Synthesis, In Vitro Evaluation and Molecular Docking of the 5-Acetyl-2-aryl-6-hydroxybenzo[b]furans against Multiple Targets Linked to Type 2 Diabetes


Abstract
:
1. Introduction
2. Materials and Methods
2.1. General
2.2. Typical Procedure for the Synthesis of Compounds 2a–h
2.2.1. 1-[6-Hydroxy-2-phenylbenzofuran-5-yl]ethanone (2a)
2.2.2. 1-[2-(3-Fluorophenyl)-6-hydroxybenzofuran-5-yl]ethanone (2b)
2.2.3. 1-[2-(4-Fluorophenyl)-6-hydroxybenzofuran-5-yl]ethanone (2c)
2.2.4. 1-[2-(3-Chlorophenyl)-6-hydroxybenzofuran-5-yl]ethanone (2d)
2.2.5. 1-[2-(4-Chlorophenyl)-6-hydroxybenzofuran-5-yl]ethanone (2e)
2.2.6. 1-[6-Hydroxy-2-(4-tolyl)benzofuran-5-yl]ethanone (2f)
2.2.7. 1-[6-Hydroxy-2-(4-methoxyphenyl)benzofuran-5-yl]ethanone (2g)
2.2.8. 1-[6-Hydroxy-2-(3,5-dimethoxyphenyl)benzofuran-5-yl]ethanone (2h)
2.3. Bioassays
2.3.1. Inhibition of α-Glucosidase Activity by Compounds by 2a–h
2.3.2. Inhibition PTP1B Activity by Compounds 2a–h
2.3.3. In Vitro β-Secretase (BACE-1) Enzyme Assay
2.3.4. DPPH Radical Scavenging Activity of Compounds 2a–h
2.3.5. COX-2 Inhibitory Assays of 2c, 2g and 2h
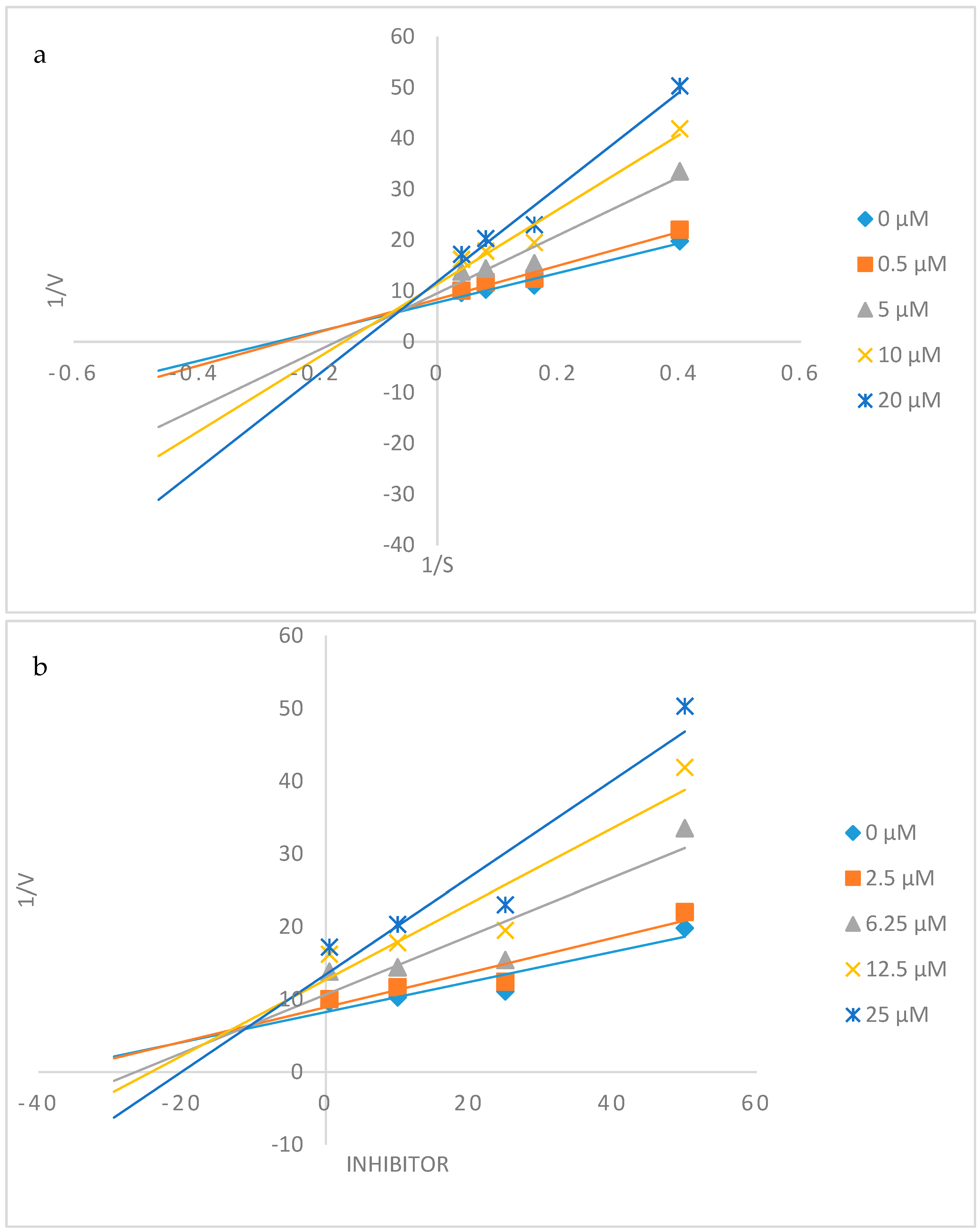
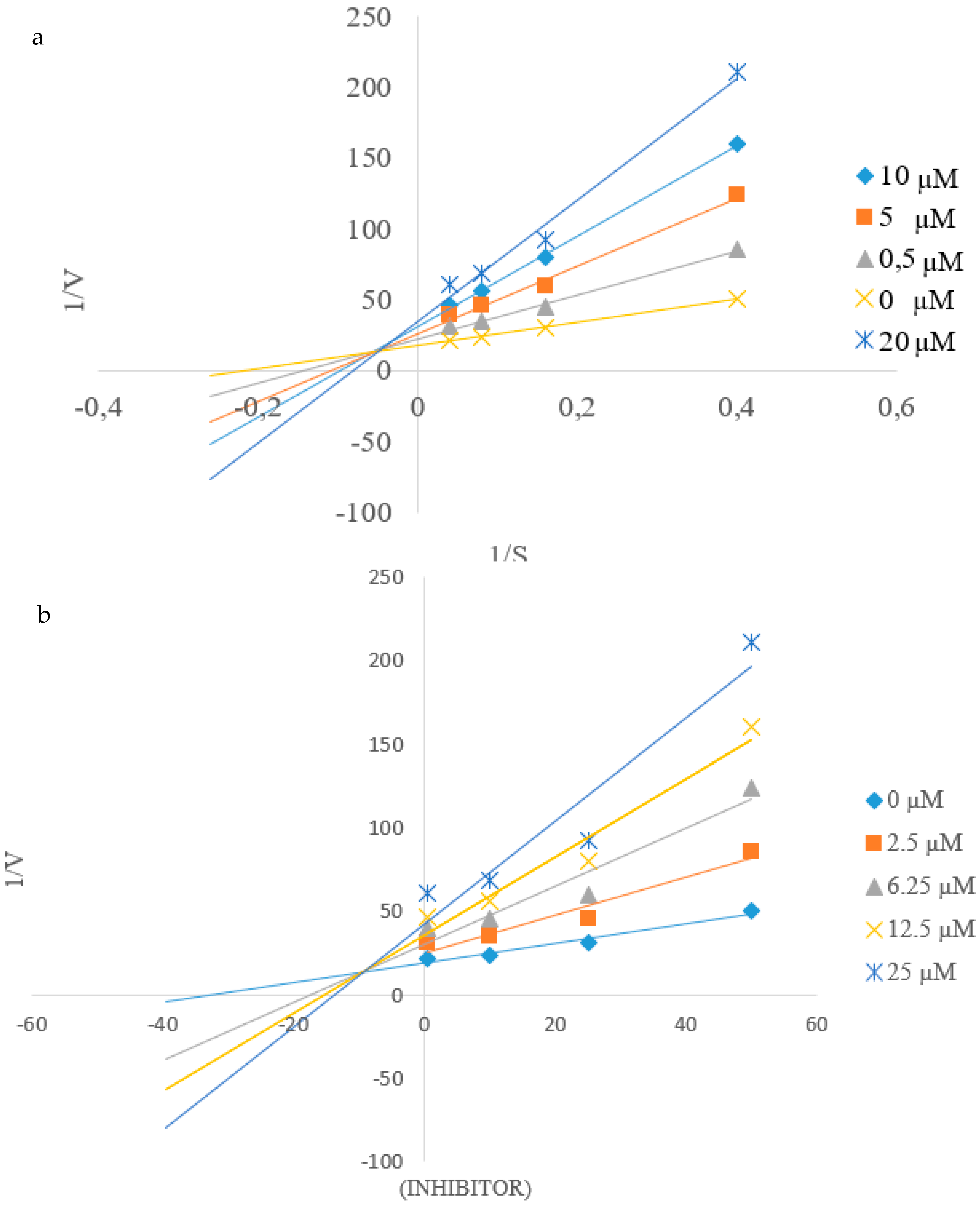
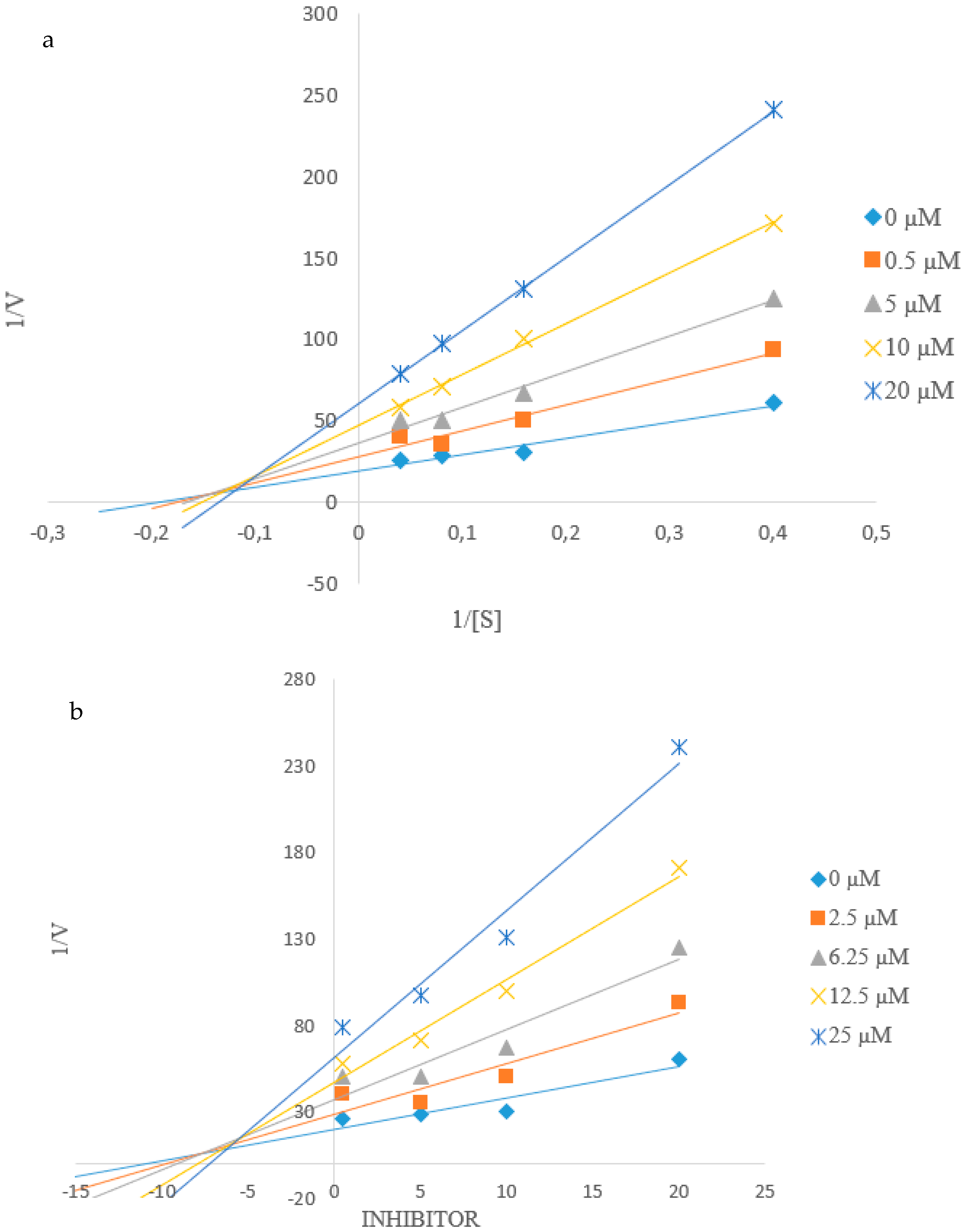
2.4. Kinetic Studies of 2c and 2h towards α-Glucosidase and PTP1B Inhibition
2.4.1. Kinetic Studies of 2c and 2h towards α-Glucosidase Inhibition
2.4.2. Kinetic Studies of 2c and 2h towards PTP1B Inhibition
2.5. Molecular Docking Studies into α-Glucosidase and PTP1B Active Sites
2.5.1. Protein Structure
2.5.2. Ligand Structure
2.5.3. Molecular Docking Simulation
3. Results and Discussion
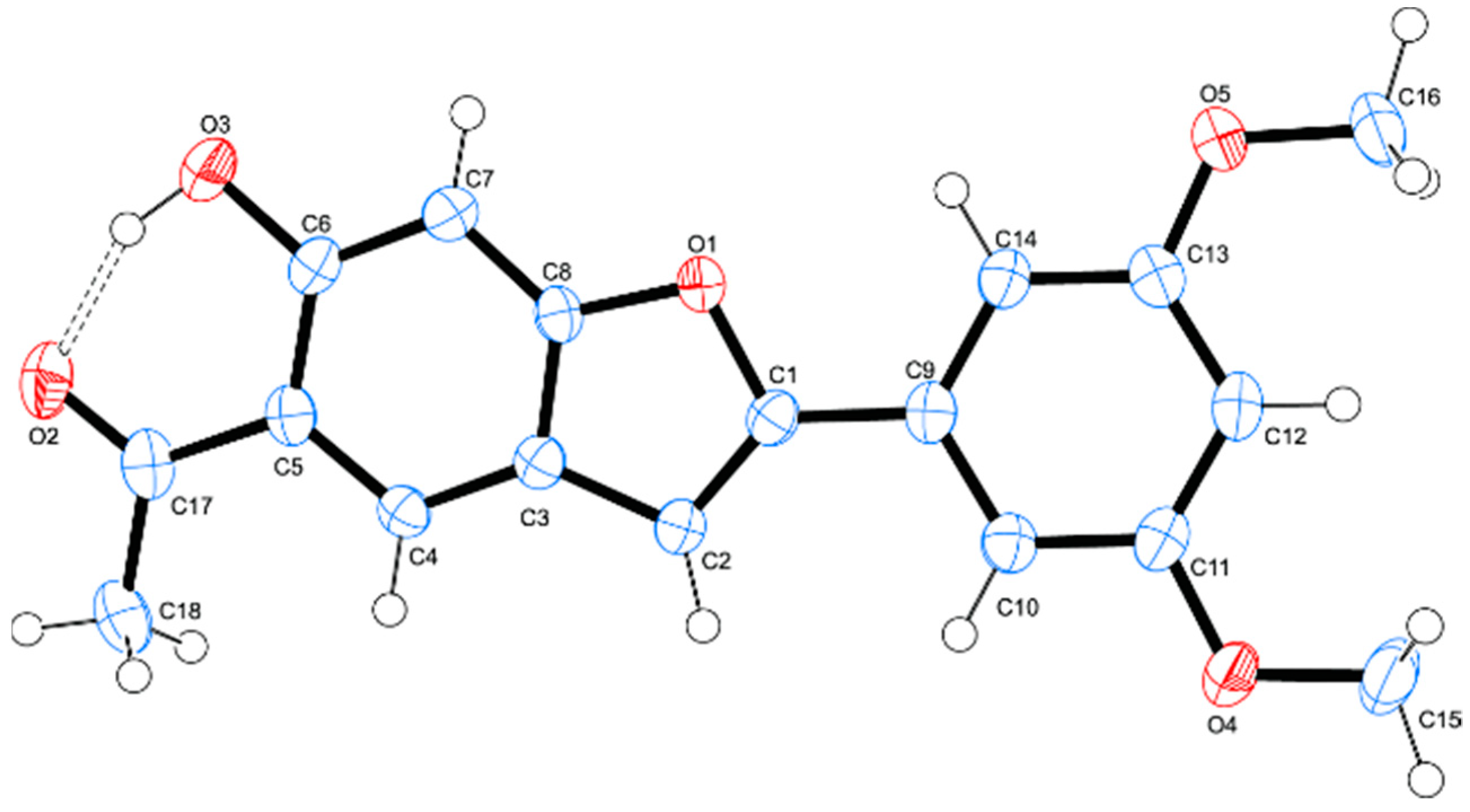
3.1. Chemistry
3.2. Biology
3.2.1. Inhibition of α-Glucosidase, PTP1B and β-Secretase and Antioxidant Activity of 2a–h
3.2.2. Inhibitory Assays of 2c, 2g and 2h against COX-2 Activity
3.2.3. Kinetic Studies on 2c and 2h against α-Glucosidase and PTP1B Activity
3.3. Molecular Docking Studies
3.3.1. Molecular Docking of 2a–h into α-Glucosidase Binding Site
3.3.2. Molecular Docking of 2a–h into PTP1B Binding Sites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Z.; Ma, S. Recent advances in synthetic α-glucosidase inhibitors. MedChemChem 2017, 12, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.F.; Gutierrez, A.; Castro, M.; Tsewang, D.; Schade, D.S. Targeting postprandial hyperglycaemia: A comparative study of insulin tropic agents in type 2 diabetes. J. Clin. Endocrinol. Metab. 2003, 88, 5248–5254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanak, S.; Saad, B.; Zaid, H. Metabolic and epigenetic action mechanisms of antidiabetic medicinal plants. Evid. Based Complement. Alternat. Med. 2019, 2019. [Google Scholar] [CrossRef]
- Seong, S.H.; Roy, A.; Jung, H.A.; Jung, H.J.; Choi, J.S. Protein tyrosine phosphatase 1B and α-glucosidase inhibitory activities of Pueraria lobata root and its constituents. J. Ethnopharmacol. 2016, 194, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, H. The mechanism of alpha-glucosidase inhibition in the management of diabetes. Clin. Investig. Med. 1995, 18, 303–311. [Google Scholar]
- Norrisa, K.; Norrisa, F.; Konod, D.H.; Vestergaardc, H.; Pedersenc, O.; Theofilopoulosd, A.N.; Mollerb, N.P.H. Expression of protein-tyrosine phosphatases in the major insulin target tissues. FEBS Lett. 1997, 415, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Trevillyan, J.M. Protein tyrosine phosphatase 1B as a target for the treatment of impaired glucose tolerance and type II diabetes. Curr. Opin. Investig. Drugs 2002, 3, 1608–1616. [Google Scholar]
- Tonks, N.K.; Neel, B.G. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 2001, 13, 182–195. [Google Scholar] [CrossRef]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924. [Google Scholar] [CrossRef]
- Song, Y.H.; Uddin, Z.; Jin, Y.M.; Li, Z.; Curtis-Long, M.S.; Kim, K.D.; Cho, J.K.; Park, K.H. Inhibition of protein tyrosine phosphatase (PTP1B) and α-glucosidase by geranylated flavonoids from Paulownia tomentosa. J. Enzyme Inhib. Med. Chem. 2017, 32, 1195–1202. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Mezzapesa, A.; Benabou, E.; Haas, M.E.; Bonardo, B.; Grino, M.; Brunel, J.-M.; Desbois-Mouthon, C.; Biddinger, S.B.; Govers, R.; et al. The beta secretase BACE1 regulates the expression of insulin receptor in the liver. Nat. Commun. 2018, 9, 1306–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, D.; Kaur, A.; Goyal, B. Benzofuran and indole: Promising scaffolds for drug development in Alzheimer’s disease. ChemMedChem 2018, 13, 1275–1299. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Signh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Picu, A.; Petcu, L.; Ştefan, S.; Mitu, M.; Lixandru, D.; Ionescu-Tîrgovişte, C.; Pîrcălăbioru, G.G.; Ciulu-Costinescu, V.; Bubulica, M.V.; Chifiriuc, M.C. Markers of oxidative stress and antioxidant defence in Romanian patients with type 2 diabetes mellitus and obesity. Molecules 2017, 22, 714. [Google Scholar] [CrossRef]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes mellitus and inflammation. Curr. Diabetes Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef]
- Khanan, H.; Shamsuzzaman. Bioactive benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef]
- Naik, R.; Harmalkar, D.S.; Xu, X.; Jang, K.; Lee, K. Bioactive benzofuran derivatives: Moraceae A–Z in medicinal chemistry. Eur. J. Med. Chem. 2015, 90, 379–393. [Google Scholar] [CrossRef]
- Guo, F.; Zhang, S.; Yan, X.; Dan, Y.; Wang, J.; Zhao, Y.; Yu, Z. Bioassay-guided isolation of antioxidant and α-glucosidase inhibitory constituents from stem of Vigna angularis. Bioorg. Chem. 2019, 87, 312–320. [Google Scholar] [CrossRef]
- Dan, W.-J.; Zhang, Q.; Zhang, F.; Wang, W.-W.; Gao, J.-M. Benzoate derivatives of acetophenone as potent α-glucosidase inhibitors: Synthesis, structure–activity relationship and mechanism. J. Enzyme Inhib. Med. Chem. 2019, 34, 937–945. [Google Scholar] [CrossRef] [Green Version]
- Spasov, A.A.; Babkov, D.A.; Prokhorova, T.Y.; Sturova, E.A.; Muleeva, D.R.; Demidov, M.R.; Osipov, D.V.; Osyanin, V.A.; Klimochkin, Y.N. Synthesis and biological evaluation of 2-acylbenzofuranes as novel α-glucosidase inhibitors with hypoglycemic activity. Chem. Biol. Drug Des. 2017, 90, 1184–1189. [Google Scholar] [CrossRef]
- Dixit, M.; Tripathi, B.K.; Srivastava, A.K.; Goel, A. Synthesis of functionalized acetophenones as protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3394–3397. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Seong, S.H.; Park, S.G.; Min, B.S.; Jung, H.A.; Choi, J.S. Insight into the PTP1B inhibitory activity of arylbenzofurans: An in vitro and in silico study. Molecules 2019, 24, 2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mphahlele, M.J.; Olomola, T.O. Synthesis and transformation of 5-acetyl-2-aryl-6-hydroxybenzofurans into furanoflavanone derivatives. Synthesis 2019, 51, 3431–3442. [Google Scholar] [CrossRef]
- Shi, Z.L.; Liu, Y.D.; Yuan, Y.Y.; Song, D.; Qi, M.F.; Yang, X.J.; Wang, P.; Li, X.Y.; Shang, J.H.; Yang, Z.X. In vitro and in vivo effects of Norathyriol and Mangiferin on α-glucosidase. Biochem. Res. Int. 2017, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, B.J.; Bittner-Kowalezyk, A.; White, M.F.; Harbeck, M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the GRB2 adaptor protein. J. Biol. Chem. 2000, 275, 4283–4289. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Zhou, H.; Qian, H. Antioxidant and free radical-scavenging activities of wheat germ protein hydrolysates (WGPH) prepared with alcalase. Process Biochem. 2006, 41, 1296–1302. [Google Scholar] [CrossRef]
- Morgat, A.; Lombardot, T.; Coudert, E.; Axelsen, K.; Neto, T.B.; Gehant, S.; Bansal, P.; Bolleman, J.; Gasteiger, E.; de Castro, E.; et al. Enzyme annotation in UniProtKB using Rhea. Bioinformatics 2019, 1–6. [Google Scholar] [CrossRef]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Pollastri, G.; McLysaght, A. Porter: A new, accurate server for protein secondary structure prediction. Bioinformatics 2005, 21, 1719–1720. [Google Scholar] [CrossRef]
- Rost, B.; Yachdav, G.; Liu, J. The PredictProtein server. Nucleic Acids Res. 2004, 32, W321–W326. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.; Simossis, V.A.; Taylor, W.R.; Heringa, J. A simple and fast secondary structure prediction algorithm using Hidden Neural Networks. Bioinformatics 2005, 21, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Šali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar]
- Liithy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Szczepankiewicz, B.G.; Liu, G.; Hajduk, P.J.; Abad-Zapatero, C.; Pei, Z.; Xin, Z.; Lubben, T.H.; Trevillyan, J.M.; Stashko, M.A.; Ballaron, S.J.; et al. Discovery of a potent, selective protein tyrosine phosphatase 1B inhibitor using a linked-fragment strategy. J. Am. Chem. Soc. 2003, 125, 4087–4096. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Ag, H.B.B. Pharmacology of α-glucosidase inhibition. Eur. J. Clin. Investig. 1994, 24, 3–10. [Google Scholar] [CrossRef]
- Zhang, W.; Kim, D.; Philip, E.; Miyan, Z.; Barykina, I.; Schmidt, B.; Stein, H. A multinational, observational study to investigate the efficacy, safety and tolerability of acarbose as add-on or monotherapy in a range of patients: The glucoVIP study. Clin. Drug Investig. 2013, 33, 263–274. [Google Scholar] [CrossRef]
- Rosak, C.; Mertes, G. Critical evaluation of the role of acarbose in the treatment of diabetes: Patient considerations. Diabetes Metab. Syndr. Obes. 2012, 5, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Böhm, H.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef]
- Hsieh, J.-F.; Lin, W.-J.; Huang, K.-F.; Liao, J.-H.; Don, M.-W.; Shen, C.-C.; Shiao, Y.-J.; Li, W.-T. Antioxidant activity and inhibition of α-glucosidase by hydroxyl-functionalized 2-arylbenzo[b]furans. Eur. J. Med. Chem. 2015, 93, 443–451. [Google Scholar] [CrossRef]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.B.; Sharpe, A.H.; et al. Increased energy expenditure, decreased adiposity and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Hauser, C.; Kahn, B.B.; Haj, F.G.; Neel, B.G.; Tonks, N.K.; Tiganis, T. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol. Cell. Biol. 2005, 25, 819–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, N.; Li, J.; Shechter, Y. Vanadium salts as insulin substitutes: Mechanisms of action, a scientific and therapeutic tool in diabetes mellitus research. Crit. Rev. Biochem. Mol. Biol. 1996, 31, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Bashan, N.; Kovsan, J.; Kjachko, I.; Ovadia, H.; Rudich, A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol. Rev. 2009, 89, 27–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Famuyiwa, S.O.; Sanusi, K.; Faloye, K.O.; Yilmaz, Y.; Ceylan, U. Antidiabetic and antioxidant activities: Is there any link between them? New J. Chem. 2019, 43, 13326–13329. [Google Scholar] [CrossRef]
- Ahangarpour, A.; Sayahi, M.; Sayahi, M. The antidiabetic and antioxidant properties of some phenolic phytochemicals: A review study. Diabetes Metab. Syndr. Rev. 2019, 13, 854–857. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Aispuro-Pérez, A.; López-Ávalos, J.; García-Páez, F.; Montes-Avila, J.; Picos-Corrales, L.A.; Ochoa-Terán, A.; Bastidas, P.; Montaño, S.; Calderón-Zamora, L.; Osuna-Martínez, U.; et al. Synthesis and molecular docking studies of imines as α-glucosidase and α-amylase inhibitors. Bioorg. Chem. 2020, 94, 103491. [Google Scholar] [CrossRef]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randall, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struc. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Lu, S.; Huang, W.; Geng, L.; Shen, Q.; Zhang, J. The mechanism of allosteric inhibition of protein tyrosine phosphatase 1B. PLoS ONE 2014, 9, e97668. [Google Scholar] [CrossRef] [Green Version]
- Shinde, R.N.; Kumar, G.S.; Eqbal, S.; Sobhia, M.E. Screening and identification of potential PTP1B allosteric inhibitors using in silico and in vitro approaches. PLoS ONE 2018, 13, e0199020. [Google Scholar] [CrossRef]
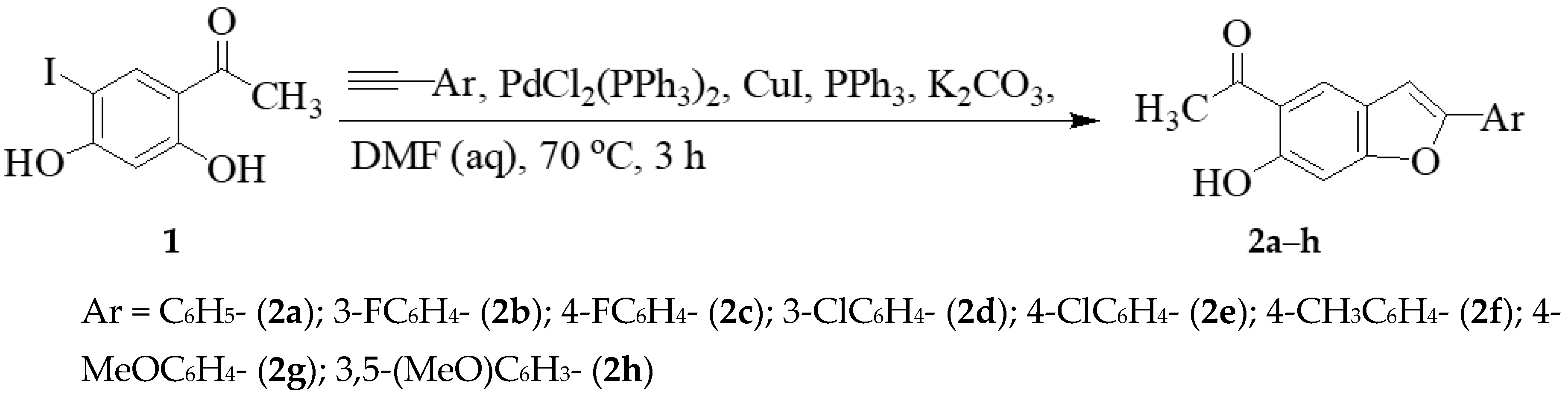




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µM) | |||
|---|---|---|---|---|
| α-Glucosidase | PTP1B | β-Secretase | DPPH | |
| 2a | >100 | 22.99 ± 0.23 | 63.15 ± 0.07 | 12.15 ± 2.10 |
| 2b | 4.65 ± 0.13 | 24.50 ± 0.29 | >100 | 46.50 ± 0.24 |
| 2c | 0.11 ± 0.04 | 17.75 ± 0.84 | >100 | 9.82 ± 0.86 |
| 2d | >100 | 29.29 ± 0.23 | >100 | 15.48 ± 0.21 |
| 2e | 64.90 ± 0.05 | 21.46 ± 0.27 | > 100 | 11.34 ± 1.20 |
| 2f | 14.05 ± 0.07 | 26.67 ± 0.30 | > 100 | 26.67 ± 0.62 |
| 2g | 0.56 ± 0.24 | 31.88 ± 0.46 | 27.30 ± 1.10 | 16.84 ± 0.38 |
| 2h | 0.78 ± 0.31 | 11.90 ± 0.35 | 25.80 ± 0.08 | 6.28 ± 0.33 |
| Acarbose | 0.01 ± 0.02 | - | - | - |
| Na2VO4 | - | 38.42 ± 0.28 | - | - |
| Quercetin | - | - | 8.76 ± 0.10 | - |
| Ascorbic acid | - | - | - | 10.72 ± 0.42 |
| Compounds | COX-2 (IC50 µM) |
|---|---|
| 2c | 2.81 ± 0.70 |
| 2g | 2.33 ± 1.52 |
| 2h | 99.55 ± 2.60 |
| Celecoxib | 1.02 ± 1.53 |
| Compound | α-Glucosidase |
|---|---|
| Control | −5.94 |
| 2a | −6.49 |
| 2b | −6.43 |
| 2c | −6.27 |
| 2d | −6.58 |
| 2e | −6.11 |
| 2f | −6.70 |
| 2g | −6.92 |
| 2h | −6.92 |
| Compound | PTB1B | |
|---|---|---|
| Catalytic Site | Allosteric Site | |
| Control | −7.81 | −11.20 |
| 2a | −5.45 | −6.85 |
| 2b | −5.43 | −6.85 |
| 2c | −5.42 | −6.82 |
| 2d | −5.60 | −7.09 |
| 2e | −5.64 | −7.10 |
| 2f | −5.58 | −7.05 |
| 2g | −5.35 | −6.82 |
| 2h | −5.43 | −7.50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mphahlele, M.J.; Choong, Y.S.; Maluleka, M.M.; Gildenhuys, S. Synthesis, In Vitro Evaluation and Molecular Docking of the 5-Acetyl-2-aryl-6-hydroxybenzo[b]furans against Multiple Targets Linked to Type 2 Diabetes. Biomolecules 2020, 10, 418. https://doi.org/10.3390/biom10030418
Mphahlele MJ, Choong YS, Maluleka MM, Gildenhuys S. Synthesis, In Vitro Evaluation and Molecular Docking of the 5-Acetyl-2-aryl-6-hydroxybenzo[b]furans against Multiple Targets Linked to Type 2 Diabetes. Biomolecules. 2020; 10(3):418. https://doi.org/10.3390/biom10030418
Chicago/Turabian StyleMphahlele, Malose J., Yee Siew Choong, Marole M. Maluleka, and Samantha Gildenhuys. 2020. "Synthesis, In Vitro Evaluation and Molecular Docking of the 5-Acetyl-2-aryl-6-hydroxybenzo[b]furans against Multiple Targets Linked to Type 2 Diabetes" Biomolecules 10, no. 3: 418. https://doi.org/10.3390/biom10030418