Cyclin-Dependent Kinase 5 Inhibitor Butyrolactone I Elicits a Partial Agonist Activity of Peroxisome Proliferator-Activated Receptor γ

 , , ,
, , , 
Abstract
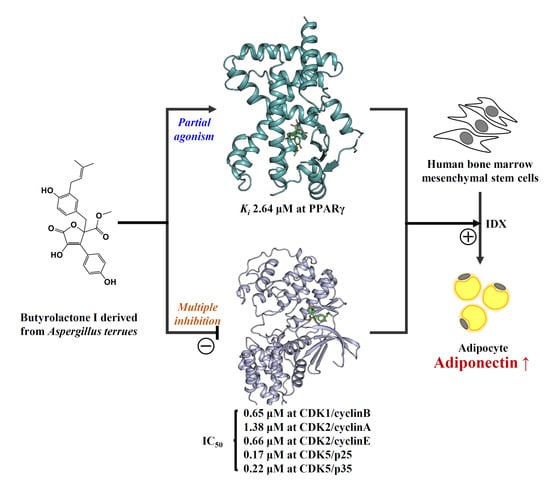
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture, Adipocyte Differentiation, Adiponectin Enzyme-Linked Immunosorbent Assay (ELISA), and Oil Red O Staining
2.3. Total RNA Isolation and Quantitiative Real-Time PCR
2.4. Nuclear Receptor Binding Assays
2.5. Cloning, Protein Expression, and Purification
2.6. Crystallization
2.7. X-Ray Data Collection, Structure Determination, and Refinement
2.8. In vitro Kinase Assay
2.9. Luciferase Reporter Gene Assay
2.10. Statistical Analyses
2.11. Accession Codes
3. Results
3.1. Adiponectin Production-Enhancing Activity of Butyrolactone I in hBM-MSCs Adipogenic Model
3.2. The Adiponectin Production-Enhancing Activity of Butyrolactone I did not Correlate with the Potency of CDK5 Inhibition
3.3. A PPARγ Partial Agonist Activity of Butyrolactone I
3.4. The Crystal Structure of PPARγ LBD in Complex with Butyrolactone I
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ahn, S.; Kim, H.J.; Lee, M.; Ahn, S.; Kim, J.; Jin, S.H.; Lee, E.; Kim, G.; Cheong, J.H.; et al. Polypharmacology of N 6-(3-Iodobenzyl) adenosine-5′-N-methyluronamide (IB-MECA) and related A3 adenosine receptor ligands: Peroxisome proliferator activated receptor (PPAR) γ partial agonist and PPARδ antagonist activity suggests their antidiabetic potential. J. Med. Chem. 2017, 60, 7459–7475. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Ma, C.T.; Choi, J.M.; An, S.; Lee, M.; Le, T.H.V.; Pyo, J.J.; Lee, J.; Choi, M.; Kwon, S.W.; et al. Adiponectin-secretion-promoting phenylethylchromones from the agarwood of Aquilaria malaccensis. J. Nat. Prod. 2019, 82, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 2006, 116, 1784–1792. [Google Scholar] [CrossRef]
- Katsiki, N.; Mantzoros, C.; Mikhailidis, D.P. Adiponectin, lipids and atherosclerosis. Curr. Opin. Lipidol. 2017, 28, 347–354. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef]
- Mantzoros, C.; Petridou, E.; Dessypris, N.; Chavelas, C.; Dalamaga, M.; Alexe, D.M.; Papadiamantis, Y.; Markopoulos, C.; Spanos, E.; Chrousos, G.; et al. Adiponectin and breast cancer risk. J. Clin. Endocrinol. Metab. 2004, 89, 1102–1107. [Google Scholar] [CrossRef]
- Petridou, E.; Mantzoros, C.S.; Dessypris, N.; Dikalioti, S.K.; Trichopoulos, D. Adiponectin in relation to childhood myeloblastic leukaemia. Br. J. Cancer 2006, 94, 156–160. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kitayama, J.; Kazama, S.; Hiramatsu, T.; Hatano, K.; Nagawa, H. Plasma adiponectin and gastric cancer. Clin. Cancer Res. 2005, 11, 466–472. [Google Scholar]
- Wei, E.K.; Giovannucci, E.; Fuchs, C.S.; Willett, W.C.; Mantzoros, C.S. Low plasma adiponectin levels and risk of colorectal cancer in men: A prospective study. J. Natl. Cancer Inst. 2005, 97, 1688–1694. [Google Scholar] [CrossRef] [PubMed]
- Finucane, F.M.; Luan, J.; Wareham, N.J.; Sharp, S.J.; O’rahilly, S.; Balkau, B.; Flyvbjerg, A.; Walker, M.; Højlund, K.; Nolan, J.J.; et al. Correlation of the leptin: Adiponectin ratio with measures of insulin resistance in non-diabetic individuals. Diabetologia 2009, 52, 2345–2349. [Google Scholar] [CrossRef]
- Oda, N.; Imamura, S.; Fujita, T.; Uchida, Y.; Inagaki, K.; Kakizawa, H.; Hayakawa, N.; Suzuki, A.; Takeda, J.; Horikawa, Y.; et al. The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metabolism 2008, 57, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Kusminski, C.M.; Scherer, P.E. Adiponectin in health and disease: Evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol. Sci. 2009, 30, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Lam, K.S.; Vanhoutte, P.M.; Xu, A. Adiponectin and cardiovascular health: An update. Br. J. Pharmacol. 2012, 165, 574–590. [Google Scholar] [CrossRef] [PubMed]
- Fisman, E.Z.; Tenenbaum, A. Adiponectin: A manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovasc. Diabetol. 2014, 13, 103. [Google Scholar] [CrossRef]
- Rizos, C.V.; Kei, A.; Elisaf, M.S. The current role of thiazolidinediones in diabetes management. Arch. Toxicol. 2016, 90, 1861–1881. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, M.; An, S.; Hyun, S.; Hwang, J.; Lee, J.; Noh, M. 2-Formyl-komarovicine promotes adiponectin production in human mesenchymal stem cells through PPARγ partial agonism. Bioorg. Med. Chem. 2018, 26, 1069–1075. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Lenhard, J.M.; Oliver, B.B.; Ringold, G.M.; Kliewer, S.A. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997, 272, 3406–3410. [Google Scholar] [CrossRef]
- Ahn, S.; Kim, J.; An, S.; Pyo, J.J.; Jung, D.; Lee, J.; Hwang, S.Y.; Gong, J.; Shin, I.; Kim, H.P.; et al. 2-Phenyl-8-(1-phenylallyl)-chromenone compounds have a pan-PPAR modulator pharmacophore. Bioorg. Med. Chem. 2019, 27, 2948–2958. [Google Scholar] [CrossRef]
- Hummasti, S.; Laffitte, B.A.; Watson, M.A.; Galardi, C.; Chao, L.C.; Ramamurthy, L.; Moore, J.T.; Tontonoz, P. Liver X receptors are regulators of adipocyte gene expression but not differentiation identification of apoD as a direct target. J. Lipid Res. 2004, 45, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Hoppmann, J.; Perwitz, N.; Meier, B.; Fasshauer, M.; Hadaschik, D.; Lehnert, H.; Klein, J. The balance between gluco-and mineralo-corticoid action critically determines inflammatory adipocyte responses. J. Endocrinol. 2010, 204, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Pye, C.R.; Bertin, M.J.; Lokey, R.S.; Gerwick, W.H.; Linington, R.G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. USA 2017, 114, 5601–5606. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Liao, L.; Hong, S.H.; Park, W.; Kwon, D.I.; Lee, J.; Noh, M.; Oh, D.C.; Oh, K.B.; Shin, J. Lumazine peptides from the marine-derived fungus Aspergillus terreus. Mar. Drugs 2015, 13, 1290–1303. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Uchiyama, M.; Okumura, E.; Saito, T.; Ishiguro, K.; Uchida, T.; Okuyama, A.; Kishimoto, T.; Hisanaga, S.I. Evidence for cdk5 as a major activity phosphorylating tau protein in porcine brain extract. J. Biochem. 1995, 117, 741–749. [Google Scholar] [CrossRef]
- Kitagawa, M.; Higashi, H.; Suzuki-Takahashi, I.; Segawa, K.; Hanks, S.K.; Taya, Y.; Nishimura, S.; Okuyama, A. Phosphorylation of E2F-1 by cyclin A-cdk2. Oncogene 1995, 10, 229–236. [Google Scholar]
- Furukawa, Y.; Iwase, S.; Terui, Y.; Kikuchi, J.; Sakai, T.; Nakamura, M.; Kitagawa, S.; Kitagawa, M. Transcriptional activation of the cdc2 gene is associated with Fas-induced apoptosis of human hematopoietic cells. J. Biol. Chem. 1996, 271, 28469–28477. [Google Scholar] [CrossRef][Green Version]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Boström, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Blüher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature 2010, 466, 451. [Google Scholar] [CrossRef]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef]
- Ahn, S.; An, S.; Lee, M.; Lee, E.; Pyo, J.J.; Kim, J.H.; Ki, M.W.; Jin, S.H.; Ha, J.; Noh, M. A long-wave UVA filter avobenzone induces obesogenic phenotypes in normal human epidermal keratinocytes and mesenchymal stem cells. Arch. Toxicol. 2019, 93, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Kim, H.J.; Jeong, J.W.; Park, C.; Kim, B.W.; Choi, Y.H. Inhibition of adipocyte differentiation by anthocyanins isolated from the fruit of Vitis coignetiae pulliat is associated with the activation of AMPK signaling pathway. Toxicol. Res. 2018, 34, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Q.; Lin, J.L.; Wang, Y.; Zhang, R.X.; Hou, J.B.; Yu, B. Recombinant human thioredoxin-1 protects macrophages from oxidized low-density lipoprotein-induced foam cell formation and cell apoptosis. Biomol. Ther. 2018, 26, 121. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Brunger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Jang, J.Y.; Bae, H.; Lee, Y.J.; Choi, Y.I.; Kim, H.J.; Park, S.B.; Suh, S.W.; Kim, S.W.; Han, B.W. Structural Basis for the Enhanced Anti-Diabetic Efficacy of Lobeglitazone on PPARgamma. Sci. Rep. 2018, 8, 31. [Google Scholar] [CrossRef]
- Niu, X.; Dahse, H.M.; Menzel, K.D.; Lozach, O.; Walther, G.; Meijer, L.; Grabley, S.; Sattler, I. Butyrolactone I derivatives from Aspergillus terreus carrying an unusual sulfate moiety. J. Nat. Prod. 2008, 71, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Higashi, H.; Takahashi, I.S.; Okabe, T.; Ogino, H.; Taya, Y.; Hishimura, S.; Okuyama, A. A cyclin-dependent kinase inhibitor, butyrolactone I, inhibits phosphorylation of RB protein and cell cycle progression. Oncogene 1994, 9, 2549–2557. [Google Scholar] [PubMed]
- Yamamoto, H.; Monden, T.; Miyoshi, H.; Izawa, H.; Ikeda, K.; Tsujie, M.; Ohnishi, T.; Sekimoto, M.; Tomita, N.; Monden, M. Cdk2/cdc2 expression in colon carcinogenesis and effects of cdk2/cdc2 inhibitor in colon cancer cells. Int. J. Oncol. 1998, 13, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Rosen, C.J. PPARγ: A circadian transcription factor in adipogenesis and osteogenesis. Nat. Rev. Endocrinol. 2010, 6, 629. [Google Scholar] [CrossRef]
- Zhuang, K.; Zhang, J.; Xiong, M.; Wang, X.; Luo, X.; Han, L.; Meng, Y.; Zhang, Y.; Liao, W.; Liu, S. CDK5 functions as a tumor promoter in human colorectal cancer via modulating the ERK5–AP-1 axis. Cell Death Dis. 2016, 7, e2415. [Google Scholar] [CrossRef]
- Zeng, J.; Xie, S.; Liu, Y.; Shen, C.; Song, X.; Zhou, G.L.; Wang, C. CDK5 Functions as a Tumor Promoter in Human Lung Cancer. J. Cancer 2018, 9, 3950. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial agonists activate PPARγ using a helix 12 independent mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef]
- Nishio, K.; Ishida, T.; Arioka, H.; Kurokawa, H.; Fukuoka, K.; Nomoto, T.; Fukumoto, H.; Yokote, H.; Saijo, N. Antitumor effects of butyrolactone I, a selective CDC2 kinase inhibitor, on human lung cancer cell lines. Anticancer Res. 1996, 16, 3387–3395. [Google Scholar]
- Pinzi, L.; Caporuscio, F.; Rastelli, G. Selection of protein conformations for structure-based polypharmacology studies. Drug Discov. Today 2018, 23, 1889–1896. [Google Scholar] [CrossRef] [PubMed]
- Sumi, N.J.; Ctortecka, C.; Hu, Q.; Bryant, A.T.; Fang, B.; Rix, L.L.R.; Ayaz, M.; Kinose, F.; Welsh, E.A.; Eschrich, S.A.; et al. Divergent Polypharmacology-Driven Cellular Activity of Structurally Similar Multi-Kinase Inhibitors through Cumulative Effects on Individual Targets. Cell Chem. Biol. 2019, 26, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Khandekar, M.J.; Banks, A.S.; Laznik-Bogoslavski, D.; White, J.P.; Choi, J.H.; Kazak, L.; Lo, J.C.; Cohen, P.; Wong, K.K.; Kamenecka, T.M.; et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2018, 115, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Gowda, M.K.B.; Lee, M.; Masagalli, J.N.; Mailar, K.; Choi, W.J.; Noh, M. Novel linked butanolide dimer compounds increase adiponectin production during adipogenesis in human mesenchymal stem cells through peroxisome proliferator-activated receptor γ modulation. Eur. J. Med. Chem. 2020, 187, 111969. [Google Scholar] [CrossRef] [PubMed]
- Lapillonne, H.; Konopleva, M.; Tsao, T.; Gold, D.; McQueen, T.; Sutherland, R.L.; Madden, T.; Andreeff, M. Activation of peroxisome proliferator-activated receptor γ by a novel synthetic triterpenoid 2-cyano-3, 12-dioxooleana-1, 9-dien-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Cancer Res. 2003, 63, 5926–5939. [Google Scholar]
- Youssef, J.; Badr, M. Peroxisome proliferator-activated receptors and cancer: Challenges and opportunities. Br. J. Pharmacol. 2011, 164, 68–82. [Google Scholar] [CrossRef]
- Liu, H.J.; Zhang, C.Y.; Song, F.; Xiao, T.; Meng, J.; Zhang, Q.; Liang, C.L.; Li, S.; Wang, J.; Zhang, B.; et al. A novel partial agonist of peroxisome proliferator-activated receptor γ with excellent effect on insulin resistance and type 2 diabetes. J. Pharmacol. Exp. Ther. 2015, 353, 573–581. [Google Scholar] [CrossRef]
- Hughes, T.S.; Chalmers, M.J.; Novick, S.; Kuruvilla, D.S.; Chang, M.R.; Kamenecka, T.M.; Rance, M.; Johnson, B.A.; Burris, T.P.; Griffin, P.R.; et al. Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Structure 2012, 20, 139–150. [Google Scholar] [CrossRef]
- Bae, H.; Jang, J.Y.; Choi, S.S.; Lee, J.J.; Kim, H.; Jo, A.; Lee, K.J.; Choi, J.H.; Suh, S.W.; Park, S.B. Mechanistic elucidation guided by covalent inhibitors for the development of anti-diabetic PPARγ ligands. Chem. Sci. 2016, 7, 5523–5529. [Google Scholar] [CrossRef]
- Jang, J.Y.; Kim, H.; Kim, H.J.; Suh, S.W.; Park, S.B.; Han, B.W. Structural basis for the inhibitory effects of a novel reversible covalent ligand on PPARγ phosphorylation. Sci. Rep. 2019, 9, 11168. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Kim, H.J.; Han, B.W. Structural Basis for the Regulation of PPARγ Activity by Imatinib. Molecules 2019, 24, 3562. [Google Scholar] [CrossRef] [PubMed]
- Guasch, L.; Sala, E.; Castell-Auví, A.; Cedó, L.; Liedl, K.R.; Wolber, G.; Muehlbacher, M.; Mulero, M.; Pinent, M.; Ardévol, A.; et al. Identification of PPARγ partial agonists of natural origin (I): Development of a virtual screening procedure and in vitro validation. PLoS ONE 2012, 7, e50816. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Name | Butyrolactone I-Bound PPARγ LBD | Ligand-Free PPARγ LBD |
|---|---|---|
| Data collection | ||
| X-ray source | PLS-5C | PLS-11C |
| X-ray wavelength (Å) | 1.0000 | 0.9794 |
| Space group | C2 | C2 |
| Unit cell parameters | ||
| a, b, c (Å) | 93.62, 61.12, 119.76 | 93.15, 62.26, 119.50 |
| α, β, γ (⁰) | 90.00, 103.64, 90.00 | 90.00, 102.51, 90.00 |
| Resolution range (Å) | 50.00–2.10 (2.14–2.10)a | 30.00–2.10 (2.14–2.10)a |
| Total/unique reflections | 177,451/38,329 | 174,642/38,322 |
| Redundancy | 4.6 (4.4) | 4.6 (4.5) |
| Completeness (%) | 99.3 | 98.0 |
| <I/σI> | 39.3 (2.0)a | 12.6 (2.3)a |
| Rmerge b(%) | 3.2 (71.0)a | 10.1 (58.3)a |
| CC1/2 | 1.00 (0.81)a | 0.99 (0.82)a |
| Model refinement | ||
| Resolution range (Å) | 50.00–2.10 | 30.00–2.10 |
| Rwork/Rfreec (%) | 22.8/26.8 | 21.8/26.8 |
| No. of non-hydrogen atoms | ||
| Protein | 4129 | 4230 |
| Ligand (butyrolactone I) | 62 | - |
| water | 150 | 159 |
| Average B factor (Å2) | ||
| Protein | 35.4 | 31.5 |
| Ligand (butyrolactone I) | 68.7 | - |
| Water | 31.2 | 28.3 |
| R.m.s. deviations from ideal geometry | ||
| Bond lengths (Å) | 0.0042 | 0.0034 |
| Bond angles (⁰) | 1.2587 | 1.2024 |
| Ramachandran plotd | ||
| Favored/Outliers (%) | 98.4/0 | 98.6/0 |
| Poor rotamersd (%) | 0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, S.; Jang, D.M.; Park, S.C.; An, S.; Shin, J.; Han, B.W.; Noh, M. Cyclin-Dependent Kinase 5 Inhibitor Butyrolactone I Elicits a Partial Agonist Activity of Peroxisome Proliferator-Activated Receptor γ. Biomolecules 2020, 10, 275. https://doi.org/10.3390/biom10020275
Ahn S, Jang DM, Park SC, An S, Shin J, Han BW, Noh M. Cyclin-Dependent Kinase 5 Inhibitor Butyrolactone I Elicits a Partial Agonist Activity of Peroxisome Proliferator-Activated Receptor γ. Biomolecules. 2020; 10(2):275. https://doi.org/10.3390/biom10020275
Chicago/Turabian StyleAhn, Sungjin, Dong Man Jang, Sung Chul Park, Seungchan An, Jongheon Shin, Byung Woo Han, and Minsoo Noh. 2020. "Cyclin-Dependent Kinase 5 Inhibitor Butyrolactone I Elicits a Partial Agonist Activity of Peroxisome Proliferator-Activated Receptor γ" Biomolecules 10, no. 2: 275. https://doi.org/10.3390/biom10020275
APA StyleAhn, S., Jang, D. M., Park, S. C., An, S., Shin, J., Han, B. W., & Noh, M. (2020). Cyclin-Dependent Kinase 5 Inhibitor Butyrolactone I Elicits a Partial Agonist Activity of Peroxisome Proliferator-Activated Receptor γ. Biomolecules, 10(2), 275. https://doi.org/10.3390/biom10020275