. Natl. Acad. Sci. USA 2004, 101, 9648–9653. [Google Scholar] [CrossRef] [Green Version]
- Bruderek, M.; Jaworek, W.; Wilkening, A.; Rüb, C.; Cenini, G.; Förtsch, A.; Sylvester, M.; Voos, W. IMiQ: A novel protein quality control compartment protecting mitochondrial functional integrity. Mol. Biol. Cell 2018, 29, 256–269. [Google Scholar] [CrossRef]
- Voos, W. Chaperone-protease networks in mitochondrial protein homeostasis. Biochim. Biophys. Acta 2013, 1833, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Von Janowsky, B.; Major, T.; Knapp, K.; Voos, W. The disaggregation activity of the mitochondrial ClpB homolog Hsp78 maintains Hsp70 function during heat stress. J. Mol. Biol. 2006, 357, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Bender, T.; Lewrenz, I.; Franken, S.; Baitzel, C.; Voos, W. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol. Biol. Cell 2011, 22, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Wilkening, A.; Rüb, C.; Sylvester, M.; Voos, W. Analysis of heat-induced protein aggregation in human mitochondria. J. Biol. Chem. 2018, 293, 11537–11552. [Google Scholar] [CrossRef] [Green Version]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017, 543, 443–446. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsell, D.A.; Kowal, A.S.; Singer, M.A.; Lindquist, S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 1994, 372, 475–478. [Google Scholar] [CrossRef]
- Van Dyck, L.; Neupert, W.; Langer, T. The ATP-dependent PIM1 protease is required for the expression of intron-containing genes in mitochondria. Genes Dev. 1998, 12, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Rep, M.; van Dijl, J.M.; Suda, K.; Schatz, G.; Grivell, L.A.; Suzuki, C.K. Promotion of mitochondrial membrane complex assembly by a proteolytically inactive yeast Lon. Science 1996, 274, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Arlt, H.; Steglich, G.; Perryman, R.; Guiard, B.; Neupert, W.; Langer, T. The formation of respiratory chain complexes in mitochondria is under the proteolytic control of the m-AAA protease. EMBO J. 1998, 17, 4837–4847. [Google Scholar] [CrossRef] [Green Version]
- Pareek, G.; Thomas, R.E.; Vincow, E.S.; Morris, D.R.; Pallanck, L.J. Lon protease inactivation in Drosophila causes unfolded protein stress and inhibition of mitochondrial translation. Cell Death Discov. 2018, 4, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bota, D.A.; Ngo, J.K.; Davies, K.J. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic. Biol. Med. 2005, 38, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, G.K.; Markovitz, D.M. The human LON protease binds to mitochondrial promoters in a single-stranded, site-specific, strand-specific manner. Biochemistry 1998, 37, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lu, B.; Lee, I.; Ondrovicova, G.; Kutejova, E.; Suzuki, C.K. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J. Biol. Chem. 2004, 279, 13902–13910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.; Hamasaki, N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: Overview of its multiple roles. Ann. NY Acad. Sci. 2005, 1042, 101–108. [Google Scholar] [CrossRef]
- Matsushima, Y.; Goto, Y.; Kaguni, L.S. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proc. Natl. Acad. Sci. USA 2010, 107, 18410–18415. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Molecular cell 2013, 49, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Bulteau, A.L.; Szweda, L.I.; Friguet, B. Mitochondrial protein oxidation and degradation in response to oxidative stress and aging. Exp. Gerontol. 2006, 41, 653–657. [Google Scholar] [CrossRef]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox. Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.; Gibellini, L.; Liu, Y.; Xu, S.; Lu, B.; Cossarizza, A. Mitochondrial Lon protease at the crossroads of oxidative stress, ageing and cancer. Cell. Mol. Life Sci. 2015, 72, 4807–4824. [Google Scholar] [CrossRef]
- Lindquist, S. The heat-shock response. Annu. Rev. Biochem. 1986, 55, 1151–1191. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, J.K.; Davies, K.J. Mitochondrial Lon protease is a human stress protein. Free Radic. Biol. Med. 2009, 46, 1042–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomatto, L.C.D.; Carney, C.; Shen, B.; Wong, S.; Halaszynski, K.; Salomon, M.P.; Davies, K.J.A.; Tower, J. The mitochondrial Lon protease is required for age-specific and sex-specific adaptation to oxidative stress. Curr. Biol. 2017, 27, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bahat, A.; Perlberg, S.; Melamed-Book, N.; Isaac, S.; Eden, A.; Lauria, I.; Langer, T.; Orly, J. Transcriptional activation of LON Gene by a new form of mitochondrial stress: A role for the nuclear respiratory factor 2 in StAR overload response (SOR). Mol. Cell. Endocrinol. 2015, 408, 62–72. [Google Scholar] [CrossRef]
- Zurita Rendon, O.; Shoubridge, E.A. LONP1 is required for maturation of a subset of mitochondrial proteins, and its loss elicits an integrated stress response. Mol. Cell Biol. 2018, 38, e00412–e00417. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Hori, O.; Ichinoda, F.; Tamatani, T.; Yamaguchi, A.; Sato, N.; Ozawa, K.; Kitao, Y.; Miyazaki, M.; Harding, H.P.; Ron, D.; et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: Enhanced expression of Lon protease. J. Cell Biol. 2002, 157, 1151–1160. [Google Scholar] [CrossRef]
- Polo, M.; Alegre, F.; Moragrega, A.B.; Gibellini, L.; Marti-Rodrigo, A.; Blas-Garcia, A.; Esplugues, J.V.; Apostolova, N. Lon protease: A novel mitochondrial matrix protein in the interconnection between drug-induced mitochondrial dysfunction and endoplasmic reticulum stress. Br. J. Pharmacol. 2017, 174, 4409–4429. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Schmidt, A.; Malmstroem, J.; Claassen, M.; Ori, A.; Szymborska, A.; Herzog, F.; Rinner, O.; Ellenberg, J.; Aebersold, R. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011, 7, 549. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkurede, U.; Miller, R.A. Improved mitochondrial stress response in long-lived Snell dwarf mice. Aging Cell 2019, 18, e13030. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.A.; Van Remmen, H.; Davies, K.J.A. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett. 2002, 532, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [Green Version]
- Erjavec, N.; Bayot, A.; Gareil, M.; Camougrand, N.; Nyström, T.; Friguet, B.; Bulteau, A.L. Deletion of the mitochondrial Pim1/Lon protease in yeast results in accelerated aging and impairment of the proteasome. Free Radic. Biol. Med. 2013, 56, 9–16. [Google Scholar] [CrossRef]
- Ngo, J.K.; Pomatto, L.C.; Bota, D.A.; Koop, A.L.; Davies, K.J. Impairment of lon-induced protection against the accumulation of oxidized proteins in senescent wi-38 fibroblasts. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Luce, K.; Osiewacz, H.D. Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat. Cell Biol. 2009, 11, 852–858. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibellini, L.; Losi, L.; De Biasi, S.; Nasi, M.; Lo Tartaro, D.; Pecorini, S.; Patergnani, S.; Pinton, P.; De Gaetano, A.; Carnevale, G.; et al. LonP1 differently modulates mitochondrial function and bioenergetics of primary versus metastatic colon cancer cells. Front. Oncol. 2018, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death. Dis. 2013, 4, e681. [Google Scholar] [CrossRef]
- Liu, Y.; Lan, L.; Huang, K.; Wang, R.; Xu, C.; Shi, Y.; Wu, X.; Wu, Z.; Zhang, J.; Chen, L.; et al. Inhibition of Lon blocks cell proliferation, enhances chemosensitivity by promoting apoptosis and decreases cellular bioenergetics of bladder cancer: Potential roles of Lon as a prognostic marker and therapeutic target in baldder cancer. Oncotarget 2014, 5, 11209–11224. [Google Scholar] [CrossRef] [Green Version]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.Y.; Chiu, Y.C.; Lee, A.Y.; Hwang, T.L. Mitochondrial Lon protease controls ROS-dependent apoptosis in cardiomyocyte under hypoxia. Mitochondrion 2015, 23, 7–16. [Google Scholar] [CrossRef]
- Sepuri, N.B.V.; Angireddy, R.; Srinivasan, S.; Guha, M.; Spear, J.; Lu, B.; Anandatheerthavarada, H.K.; Suzuki, C.K.; Avadhani, N.G. Mitochondrial LON protease-dependent degradation of cytochrome c oxidase subunits under hypoxia and myocardial ischemia. Biochim. Biophys. Acta 2017, 1858, 519–528. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voos, W.; Pollecker, K. The Mitochondrial Lon Protease: Novel Functions off the Beaten Track? Biomolecules 2020, 10, 253. https://doi.org/10.3390/biom10020253
Voos W, Pollecker K. The Mitochondrial Lon Protease: Novel Functions off the Beaten Track? Biomolecules. 2020; 10(2):253. https://doi.org/10.3390/biom10020253
Chicago/Turabian StyleVoos, Wolfgang, and Karen Pollecker. 2020. "The Mitochondrial Lon Protease: Novel Functions off the Beaten Track?" Biomolecules 10, no. 2: 253. https://doi.org/10.3390/biom10020253