Hypothalamic NAD+-Sirtuin Axis: Function and Regulation


Abstract
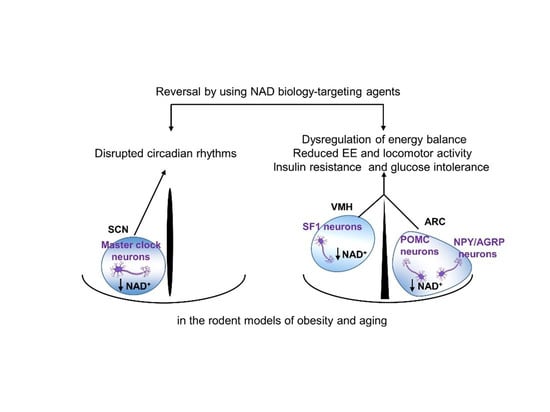
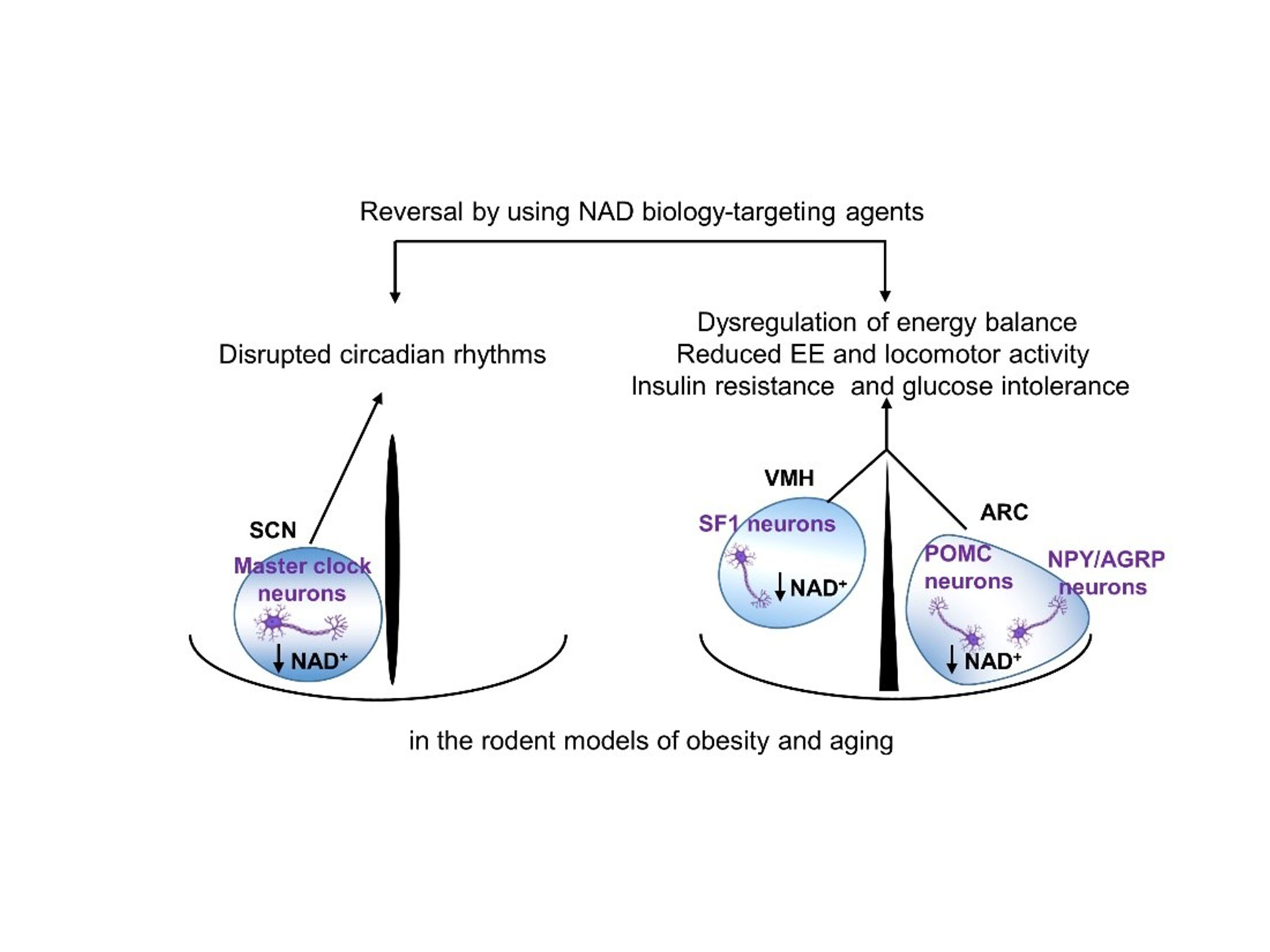
:
{kind=link}
{kind=link}
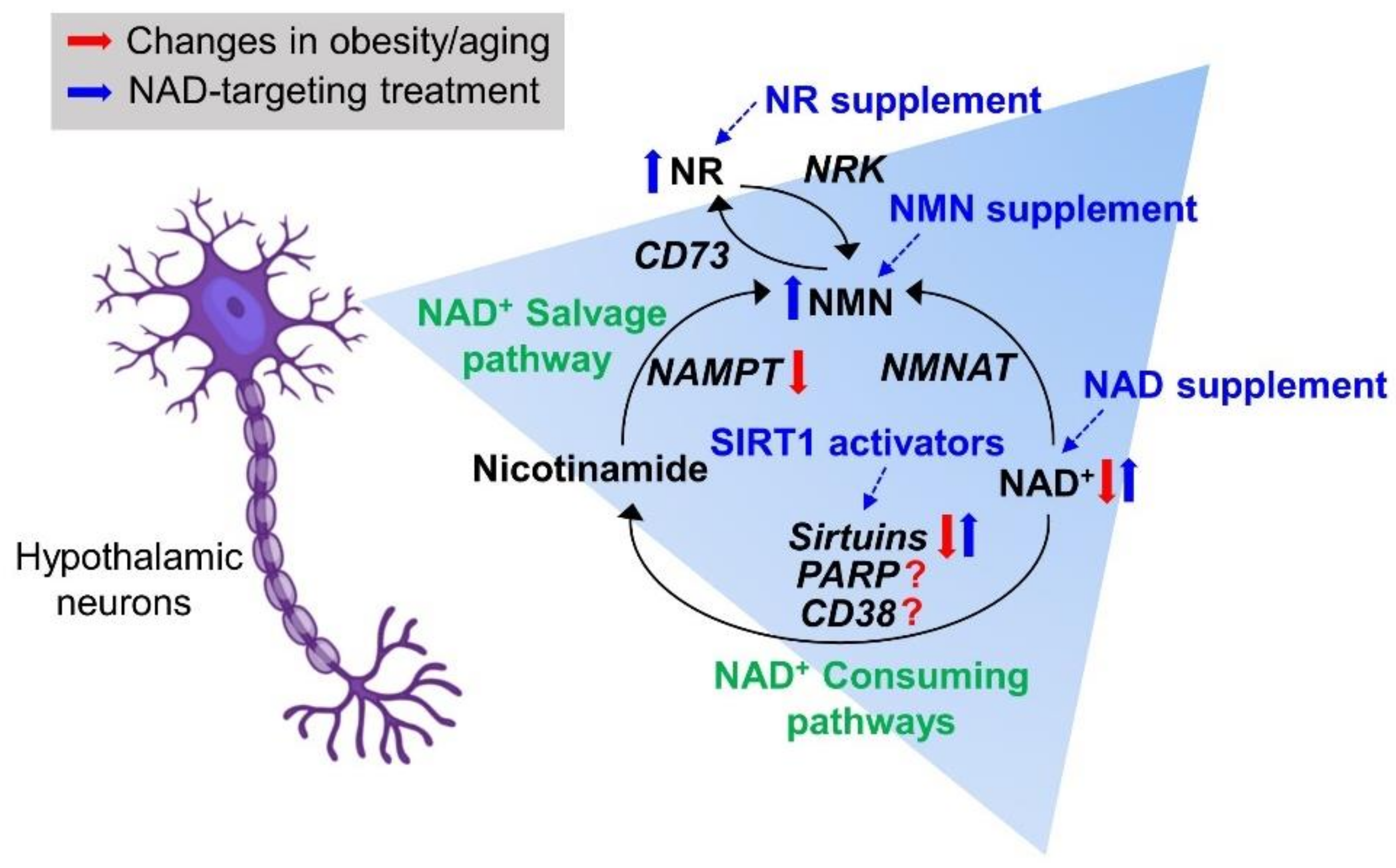
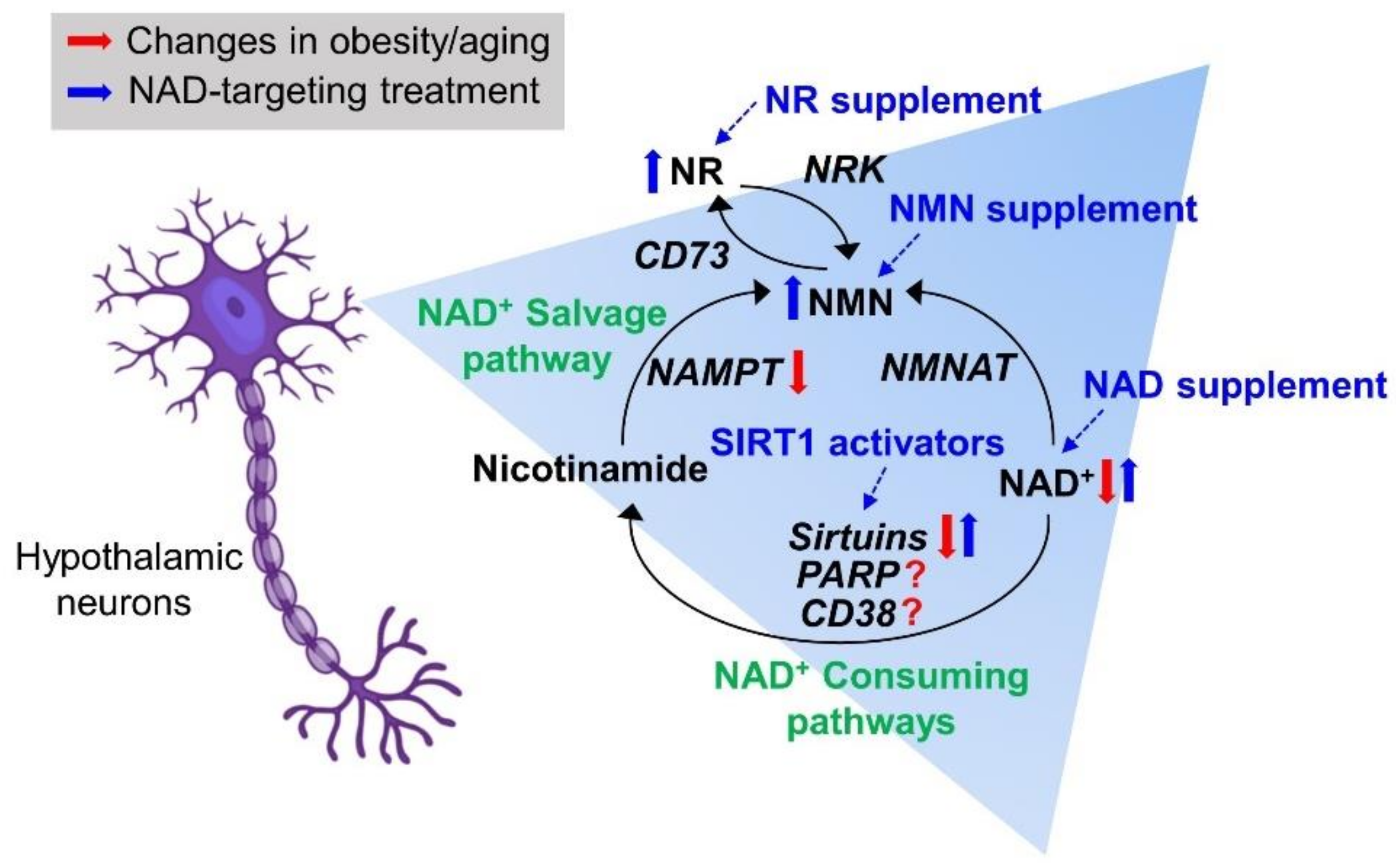{kind=link}
1. Introduction
2. NAD Biosynthetic Pathways
3. NAD+-Consuming Pathways
4. Hypothalamic Regulation of Energy Homeostasis and the Circadian Rhythm
5. Regulatory Roles of the Hypothalamic NAD+-Sirtuin Axis in Normal Physiological Conditions
6. Disrupted Hypothalamic NAD+ Biology in Obesity and Aging
7. Therapeutic Trials Targeting Hypothalamic NAD+ Biology
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [Green Version]
- Roh, E.; Kim, M.-S. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp. Mol. Med. 2016, 48, e216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, F.; Stebbins, R.; Chang, S.Z.; McPeek, M.A.; Brenner, C. Microbial NAD metabolism: lessons from comparative genomics. Microbiol. Mol. Biol. Rev. 2009, 73, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revollo, J.R.; Grimm, A.A.; Imai, S.-i. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Zhang, X.; Bheda, P.; Revollo, J.R.; Imai, S.I.; Wolberger, C. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat. Struct. Mol. Biol. 2006, 13, 661–662. [Google Scholar] [CrossRef]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef] [Green Version]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.-I.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- De Flora, A.; Zocchi, E.; Guida, L.; Franco, L.; Bruzzone, S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N. Y. Acad. Sci. 2004, 1028, 176–191. [Google Scholar]
- Bai, P.; Cantó, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: implications in aging and longevity. Ageing Res. Rev. 2018, 47, 1–17. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.-i.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Tarragó, M.G.; Chini, C.C.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab. 2018, 27, 1081–1095.e1010. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Landry, J.; Sutton, A.; Tafrov, S.T.; Heller, R.C.; Stebbins, J.; Pillus, L.; Sternglanz, R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 5807–5811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Feldman, J.L.; Baeza, J.; Denu, J.M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, K.G.; Landry, J.; Sternglanz, R.; Denu, J.M. Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. USA 2000, 97, 14178–14182. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Voelter-Mahlknecht, S.; Mahlknecht, U. Cloning, chromosomal characterization and mapping of the NAD-dependent histone deacetylases gene sirtuin 1. Int. J. Mol. Med. 2006, 17, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; de Oliveira, R.M.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Gomes, P.; Outeiro, T.F.; Cavadas, C. Emerging role of sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [Green Version]
- McCord, R.A.; Michishita, E.; Hong, T.; Berber, E.; Boxer, L.D.; Kusumoto, R.; Guan, S.; Shi, X.; Gozani, O.; Burlingame, A.L. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 2009, 1, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Shi, L.; Yang, S.; Yan, R.; Zhang, D.; Yang, J.; He, L.; Li, W.; Yi, X.; Sun, L. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly (ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Dregalla, R.C.; Zhou, J.; Idate, R.R.; Battaglia, C.L.; Liber, H.L.; Bailey, S.M. Regulatory roles of tankyrase 1 at telomeres and in DNA repair: suppression of T-SCE and stabilization of DNA-PKcs. Aging (Albany NY) 2010, 2, 691–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly (ADP-ribosyl) ation by PARP-1:PAR-laying’NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, W.M.; Amé, J.-C.; Wilson, M.V.; Wang, Z.-Q.; Koh, D.W.; Jacobson, M.K.; Jacobson, E.L. Poly (ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J. Biol. Chem. 1998, 273, 30069–30072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouquerel, E.; Sobol, R.W. ARTD1 (PARP1) activation and NAD+ in DNA repair and cell death. DNA Repair 2014, 23, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Chini, E.N. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, P.; Escande, C.; White, T.A.; Thompson, M.; Soares, S.; Benech, J.C.; Chini, E.N. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun. 2006, 349, 353–359. [Google Scholar] [CrossRef]
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 1993, 261, 1330–1333. [Google Scholar] [CrossRef]
- Morton, G.J.; Meek, T.H.; Schwartz, M.W. Neurobiology of food intake in health and disease. Nat. Rev. Neurosci. 2014, 15, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Roh, E.; Kim, M.-S. Brain regulation of energy metabolism. Endocrinol. Metab. 2016, 31, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Broadwell, R.D.; Brightman, M.W. Entry of peroxidase into neurons of the central and peripheral nervous systems from extracerebral and cerebral blood. J. Comp. Neurol. 1976, 166, 257–283. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.-K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 1997, 278, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.D.; Lu, D.; Koppula, S.; Vage, D.I.; Klungland, H.; Boston, B.; Chen, W.; Orth, D.N.; Pouton, C.; Kesterson, R.A. The melanocortin receptors: agonists, antagonists, and the hormonal control of pigmentation. Recent Prog. Horm. Res. 1996, 51, 287–317, discussion 318. [Google Scholar] [PubMed]
- Wu, Q.; Howell, M.P.; Cowley, M.A.; Palmiter, R.D. Starvation after AgRP neuron ablation is independent of melanocortin signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2687–2692. [Google Scholar] [CrossRef] [Green Version]
- Ahima, R.S.; Prabakaran, D.; Mantzoros, C.; Qu, D.; Lowell, B.; Maratos-Flier, E.; Flier, J.S. Role of leptin in the neuroendocrine response to fasting. Nature 1996, 382, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Baskin, D.G.; Bukowski, T.R.; Kuijper, J.L.; Foster, D.; Lasser, G.; Prunkard, D.E.; Porte, D.; Woods, S.C.; Seeley, R.J. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes 1996, 45, 531–535. [Google Scholar] [CrossRef] [Green Version]
- Hetherington, A.; Ranson, S. The Anatomical Record, Volume 78, 1940: Hypothalamic lesions and adiposity in the rat. Nutr. Rev. 1983, 41, 124–127. [Google Scholar] [CrossRef]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Zhao, L.; Donato, J.; Kohno, D.; Xu, Y.; Elias, C.F.; Lee, C.; Parker, K.L.; Elmquist, J.K. Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc. Natl. Acad. Sci. USA 2011, 108, 10673–10678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majdic, G.; Young, M.; Gomez-Sanchez, E.; Anderson, P.; Szczepaniak, L.S.; Dobbins, R.L.; McGarry, J.D.; Parker, K.L. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology 2002, 143, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.R. The suprachiasmatic nucleus: a 25-year retrospective. J. Biol. Rhythms. 1998, 13, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Saper, C.B.; Scammell, T.E.; Lu, J. Hypothalamic regulation of sleep and circadian rhythms. Nature 2005, 437, 1257–1263. [Google Scholar] [CrossRef]
- Bernard, S.; Gonze, D.; Čajavec, B.; Herzel, H.; Kramer, A. Synchronization-induced rhythmicity of circadian oscillators in the suprachiasmatic nucleus. PLoS Comput. Biol. 2007, 3, e68. [Google Scholar] [CrossRef] [Green Version]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [Green Version]
- Hastings, M.H.; Reddy, A.B.; Maywood, E.S. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat. Rev. Neurosci. 2003, 4, 649–661. [Google Scholar] [CrossRef]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Panda, S.; Antoch, M.P.; Miller, B.H.; Su, A.I.; Schook, A.B.; Straume, M.; Schultz, P.G.; Kay, S.A.; Takahashi, J.S.; Hogenesch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [Green Version]
- Ramadori, G.; Lee, C.E.; Bookout, A.L.; Lee, S.; Williams, K.W.; Anderson, J.; Elmquist, J.K.; Coppari, R. Brain SIRT1: anatomical distribution and regulation by energy availability. J. Neurosci. 2008, 28, 9989–9996. [Google Scholar] [CrossRef] [PubMed]
- Zakhary, S.M.; Ayubcha, D.; Dileo, J.N.; Jose, R.; Leheste, J.R.; Horowitz, J.M.; Torres, G. Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec. (Hoboken) 2010, 293, 1024–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çakir, I.; Perello, M.; Lansari, O.; Messier, N.J.; Vaslet, C.A.; Nillni, E.A. Hypothalamic Sirt1 regulates food intake in a rodent model system. PLoS ONE 2009, 4, e8322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, T.; Kim, H.-J.; Kobayashi, M.; Kitamura, Y.-I.; Yokota-Hashimoto, H.; Shiuchi, T.; Minokoshi, Y.; Kitamura, T. Induction of hypothalamic Sirt1 leads to cessation of feeding via agouti-related peptide. Endocrinology 2010, 151, 2556–2566. [Google Scholar] [CrossRef] [Green Version]
- Ramadori, G.; Fujikawa, T.; Fukuda, M.; Anderson, J.; Morgan, D.A.; Mostoslavsky, R.; Stuart, R.C.; Perello, M.; Vianna, C.R.; Nillni, E.A. SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell Metab. 2010, 12, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadori, G.; Fujikawa, T.; Anderson, J.; Berglund, E.D.; Frazao, R.; Michán, S.; Vianna, C.R.; Sinclair, D.A.; Elias, C.F.; Coppari, R. SIRT1 deacetylase in SF1 neurons protects against metabolic imbalance. Cell Metab. 2011, 14, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, M.O.; Antunes, C.; Geliang, G.; Liu, Z.-W.; Borok, E.; Nie, Y.; Xu, A.W.; Souza, D.O.; Gao, Q.; Diano, S. Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: roles for Sirt1 in neuronal firing and synaptic plasticity. J. Neurosci. 2010, 30, 11815–11825. [Google Scholar] [CrossRef] [Green Version]
- Coppari, R. Metabolic actions of hypothalamic SIRT1. Trends Endocrinol. Metab. 2012, 23, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Velásquez, D.A.; Martínez, G.; Romero, A.; Vázquez, M.J.; Boit, K.D.; Dopeso-Reyes, I.G.; López, M.; Vidal, A.; Nogueiras, R.; Diéguez, C. The central Sirtuin 1/p53 pathway is essential for the orexigenic action of ghrelin. Diabetes 2011, 60, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- De Guia, R.M.; Hassing, A.S.; Skov, L.J.; Ratner, C.; Plucinska, K.; Madsen, S.; Diep, T.A.; Cruz, G.V.D.; Trammell, S.A.; Sustarsic, E.G. Fasting-and ghrelin-induced food intake is regulated by NAMPT in the hypothalamus. Acta Physiol. 2020, e13437. [Google Scholar]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.-K.; Chong, J.L.; Buhr, E.D.; Lee, C. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Orozco-Solis, R.; Ramadori, G.; Coppari, R.; Sassone-Corsi, P. SIRT1 relays nutritional inputs to the circadian clock through the Sf1 neurons of the ventromedial hypothalamus. Endocrinology 2015, 156, 2174–2184. [Google Scholar] [CrossRef] [Green Version]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.-i. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Kawahara, T.; Fujiwara, K.; Shimpuku, M.; Sasaki, T.; Kitamura, T.; Yoshikawa, K. Necdin controls Foxo1 acetylation in hypothalamic arcuate neurons to modulate the thyroid axis. J. Neurosci. 2012, 32, 5562–5572. [Google Scholar] [CrossRef] [Green Version]
- Kolthur-Seetharam, U.; Teerds, K.; de Rooij, D.G.; Wendling, O.; McBurney, M.; Sassone-Corsi, P.; Davidson, I. The histone deacetylase SIRT1 controls male fertility in mice through regulation of hypothalamic-pituitary gonadotropin signaling. Biol. Reprod. 2009, 80, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Coussens, M.; Maresh, J.G.; Yanagimachi, R.; Maeda, G.; Allsopp, R. Sirt1 deficiency attenuates spermatogenesis and germ cell function. PLoS ONE 2008, 3, e1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBurney, M.W.; Yang, X.; Jardine, K.; Hixon, M.; Boekelheide, K.; Webb, J.R.; Lansdorp, P.M.; Lemieux, M. The mammalian SIR2α protein has a role in embryogenesis and gametogenesis. Mol. Cell Biol. 2003, 23, 38–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwer, B.; Schumacher, B.; Lombard, D.B.; Xiao, C.; Kurtev, M.V.; Gao, J.; Schneider, J.I.; Chai, H.; Bronson, R.T.; Tsai, L.-H.; et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 21790–21794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahima, R.S. Digging deeper into obesity. J. Clin. Invest. 2011, 121, 2076–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blüher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Yang, W.; Chen, C.-S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. (Lond). 2008, 32, 1431–1437. [Google Scholar] [CrossRef] [Green Version]
- Doak, C.; Wijnhoven, T.; Schokker, D.; Visscher, T.; Seidell, J. Age standardization in mapping adult overweight and obesity trends in the WHO European Region. Obes. Rev. 2012, 13, 174–191. [Google Scholar] [CrossRef]
- Parton, L.E.; Ye, C.P.; Coppari, R.; Enriori, P.J.; Choi, B.; Zhang, C.-Y.; Xu, C.; Vianna, C.R.; Balthasar, N.; Lee, C.E. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 2007, 449, 228–232. [Google Scholar] [CrossRef]
- Münzberg, H.; Flier, J.S.; Bjørbæk, C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 2004, 145, 4880–4889. [Google Scholar] [CrossRef] [Green Version]
- Enriori, P.J.; Evans, A.E.; Sinnayah, P.; Jobst, E.E.; Tonelli-Lemos, L.; Billes, S.K.; Glavas, M.M.; Grayson, B.E.; Perello, M.; Nillni, E.A. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007, 5, 181–194. [Google Scholar] [CrossRef]
- Satoh, A.; Imai, S.-i. Systemic regulation of mammalian ageing and longevity by brain sirtuins. Nat. Commun. 2014, 5, 4211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toorie, A.M.; Nillni, E.A. Minireview: Central Sirt1 regulates energy balance via the melanocortin system and alternate pathways. Mol. Endocrinol. 2014, 28, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S.-i. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet-and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, T.; Kikuchi, O.; Shimpuku, M.; Susanti, V.Y.; Yokota-Hashimoto, H.; Taguchi, R.; Shibusawa, N.; Sato, T.; Tang, L.; Amano, K. Hypothalamic SIRT1 prevents age-associated weight gain by improving leptin sensitivity in mice. Diabetologia 2014, 57, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, E.; Myoung Kang, G.; Young Gil, S.; Hee Lee, C.; Kim, S.; Hong, D.; Hoon Son, G.; Kim, M.S. Effects of Chronic NAD Supplementation on Energy Metabolism and Diurnal Rhythm in Obese Mice. Obesity 2018, 26, 1448–1456. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.-Y.; Wang, F.; Zhang, X.-Y.; Huang, P.; Lu, Y.-B.; Wei, E.-Q.; Zhang, W.-P. Nicotinamide phosphoribosyltransferase may be involved in age-related brain diseases. PLoS ONE 2012, 7, e44933. [Google Scholar] [CrossRef] [Green Version]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Zhu, X.-H.; Lu, M.; Lee, B.-Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Penke, M.; Larsen, P.S.; Schuster, S.; Dall, M.; Jensen, B.A.; Gorski, T.; Meusel, A.; Richter, S.; Vienberg, S.G.; Treebak, J.T.; et al. Hepatic NAD salvage pathway is enhanced in mice on a high-fat diet. Mol. Cell. Endocrinol. 2015, 412, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Dall, M.; Penke, M.; Sulek, K.; Matz-Soja, M.; Holst, B.; Garten, A.; Kiess, W.; Treebak, J.T. Hepatic NAD+ levels and NAMPT abundance are unaffected during prolonged high-fat diet consumption in C57BL/6JBomTac mice. Mol. Cell. Endocrinol. 2018, 473, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Auguet, T.; Terra, X.; Porras, J.A.; Orellana-Gavaldà, J.M.; Martinez, S.; Aguilar, C.; Lucas, A.; Pellitero, S.; Hernández, M.; Del Castillo, D.; et al. Plasma visfatin levels and gene expression in morbidly obese women with associated fatty liver disease. Clin. Biochem. 2013, 46, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Roldan, M.; Agerholm, M.; Nielsen, T.S.; Consitt, L.A.; Søgaard, D.; Helge, J.W.; Larsen, S.; Brandauer, J.; Houmard, J.A.; Treebak, J.T. Aerobic and resistance exercise training reverses age-dependent decline in NAD+ salvage capacity in human skeletal muscle. Physiol. Rep. 2019, 7, e14139. [Google Scholar]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 2012, 122, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Xu, A.W.; Koliwad, S.K. Hypothalamic inflammation in the control of metabolic function. Annu. Rev. Physiol. 2015, 77, 131–160. [Google Scholar] [CrossRef]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab. 2017, 26, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Pais, T.F.; Szego, E.M.; Marques, O.; Miller-Fleming, L.; Antas, P.; Guerreiro, P.; de Oliveira, R.M.; Kasapoglu, B.; Outeiro, T.F. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. 2013, 32, 2603–2616. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Minor, R.K.; Baur, J.A.; Gomes, A.P.; Ward, T.M.; Csiszar, A.; Mercken, E.M.; Abdelmohsen, K.; Shin, Y.-K.; Canto, C.; Scheibye-Knudsen, M. SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 2011, 1, 70. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol. Neurobiol. 2010, 41, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008, 8, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Conte, C.; Fontana, L.; Mittendorfer, B.; Imai, S.I.; Schechtman, K.B.; Gu, C.; Kunz, I.; Fanelli, F.R.; Patterson, B.W. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. 2012, 16, 658–664. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, D.A.; Guarente, L. Small-molecule allosteric activators of sirtuins. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 363–380. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.J.; Martin-Montalvo, A.; Mercken, E.M.; Palacios, H.H.; Ward, T.M.; Abulwerdi, G.; Minor, R.K.; Vlasuk, G.P.; Ellis, J.L.; Sinclair, D.A. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014, 6, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubota, K. The first human clinical study for NMN has started in Japan. NPJ Aging Mech. Dis. 2016, 2, 16021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poddar, S.K.; Sifat, A.E.; Haque, S.; Nahid, N.A.; Chowdhury, S.; Mehedi, I. Nicotinamide mononucleotide: exploration of diverse therapeutic applications of a potential molecule. Biomolecules 2019, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Elhassan, Y.S.; Kluckova, K.; Fletcher, R.S.; Schmidt, M.S.; Garten, A.; Doig, C.L.; Cartwright, D.M.; Oakey, L.; Burley, C.V.; Jenkinson, N. Nicotinamide riboside augments the aged human skeletal muscle NAD+ metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 2019, 28, 1717–1728.e1716. [Google Scholar] [CrossRef] [Green Version]
- Dollerup, O.L.; Christensen, B.; Svart, M.; Schmidt, M.S.; Sulek, K.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Møller, N.; Brenner, C.; Treebak, J.T.; et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 2018, 108, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Dollerup, O.L.; Trammell, S.A.; Hartmann, B.; Holst, J.J.; Christensen, B.; Møller, N.; Gillum, M.P.; Treebak, J.T.; Jessen, N. Effects of Nicotinamide Riboside on Endocrine Pancreatic Function and Incretin Hormones in Nondiabetic Men With Obesity. J. Clin. Endocrinol. Metab. 2019, 104, 5703–5714. [Google Scholar] [CrossRef]
- Dollerup, O.L.; Chubanava, S.; Agerholm, M.; Søndergård, S.D.; Altıntaş, A.; Møller, A.B.; Høyer, K.F.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Lavery, G.G.; et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J. Physiol. 2020, 598, 731–754. [Google Scholar] [CrossRef]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E. Slc12a8 is a nicotinamide mononucleotide transporter. Nature Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S.i. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014, 33, 1321–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.J.; Yoshida, M.; Johnson, S.; Takikawa, A.; Usui, I.; Tobe, K.; Nakagawa, T.; Yoshino, J.; Imai, S.-i. SIRT1-mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD+ and function in mice. Cell Metab. 2015, 21, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, E.; Park, J.W.; Kang, G.M.; Lee, C.H.; Dugu, H.; Gil, S.Y.; Song, D.K.; Kim, H.J.; Son, G.H.; Yu, R. Exogenous nicotinamide adenine dinucleotide regulates energy metabolism via hypothalamic connexin 43. Metabolism 2018, 88, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Rouach, N.; Koulakoff, A.; Abudara, V.; Willecke, K.; Giaume, C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008, 322, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, A.J.; Domínguez-Pinos, M.-D.; Guerra, M.M.; Fernández-Llebrez, P.; Pérez-Fígares, J.-M. Structure and function of the ependymal barrier and diseases associated with ependyma disruption. Tissue barriers 2014, 2, e28426. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R. Nampt/PBEF/visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell metab. 2007, 6, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.I. The NAD World 2.0: the importance of the inter-tissue communication mediated by NAMPT/NAD+/SIRT1 in mammalian aging and longevity control. NPJ Syst. Biol. Appl. 2016, 2, 16018. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.H.; Chang, D.M.; Lin, K.C.; Shin, S.J.; Lee, Y.J. Visfatin in overweight/obesity, type 2 diabetes mellitus, insulin resistance, metabolic syndrome and cardiovascular diseases: a meta-analysis and systemic review. Diabetes Metab. Res. Rev. 2011, 27, 515–527. [Google Scholar] [CrossRef]
- Blüher, M. Vaspin in obesity and diabetes: pathophysiological and clinical significance. Endocrine. 2012, 41, 176–182. [Google Scholar] [CrossRef]
- Barbosa, M.T.P.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2. 2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roh, E.; Kim, M.-S. Hypothalamic NAD+-Sirtuin Axis: Function and Regulation. Biomolecules 2020, 10, 396. https://doi.org/10.3390/biom10030396
Roh E, Kim M-S. Hypothalamic NAD+-Sirtuin Axis: Function and Regulation. Biomolecules. 2020; 10(3):396. https://doi.org/10.3390/biom10030396
Chicago/Turabian StyleRoh, Eun, and Min-Seon Kim. 2020. "Hypothalamic NAD+-Sirtuin Axis: Function and Regulation" Biomolecules 10, no. 3: 396. https://doi.org/10.3390/biom10030396
APA StyleRoh, E., & Kim, M. -S. (2020). Hypothalamic NAD+-Sirtuin Axis: Function and Regulation. Biomolecules, 10(3), 396. https://doi.org/10.3390/biom10030396