Improved Protocol for the Production of the Low-Expression Eukaryotic Membrane Protein Human Aquaporin 2 in Pichia pastoris for Solid-State NMR



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Transformation
2.3. Colony Screening
2.4. hAQP2 Expression Optimization
2.5. LT-SEVIN Large Scale Growth
2.6. Cell Breakage
2.7. Solubilization
2.8. Purification
2.9. Functional Assay of hAQP2
2.10. Lipid Reconstitution for SSNMR
2.11. Sample Preparation for SSNMR
2.12. SSNMR Spectroscopy
3. Results
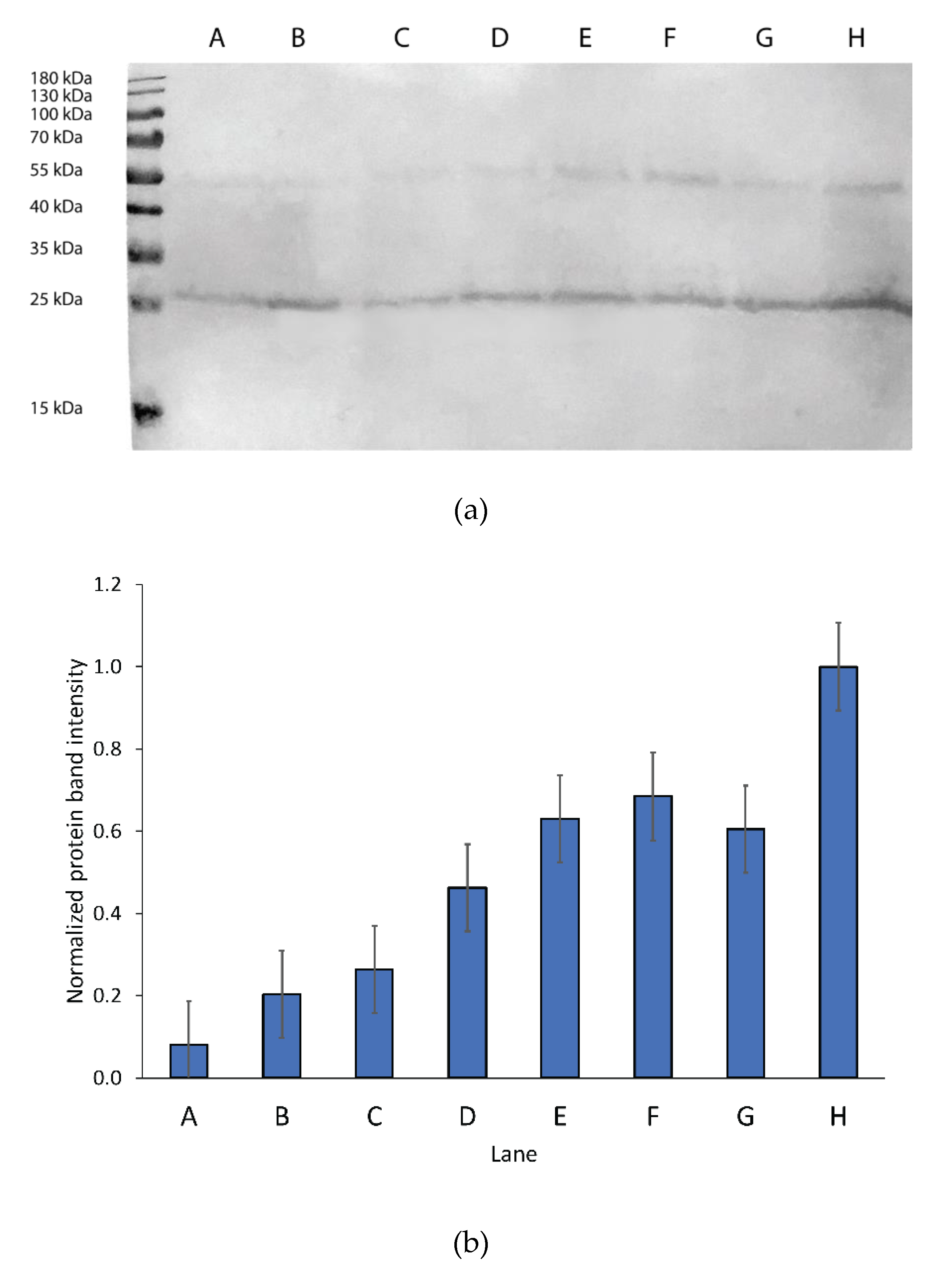
3.1. hAQP2 Expression Optimization
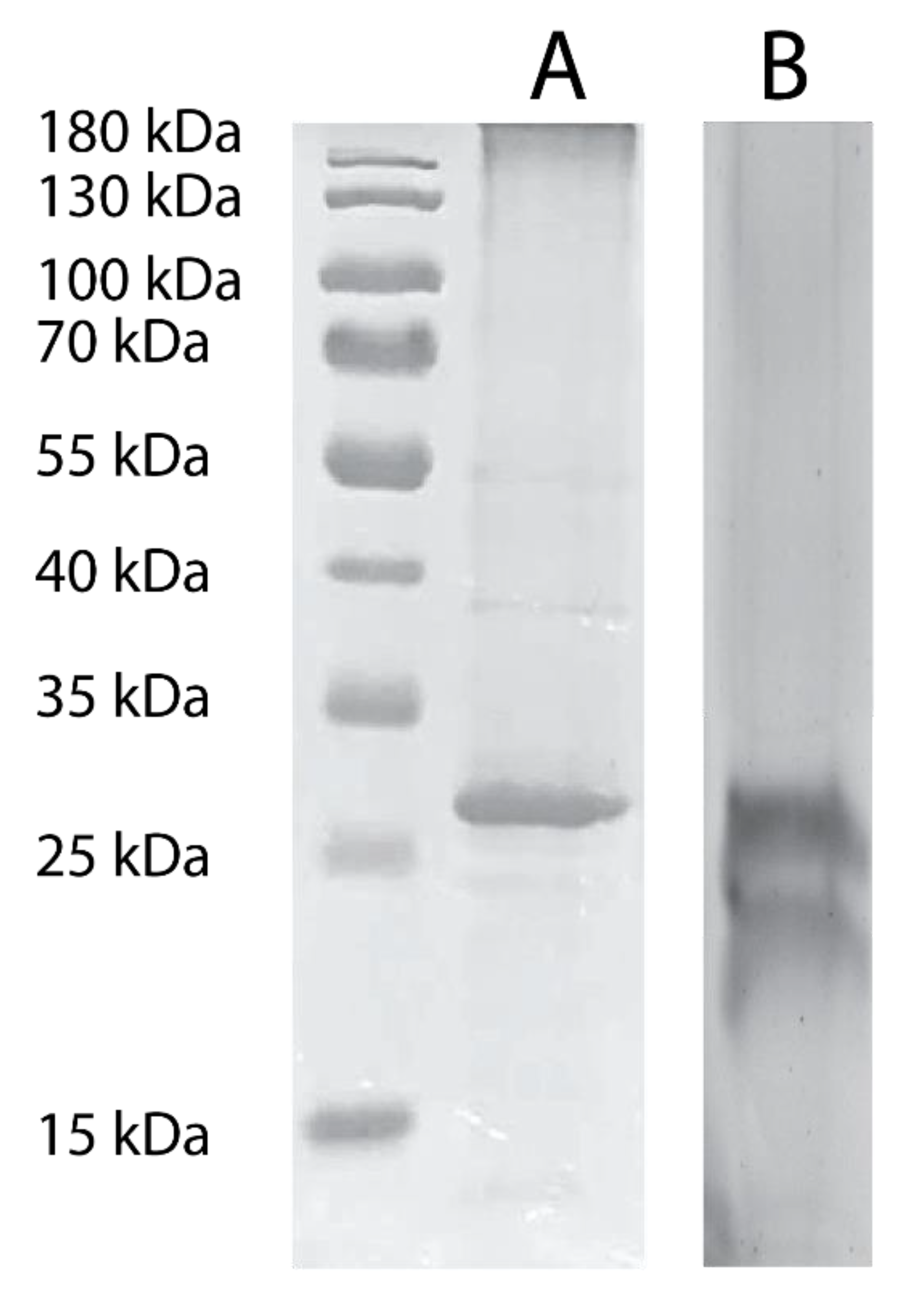
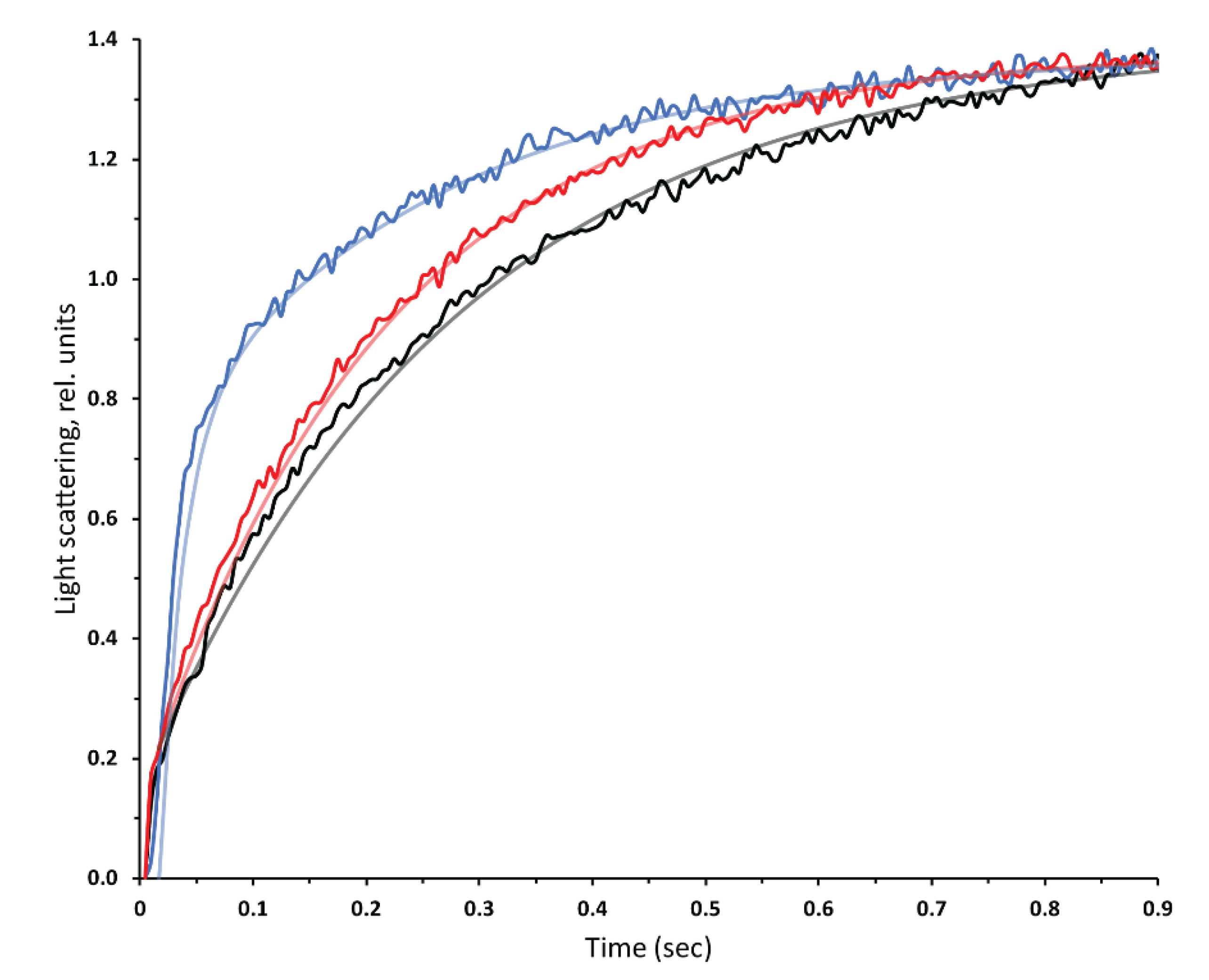
3.2. Functional Assays
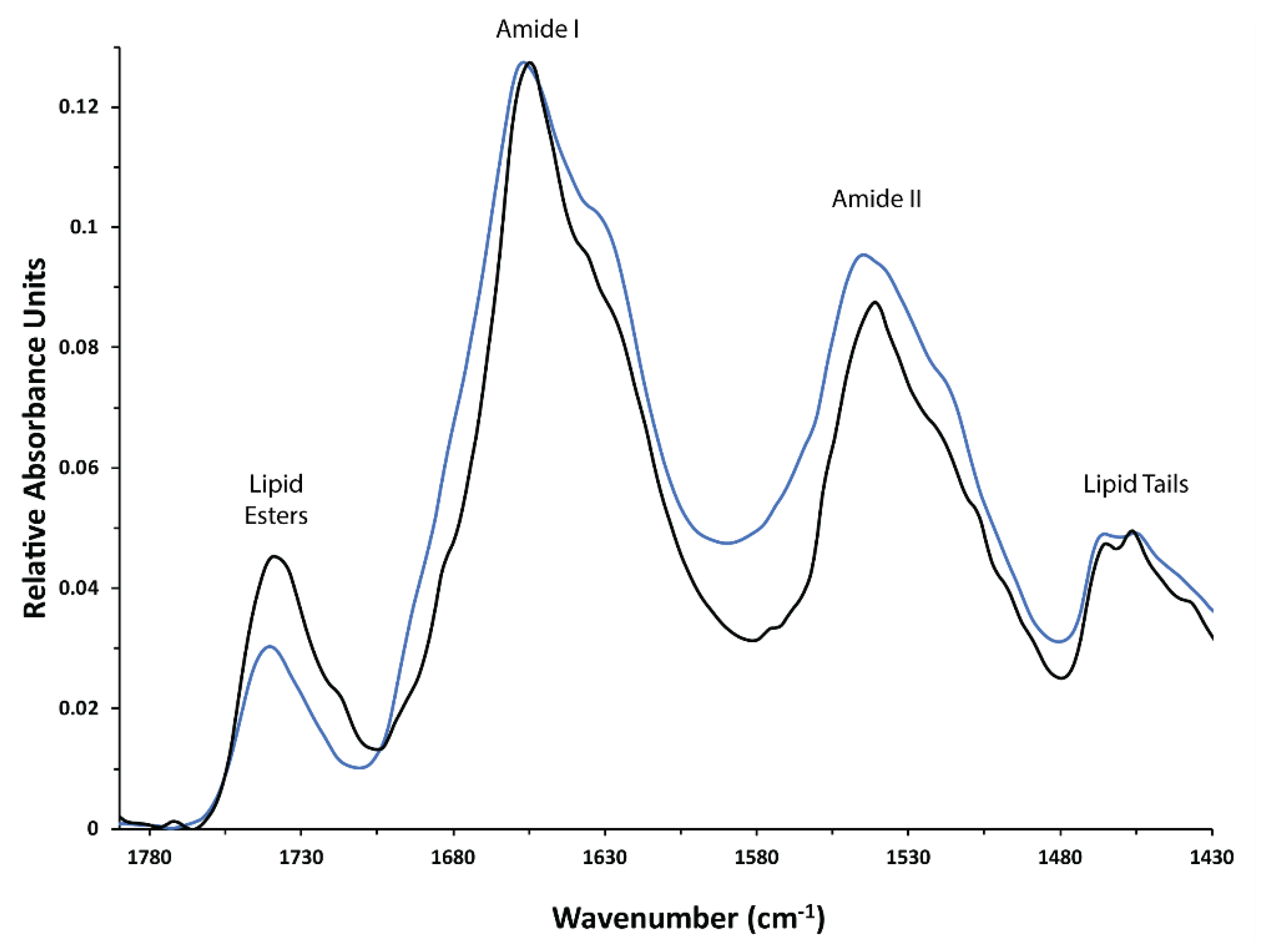
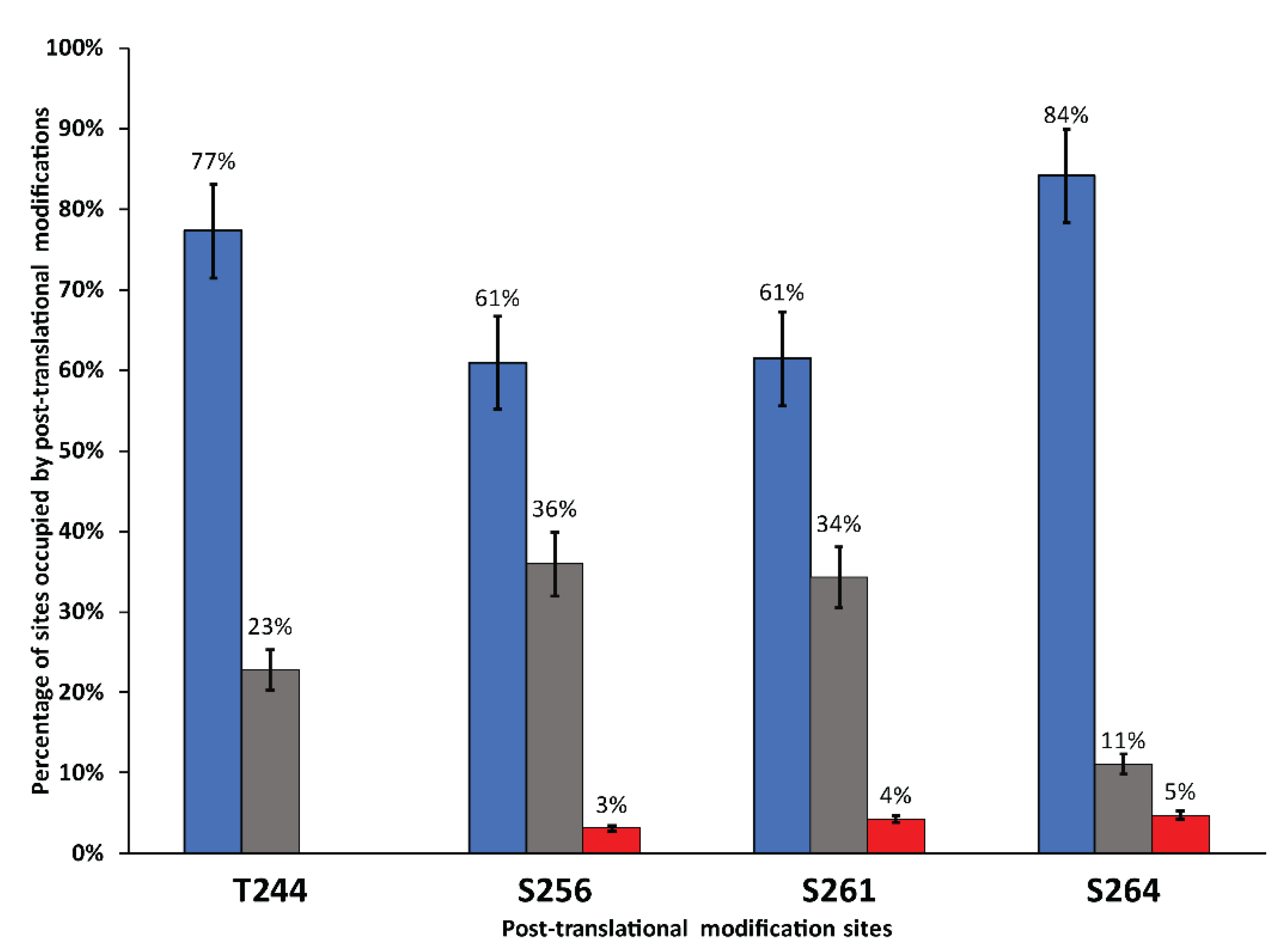
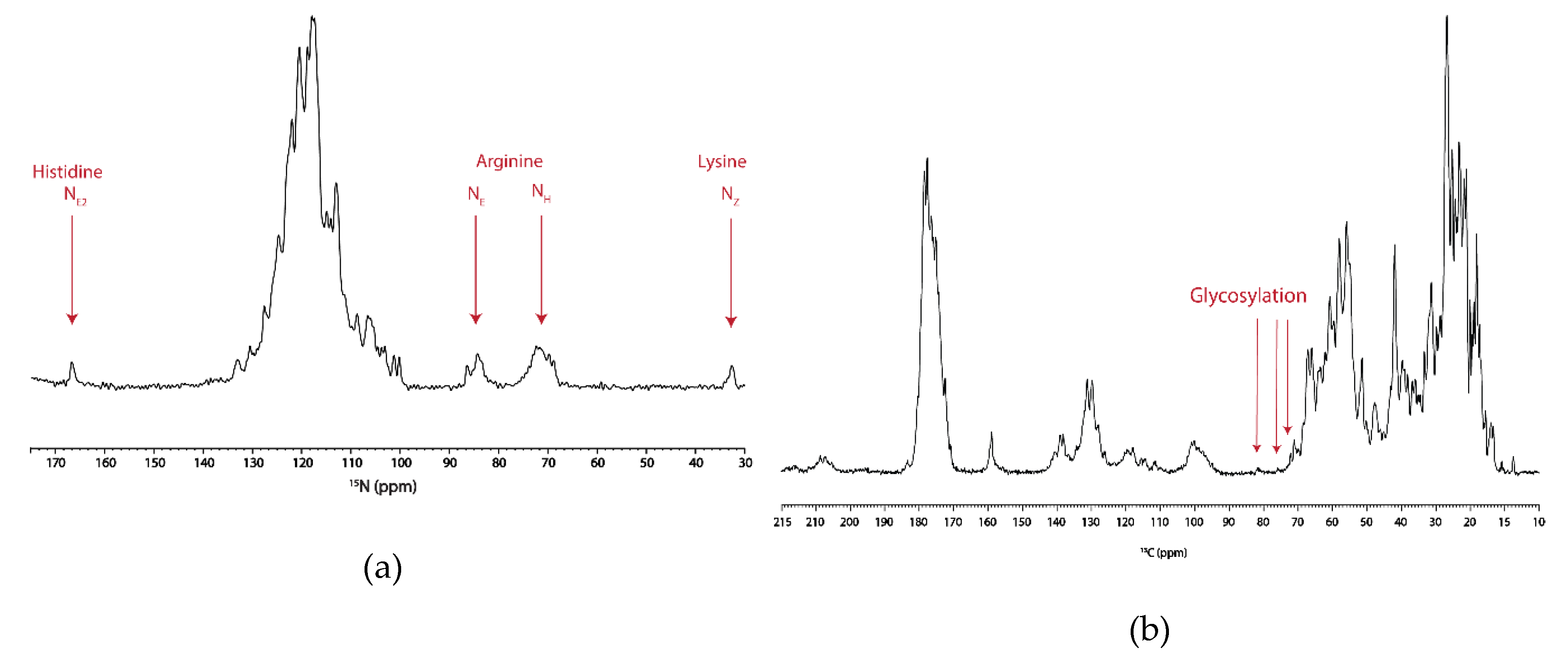
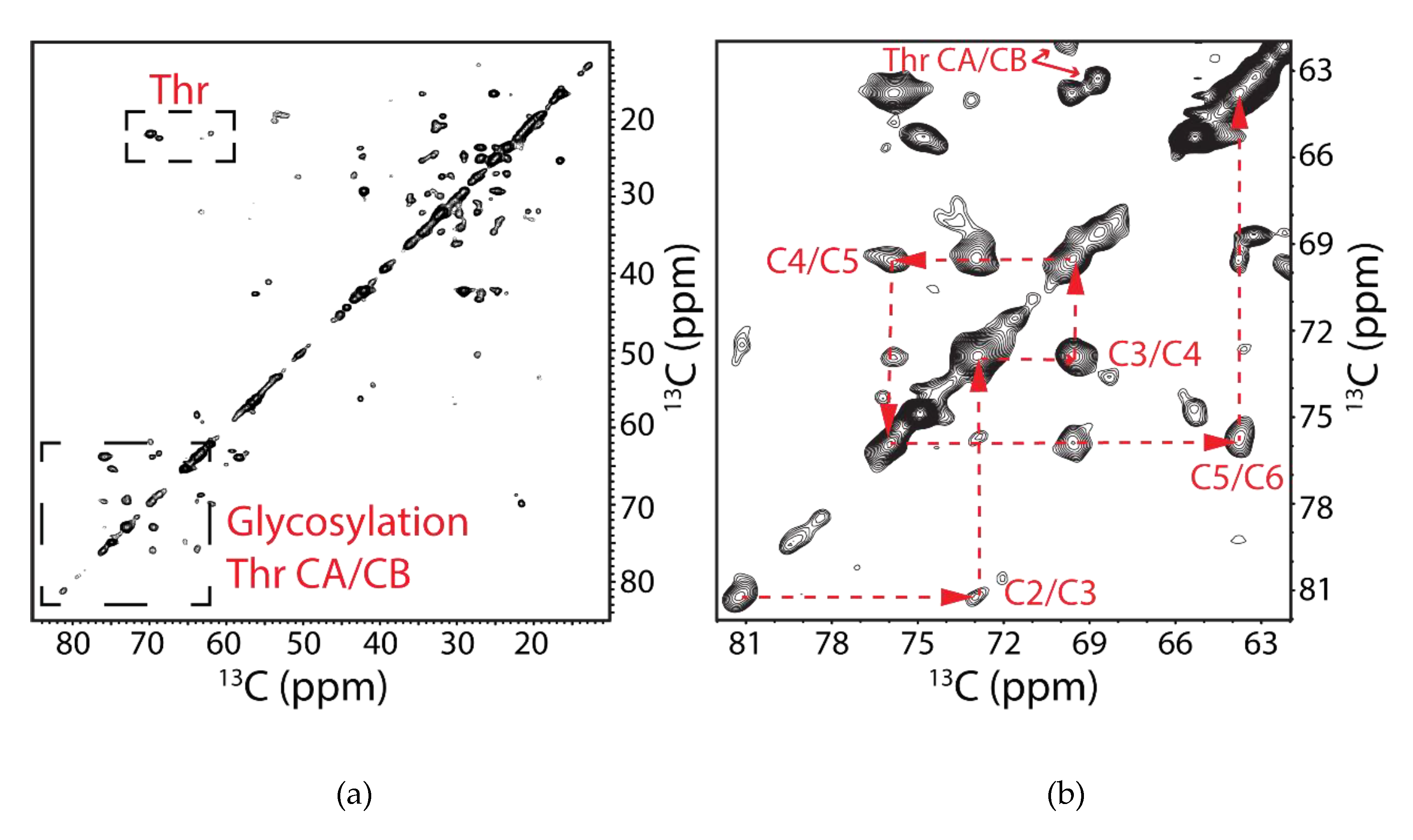
3.3. Analysis of Post-Translational Modifications by Mass Spectrometry
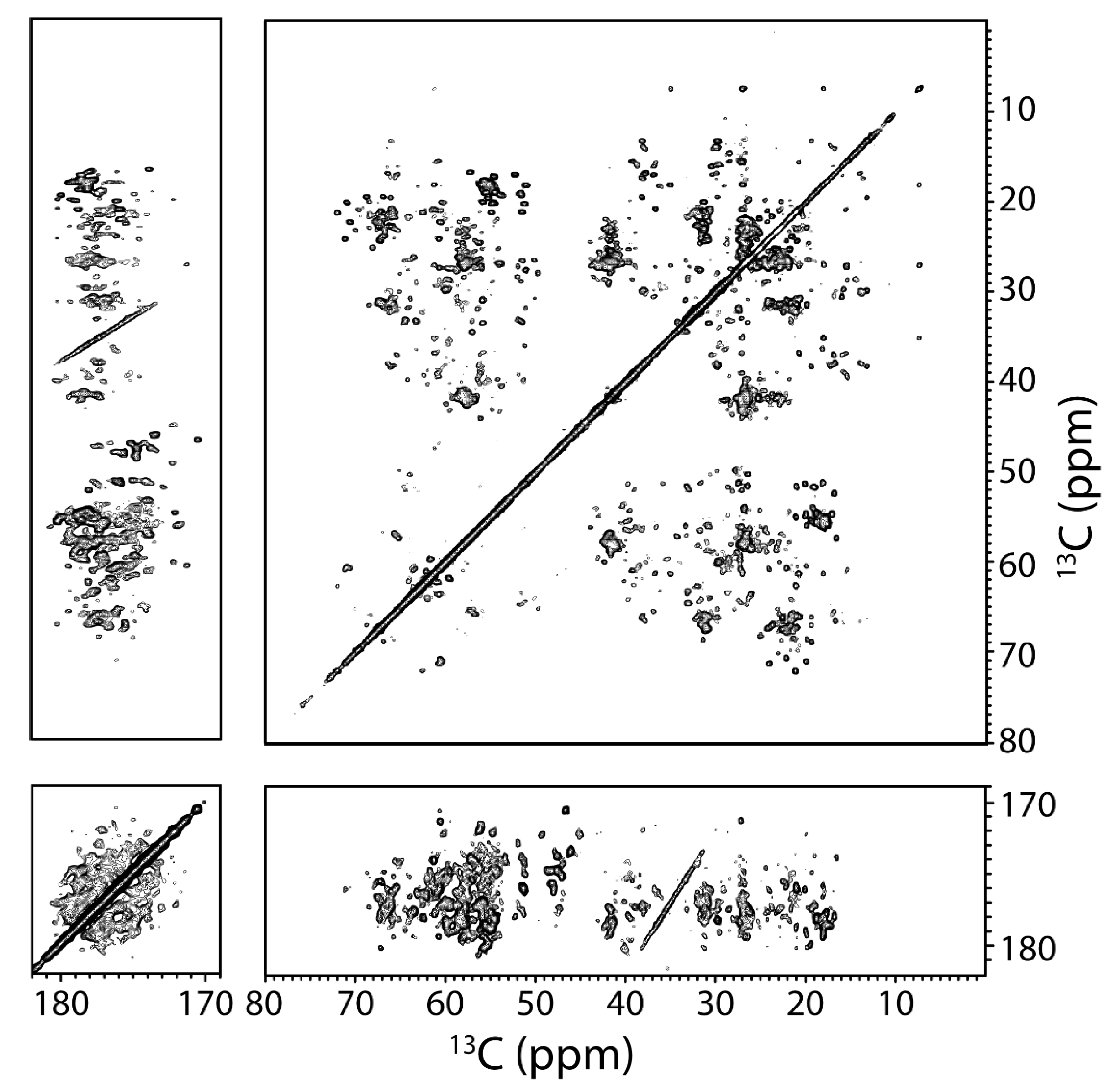
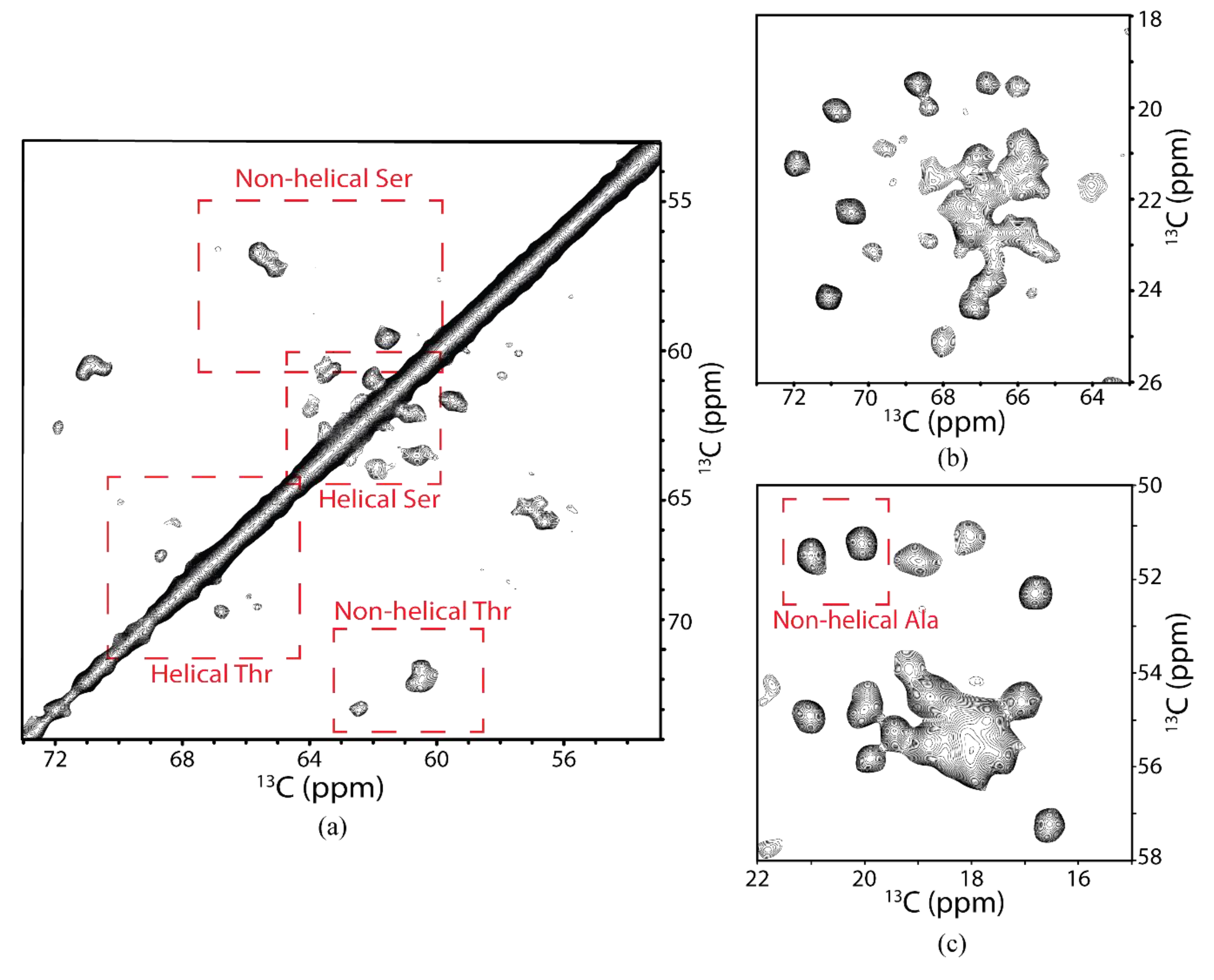
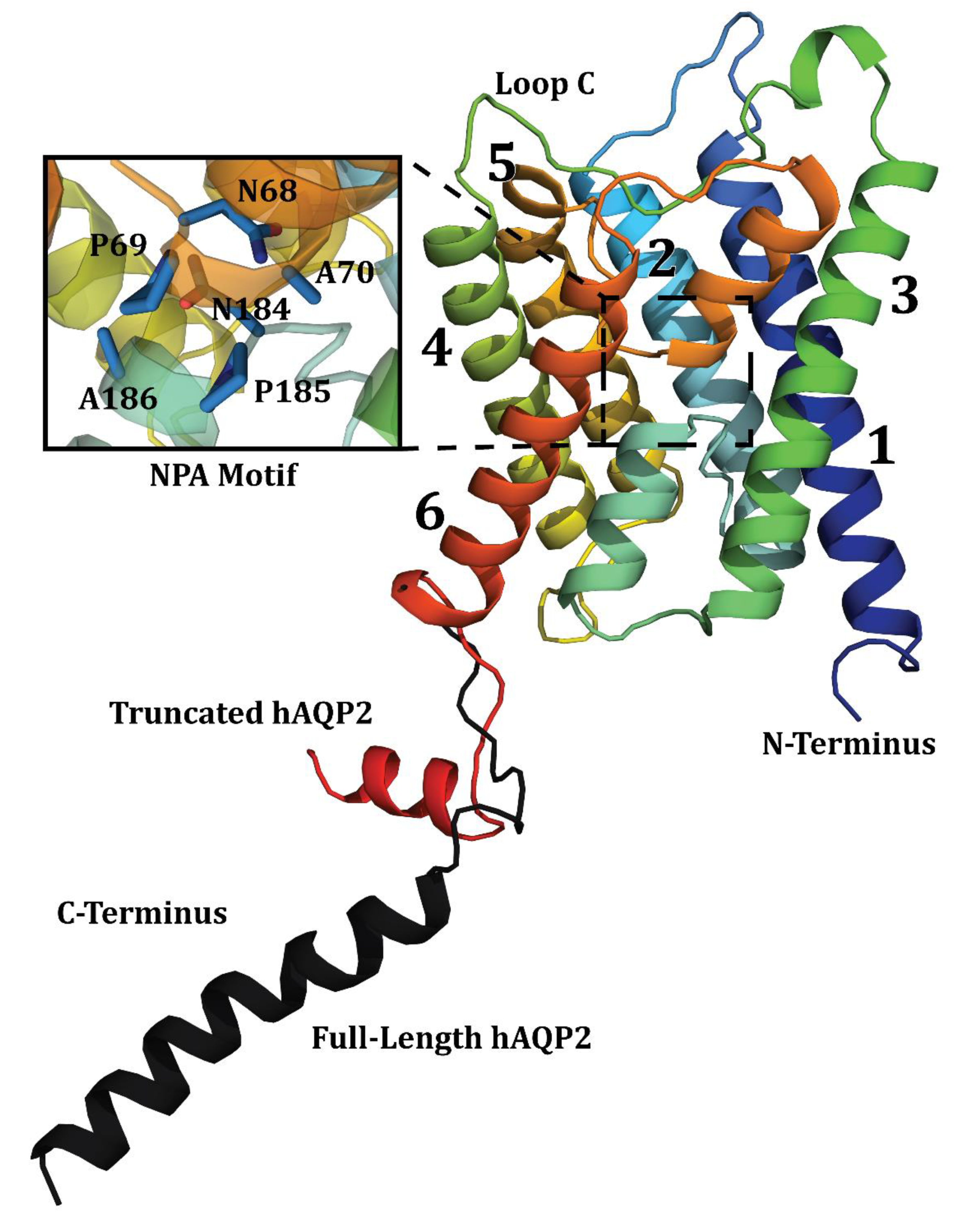
3.4. SSNMR Spectroscopic Characterization
4. Discussion
4.1. Post-Translational Modification of hAQP2
4.2. Expression of hAQP2
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- White, S. Membrane Proteins of Known 3D Structure. Available online: https://blanco.biomol.uci.edu/mpstruc/ (accessed on 24 January 2020).
- Russell, R.B.; Eggleston, D.S. New roles for structure in biology and drug discovery. Nat. Struct. Mol. Biol. 2000, 7, 928. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19. [Google Scholar] [CrossRef]
- Zhao, X. Protein structure determination by solid-state NMR. In NMR of Proteins and Small Biomolecules; Springer: Berlin/Heidelberg, Germany, 2011; pp. 187–213. [Google Scholar]
- Zhou, H.-X.; Cross, T.A. Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 2013, 42, 361–392. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Zhang, Y.; Hu, F. Membrane protein structure and dynamics from NMR spectroscopy. Annu. Rev. Phys. Chem. 2012, 63, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Judge, P.J.; Taylor, G.F.; Dannatt, H.R.W.; Watts, A. Solid-State Nuclear Magnetic Resonance Spectroscopy for Membrane Protein Structure Determination. In Structural Proteomics: High-Throughput Methods; Owens, R.J., Ed.; Springer: New York, NY, USA, 2015; pp. 331–347. [Google Scholar] [CrossRef]
- Murray, D.T.; Das, N.; Cross, T.A. Solid state NMR strategy for characterizing native membrane protein structures. Acc. Chem. Res. 2013, 46, 2172–2181. [Google Scholar] [CrossRef] [Green Version]
- Renault, M.; Tommassen-van Boxtel, R.; Bos, M.P.; Post, J.A.; Tommassen, J.; Baldus, M. Cellular solid-state nuclear magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 4863–4868. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.E.; Wang, S.; Munro, R.; Ritz, E.; Hung, I.; Gor’kov, P.L.; Jiang, Y.; Liang, H.; Brown, L.S.; Ladizhansky, V. In situ structural studies of Anabaena sensory rhodopsin in the E. coli membrane. Biophys. J. 2015, 108, 1683–1696. [Google Scholar] [CrossRef] [Green Version]
- Castellani, F.; Van Rossum, B.; Diehl, A.; Schubert, M.; Rehbein, K.; Oschkinat, H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 2002, 420, 98. [Google Scholar] [CrossRef]
- Zhou, D.H.; Shea, J.J.; Nieuwkoop, A.J.; Franks, W.T.; Wylie, B.J.; Mullen, C.; Sandoz, D.; Rienstra, C.M. Solid-State Protein-Structure Determination with Proton-Detected Triple-Resonance 3D Magic-Angle-Spinning NMR Spectroscopy. Angew. Chem. Int. Ed. 2007, 46, 8380–8383. [Google Scholar] [CrossRef]
- Loquet, A.; Bardiaux, B.; Gardiennet, C.; Blanchet, C.; Baldus, M.; Nilges, M.; Malliavin, T.; Böckmann, A. 3D structure determination of the Crh protein from highly ambiguous solid-state NMR restraints. J. Am. Chem. Soc. 2008, 130, 3579–3589. [Google Scholar] [CrossRef] [Green Version]
- Wylie, B.J.; Sperling, L.J.; Nieuwkoop, A.J.; Franks, W.T.; Oldfield, E.; Rienstra, C.M. Ultrahigh resolution protein structures using NMR chemical shift tensors. Proc. Natl. Acad. Sci. USA 2011, 108, 16974–16979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, M.J.; Pell, A.J.; Bertini, I.; Felli, I.C.; Gonnelli, L.; Pierattelli, R.; Herrmann, T.; Emsley, L.; Pintacuda, G. Structure and backbone dynamics of a microcrystalline metalloprotein by solid-state NMR. Proc. Natl. Acad. Sci. USA 2012, 109, 11095–11100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Hou, G.; Schwieters, C.D.; Ahmed, S.; Williams, J.C.; Polenova, T. Three-Dimensional Structure of CAP-Gly Domain of Mammalian Dynactin Determined by Magic Angle Spinning NMR Spectroscopy: Conformational Plasticity and Interactions with End-Binding Protein EB1. J. Mol. Biol. 2013, 425, 4249–4266. [Google Scholar] [CrossRef] [Green Version]
- Opella, S. NMR and membrane proteins. Nat. Struct. Biol. 1997, 4, 845–848. [Google Scholar]
- Griffin, R. Dipolar recoupling in MAS spectra of biological solids. Nat. Struct. Mol. Biol. 1998, 5, 508. [Google Scholar] [CrossRef]
- de Groot, H.J. Solid-state NMR spectroscopy applied to membrane proteins. Curr. Opin. Struct. Biol. 2000, 10, 593–600. [Google Scholar] [CrossRef]
- Opella, S.J.; Marassi, F.M. Applications of NMR to membrane proteins. Arch. Biochem. Biophys. 2017, 628, 92–101. [Google Scholar] [CrossRef]
- Pandey, A.; Shin, K.; Patterson, R.E.; Liu, X.-Q.; Rainey, J.K. Current strategies for protein production and purification enabling membrane protein structural biology. Biochem. Cell Biol. 2016, 94, 507–527. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Munro, R.A.; Shi, L.; Kawamura, I.; Okitsu, T.; Wada, A.; Kim, S.-Y.; Jung, K.-H.; Brown, L.S.; Ladizhansky, V. Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat. Methods 2013, 10, 1007. [Google Scholar] [CrossRef]
- Park, S.H.; Das, B.B.; Casagrande, F.; Tian, Y.; Nothnagel, H.J.; Chu, M.; Kiefer, H.; Maier, K.; De Angelis, A.A.; Marassi, F.M.; et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 2012, 491, 779–783. [Google Scholar] [CrossRef]
- Andreas, L.B.; Reese, M.; Eddy, M.T.; Gelev, V.; Ni, Q.Z.; Miller, E.A.; Emsley, L.; Pintacuda, G.; Chou, J.J.; Griffin, R.G. Structure and mechanism of the influenza A M218–60 dimer of dimers. J. Am. Chem. Soc. 2015, 137, 14877–14886. [Google Scholar] [CrossRef] [Green Version]
- Das, N.; Dai, J.; Hung, I.; Rajagopalan, M.; Zhou, H.-X.; Cross, T.A. Structure of CrgA, a cell division structural and regulatory protein from Mycobacterium tuberculosis, in lipid bilayers. Proc. Natl. Acad. Sci. USA 2015, 112, E119–E126. [Google Scholar] [CrossRef] [Green Version]
- Retel, J.S.; Nieuwkoop, A.J.; Hiller, M.; Higman, V.A.; Barbet-Massin, E.; Stanek, J.; Andreas, L.B.; Franks, W.T.; van Rossum, B.-J.; Vinothkumar, K.R.; et al. Structure of outer membrane protein G in lipid bilayers. Nat. Commun. 2017, 8, 2073. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Shimada, I. Production of isotopically labeled heterologous proteins in non-E. coli prokaryotic and eukaryotic cells. J. Biomol. NMR 2010, 46, 3. [Google Scholar] [CrossRef]
- Kim, H.J.; Howell, S.C.; Van Horn, W.D.; Jeon, Y.H.; Sanders, C.R. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 55, 335. [Google Scholar] [CrossRef] [Green Version]
- Gautier, A. Structure determination of α-helical membrane proteins by solution-state NMR: Emphasis on retinal proteins. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1837, 578–588. [Google Scholar] [CrossRef] [Green Version]
- Emami, S.; Fan, Y.; Munro, R.; Ladizhansky, V.; Brown, L.S. Yeast-expressed human membrane protein aquaporin-1 yields excellent resolution of solid-state MAS NMR spectra. J. Biomol. NMR 2013, 55, 147–155. [Google Scholar] [CrossRef]
- Fan, Y.; Emami, S.; Munro, R.; Ladizhansky, V.; Brown, L.S. Isotope Labeling of Eukaryotic Membrane Proteins in Yeast for Solid-State NMR. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2015; Volume 565, pp. 193–212. [Google Scholar]
- Fan, Y.; Shi, L.; Ladizhansky, V.; Brown, L.S. Uniform isotope labeling of a eukaryotic seven-transmembrane helical protein in yeast enables high-resolution solid-state NMR studies in the lipid environment. J. Biomol. NMR 2011, 49, 151–161. [Google Scholar] [CrossRef]
- Zech, S.G.; Wand, A.J.; McDermott, A.E. Protein structure determination by high-resolution solid-state NMR spectroscopy: Application to microcrystalline ubiquitin. J. Am. Chem. Soc. 2005, 127, 8618–8626. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor. G. Laboratory Press: Cold Spring Harbor, NY, USA, 1989; Volume 3. [Google Scholar]
- McIntosh, L.P.; Dahlquist, F.W. Biosynthetic incorporation of 15 N and 13 C for assignment and interpretation of nuclear magnetic resonance spectra of proteins. Q. Rev. Biophys. 1990, 23, 1–38. [Google Scholar] [CrossRef]
- Marley, J.; Lu, M.; Bracken, C. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 2001, 20, 71–75. [Google Scholar] [CrossRef]
- Reilly, D.; Fairbrother, W.J. A novel isotope labeling protocol for bacterially expressed proteins. J. Biomol. NMR 1994, 4, 459–462. [Google Scholar] [CrossRef]
- Makrides, S.C. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Mol. Biol. Rev. 1996, 60, 512–538. [Google Scholar] [CrossRef] [Green Version]
- Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999, 10, 411–421. [Google Scholar] [CrossRef]
- LeMaster, D.M.; Kushlan, D.M. Dynamical mapping of E. coli thioredoxin via 13C NMR relaxation analysis. J. Am. Chem. Soc. 1996, 118, 9255–9264. [Google Scholar] [CrossRef]
- Hong, M.; Jakes, K. Selective and extensive 13C labeling of a membrane protein for solid-state NMR investigations. J. Biomol. Nmr 1999, 14, 71–74. [Google Scholar] [CrossRef]
- Liu, J.; Liu, C.; Fan, Y.; Munro, R.A.; Ladizhansky, V.; Brown, L.S.; Wang, S. Sparse 13 C labelling for solid-state NMR studies of P. pastoris expressed eukaryotic seven-transmembrane proteins. J. Biomol. NMR 2016, 65, 7–13. [Google Scholar] [CrossRef]
- Verardi, R.; Traaseth, N.J.; Masterson, L.R.; Vostrikov, V.V.; Veglia, G. Isotope labeling for solution and solid-state NMR spectroscopy of membrane proteins. In Isotope Labeling in Biomolecular NMR; Springer: Berlin/Heidelberg, Germany, 2012; pp. 35–62. [Google Scholar]
- Brown, L.S.; Ladizhansky, V. Membrane proteins in their native habitat as seen by solid-state NMR spectroscopy. Protein Sci. 2015, 24, 1333–1346. [Google Scholar] [CrossRef] [Green Version]
- Quinn, C.M.; Polenova, T. Structural biology of supramolecular assemblies by magic-angle spinning NMR spectroscopy. Q. Rev. Biophys. 2017, 50. [Google Scholar] [CrossRef]
- Laage, R.; Langosch, D. Strategies for prokaryotic expression of eukaryotic membrane proteins. Traffic 2001, 2, 99–104. [Google Scholar] [CrossRef]
- Schertler, G.F. Overproduction of membrane proteins. Curr. Opin. Struct. Biol. 1992, 2, 534–544. [Google Scholar] [CrossRef]
- Kaushal, S.; Ridge, K.D.; Khorana, H.G. Structure and function in rhodopsin: The role of asparagine-linked glycosylation. Proc. Natl. Acad. Sci. USA 1994, 91, 4024–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rands, E.; Candelore, M.; Cheung, A.; Hill, W.S.; Strader, C.; Dixon, R. Mutational analysis of beta-adrenergic receptor glycosylation. J. Biol. Chem. 1990, 265, 10759–10764. [Google Scholar] [PubMed]
- Kusui, T.; Benya, R.V.; Battey, J.F.; Jensen, R.T. Glycosylation of bombesin receptors: Characterization, effect on binding, and G-protein coupling. Biochemistry 1994, 33, 12968–12980. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Smith, R.D.; Jagadeesh, G.; Baukal, A.J.; Hunyady, L.S.; Catt, K.J. N-linked glycosylation is required for optimal AT1a angiotensin receptor expression in COS-7 cells. Endocrinology 1999, 140, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Filipeanu, C.M.; Duvernay, M.T.; Wu, G. Regulation of G protein-coupled receptor export trafficking. Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 853–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rens-Domiano, S.; Reisine, T. Structural analysis and functional role of the carbohydrate component of somatostatin receptors. J. Biol. Chem. 1991, 266, 20094–20102. [Google Scholar]
- McCusker, E.C.; Bane, S.E.; O’Malley, M.A.; Robinson, A.S. Heterologous GPCR expression: A bottleneck to obtaining crystal structures. Biotechnol. Prog. 2007, 23, 540–547. [Google Scholar] [CrossRef]
- Lundstrom, K.; Wagner, R.; Reinhart, C.; Desmyter, A.; Cherouati, N.; Magnin, T.; Zeder-Lutz, G.; Courtot, M.; Prual, C.; Andre, N.; et al. Structural genomics on membrane proteins: Comparison of more than 100 GPCRs in 3 expression systems. J. Struct. Funct. Genom. 2006, 7, 77–91. [Google Scholar] [CrossRef]
- Maeda, S.; Schertler, G.F. Production of GPCR and GPCR complexes for structure determination. Curr. Opin. Struct. Biol. 2013, 23, 381–392. [Google Scholar] [CrossRef]
- de Groot, B.L.; Engel, A.; Grubmüller, H. A refined structure of human aquaporin-1. FEBS Lett. 2001, 504, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Frick, A.; Eriksson, U.K.; de Mattia, F.; Öberg, F.; Hedfalk, K.; Neutze, R.; Willem, J.; Deen, P.M.; Törnroth-Horsefield, S. X-ray structure of human aquaporin 2 and its implications for nephrogenic diabetes insipidus and trafficking. Proc. Natl. Acad. Sci. USA 2014, 111, 6305–6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamshad, M.; Rajesh, S.; Stamataki, Z.; McKeating, J.A.; Dafforn, T.; Overduin, M.; Bill, R.M. Structural characterization of recombinant human CD81 produced in Pichia pastoris. Protein Expr. Purif. 2008, 57, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsefield, R.; Nordén, K.; Fellert, M.; Backmark, A.; Törnroth-Horsefield, S.; van Scheltinga, A.C.T.; Kvassman, J.; Kjellbom, P.; Johanson, U.; Neutze, R. High-resolution x-ray structure of human aquaporin 5. Proc. Natl. Acad. Sci. USA 2008, 105, 13327–13332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Diver, M.M.; Pedi, L.; Koide, A.; Koide, S.; Long, S.B. Atomic structure of the eukaryotic intramembrane RAS methyltransferase ICMT. Nature 2018, 553, 526–529. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Montgomery, M.G.; Liu, S.; Walker, J.E. Structure of the mitochondrial ATP synthase from Pichia angusta determined by electron cryo-microscopy. Proc. Natl. Acad. Sci. USA 2016, 113, 12709–12714. [Google Scholar] [CrossRef] [Green Version]
- Oldham, M.L.; Grigorieff, N.; Chen, J. Structure of the transporter associated with antigen processing trapped by herpes simplex virus. eLife 2016, 5. [Google Scholar] [CrossRef]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. Sect. Dbiological Crystallogr. 2015, 71, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Cereghino, J.L.; Cregg, J.M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 2000, 24, 45–66. [Google Scholar] [CrossRef]
- Bill, R.M. Yeast–a panacea for the structure–function analysis of membrane proteins? Curr. Genet. 2001, 40, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, S.A. Use of Pichia pastoris for expression of recombinant proteins. Methods Enzym. 1999, 306, 154–169. [Google Scholar] [CrossRef]
- Singh, S.; Zhang, M.; Bertheleme, N.; Strange, P.G.; Byrne, B. Purification of the human G protein-coupled receptor adenosine A(2a)R in a stable and functional form expressed in Pichia pastoris. Curr. Protoc. Protein Sci. 2012, 29, 29.23.21–29.23.22. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.; Dikiy, I.; Rosenbaum, D.M.; Gardner, K.H. On the use of Pichia pastoris for isotopic labeling of human GPCRs for NMR studies. J. Biomol. NMR 2018, 71, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Sugiki, T.; Ichikawa, O.; Miyazawa-Onami, M.; Shimada, I.; Takahashi, H. Isotopic labeling of heterologous proteins in the yeast Pichia pastoris and Kluyveromyces lactis. Methods Mol. Biol. (Cliftonn. J.) 2012, 831, 19–36. [Google Scholar] [CrossRef]
- Wegner, G.H. Emerging applications of the methylotrophic yeasts. FEMS Microbiol. Rev. 1990, 7, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Hartner, F.S.; Glieder, A. Regulation of methanol utilisation pathway genes in yeasts. Microb. Cell Factories 2006, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Yurimoto, H.; Oku, M.; Sakai, Y. Yeast methylotrophy: Metabolism, gene regulation and peroxisome homeostasis. Int. J. Microbiol. 2011, 2011, 101298. [Google Scholar] [CrossRef] [Green Version]
- van der Klei, I.J.; Yurimoto, H.; Sakai, Y.; Veenhuis, M. The significance of peroxisomes in methanol metabolism in methylotrophic yeast. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.R.; Cregg, J.M. Introduction to Pichia pastoris. Methods Mol. Biol. (Cliftonn.J.) 1998, 103, 1–15. [Google Scholar] [CrossRef]
- Öberg, F.; Sjöhamn, J.; Conner, M.T.; Bill, R.M.; Hedfalk, K. Improving recombinant eukaryotic membrane protein yields in Pichia pastoris: The importance of codon optimization and clone selection. Mol. Membr. Biol. 2011, 28, 398–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clare, J.J.; Rayment, F.B.; Ballantine, S.P.; Sreekrishna, K.; Romanos, M.A. High-Level Expression of Tetanus Toxin Fragment C in Pichia Pastoris Strains Containing Multiple Tandem Integrations of the Gene. Bio/Technology 1991, 9, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Fairlie, W.D.; Russell, P.K.; Zhang, H.P.; Breit, S.N. Screening procedure for Pichia pastoris clones containing multiple copy gene inserts. BioTechniques 1999, 26, 1042–1044. [Google Scholar] [CrossRef]
- Nyblom, M.; Öberg, F.; Lindkvist-Petersson, K.; Hallgren, K.; Findlay, H.; Wikström, J.; Karlsson, A.; Hansson, Ö.; Booth, P.J.; Bill, R.M. Exceptional overproduction of a functional human membrane protein. Protein Expr. Purif. 2007, 56, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Öberg, F.; Ekvall, M.; Nyblom, M.; Backmark, A.; Neutze, R.; Hedfalk, K. Insight into factors directing high production of eukaryotic membrane proteins; production of 13 human AQPs in Pichia pastoris. Mol. Membr. Biol. 2009, 26, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Day, R.E.; Kitchen, P.; Owen, D.S.; Bland, C.; Marshall, L.; Conner, A.C.; Bill, R.M.; Conner, M.T. Human aquaporins: Regulators of transcellular water flow. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 1492–1506. [Google Scholar] [CrossRef] [Green Version]
- Yang, B. The human aquaporin gene family. Curr. Genom. 2000, 1, 91–102. [Google Scholar] [CrossRef]
- Magni, F.; Sarto, C.; Ticozzi, D.; Soldi, M.; Bosso, N.; Mocarelli, P.; Kienle, M.G. Proteomic knowledge of human aquaporins. Proteomics 2006, 6, 5637–5649. [Google Scholar] [CrossRef]
- Fushimi, K.; Uchida, S.; Hara, Y.; Hirata, Y.; Marumo, F.; Sasaki, S. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature 1993, 361, 549–552. [Google Scholar] [CrossRef]
- Schrier, R.W.; Cadnapaphornchai, M.A. Renal aquaporin water channels: From molecules to human disease. Prog. Biophys. Mol. Biol. 2003, 81, 117–131. [Google Scholar] [CrossRef]
- Ishikawa, S.E.; Schrier, R.W. Pathophysiological roles of arginine vasopressin and aquaporin-2 in impaired water excretion. Clin. Endocrinol. 2003, 58, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G.; Bockenhauer, D. Genetic forms of nephrogenic diabetes insipidus (NDI): Vasopressin receptor defect (X-linked) and aquaporin defect (autosomal recessive and dominant). Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Tamarappoo, B.; Verkman, A. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J. Clin. Investig. 1998, 101, 2257–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesche, D.; Deen, P.M.; Knoers, N.V. Congenital nephrogenic diabetes insipidus: The current state of affairs. Pediatric Nephrol. 2012, 27, 2183–2204. [Google Scholar] [CrossRef]
- Chen, C.-H.; Chen, W.-Y.; Liu, H.-L.; Liu, T.-T.; Tsou, A.-P.; Lin, C.-Y.; Chao, T.; Qi, Y.; Hsiao, K.-J. Identification of mutations in the arginine vasopressin receptor 2 gene causing nephrogenic diabetes insipidus in Chinese patients. J. Hum. Genet. 2002, 47, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, W.; Seibold, A.; Antaramian, A.; Gilbert, S.; Birnbaumer, M.; Bichet, D.; Arthus, M.; Lonergan, M. Mutations in the vasopressin V2 receptor gene in families with nephrogenic diabetes insipidus and functional expression of the Q-2 mutant. Cell. Mol. Biol. (Noisy-Le-Grandfrance) 1994, 40, 429–436. [Google Scholar]
- Mulders, S.M.; Bichet, D.G.; Rijss, J.; Kamsteeg, E.-J.; Arthus, M.-F.; Lonergan, M.; Fujiwara, M.; Morgan, K.; Leijendekker, R.; van der Sluijs, P. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J. Clin. Investig. 1998, 102, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Fenton, R.A.; Pedersen, C.N.; Moeller, H.B. New insights into regulated aquaporin-2 function. Curr. Opin. Nephrol. Hypertens. 2013, 22, 551–558. [Google Scholar] [CrossRef]
- Vahedi-Faridi, A.; Lodowski, D.; Schenk, A.; Kaptan, S.; De Groot, B.; Walz, T.; Engel, A. The structure of aquaporin. 2014; in press. [Google Scholar]
- Zwang, N.A.; Hoffert, J.D.; Pisitkun, T.; Moeller, H.B.; Fenton, R.A.; Knepper, M.A. Identification of phosphorylation-dependent binding partners of aquaporin-2 using protein mass spectrometry. J. Proteome Res. 2009, 8, 1540–1554. [Google Scholar] [CrossRef] [Green Version]
- Moeller, H.B.; Olesen, E.T.; Fenton, R.A. Regulation of the water channel aquaporin-2 by posttranslational modification. Am. J. Physiol. Ren. Physiol. 2011, 300, F1062–CF1073. [Google Scholar] [CrossRef] [Green Version]
- Hoffert, J.D.; Chou, C.L.; Knepper, M.A. Aquaporin-2 in the “-omics” era. J. Biol. Chem. 2009, 284, 14683–14687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eto, K.; Noda, Y.; Horikawa, S.; Uchida, S.; Sasaki, S. Phosphorylation of aquaporin-2 regulates its water permeability. J. Biol. Chem. 2010, 285, 40777–40784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesverova, V.; Törnroth-Horsefield, S. Phosphorylation-dependent regulation of mammalian aquaporins. Cells 2019, 8, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberg, F.; Hedfalk, K. Recombinant production of the human aquaporins in the yeast Pichia pastoris (Invited Review). Mol. Membr. Biol. 2013, 30, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Inan, M.; Meagher, M.M. Non-repressing carbon sources for alcohol oxidase (AOX1) promoter of Pichia pastoris. J. Biosci. Bioeng. 2001, 92, 585–589. [Google Scholar] [CrossRef]
- Nakagawa, T.; Inagaki, A.; Ito, T.; Fujimura, S.; Miyaji, T.; Yurimoto, H.; Kato, N.; Sakai, Y.; Tomizuka, N. Regulation of two distinct alcohol oxidase promoters in the methylotrophic yeast Pichia methanolica. Yeast 2006, 23, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Ramón, R.; Ferrer, P.; Valero, F. Sorbitol co-feeding reduces metabolic burden caused by the overexpression of a Rhizopus oryzae lipase in Pichia pastoris. J. Biotechnol. 2007, 130, 39–46. [Google Scholar] [CrossRef]
- Jungo, C.; Schenk, J.; Pasquier, M.; Marison, I.W.; von Stockar, U. A quantitative analysis of the benefits of mixed feeds of sorbitol and methanol for the production of recombinant avidin with Pichia pastoris. J. Biotechnol. 2007, 131, 57–66. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Zhang, D.; Li, J.; Hua, Z.; Du, G.; Chen, J. Enhancement of cell viability and alkaline polygalacturonate lyase production by sorbitol co-feeding with methanol in Pichia pastoris fermentation. Bioresour. Technol. 2010, 101, 1318–1323. [Google Scholar] [CrossRef]
- Çelik, E.; Çalık, P.; Oliver, S.G. Fed-batch methanol feeding strategy for recombinant protein production by Pichia pastoris in the presence of co-substrate sorbitol. Yeast 2009, 26, 473–484. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, J.; Viroonchatapan, N.; Samson, A.; Chill, J.; Rothe, E.; Anglister, J.; Wang, Z.-Z. Yeast expression and NMR analysis of the extracellular domain of muscle nicotinic acetylcholine receptor α subunit. J. Biol. Chem. 2002, 277, 12613–12621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, H.; Jost, L.; Pirlot, N.; Sassi, H.; Daukandt, M.; Rodriguez, C.; Fickers, P. A quantitative study of methanol/sorbitol co-feeding process of a Pichia pastoris Mut+/pAOX1-lacZ strain. Microb. Cell Factories 2013, 12, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öberg, F.; Sjöhamn, J.; Fischer, G.; Moberg, A.; Pedersen, A.; Neutze, R.; Hedfalk, K. Glycosylation increases the thermostability of human aquaporin 10 protein. J. Biol. Chem. 2011, 286, 31915–31923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laizé, V.; Ripoche, P.; Tacnet, F. Purification and Functional Reconstitution of the Human CHIP28 Water Channel Expressed inSaccharomyces cerevisiae. Protein Expr. Purif. 1997, 11, 284–288. [Google Scholar] [CrossRef]
- Rigaud, J.; Bluzat, A.; Buschlen, S. Incorporation of bacteriorhodopsin into large unilamellar liposomes by reverse phase evaporation. Biochem. Biophys. Res. Commun. 1983, 111, 373–382. [Google Scholar] [CrossRef]
- Schenk, A.D.; Werten, P.J.; Scheuring, S.; de Groot, B.L.; Müller, S.A.; Stahlberg, H.; Philippsen, A.; Engel, A. The 4.5 Å structure of human AQP2. J. Mol. Biol. 2005, 350, 278–289. [Google Scholar] [CrossRef]
- Ammann, C.; Meier, P.; Merbach, A. A simple multinuclear NMR thermometer. J. Magn. Reson. (1969) 1982, 46, 319–321. [Google Scholar] [CrossRef]
- Pines, A.; Gibby, M.G.; Waugh, J.S. Proton-Enhanced Nuclear Induction Spectroscopy. A Method for High Resolution NMR of Dilute Spins in Solids. J. Chem. Phys. 1972, 56, 1776–1777. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, S.R.; Hahn, E.L. Nuclear Double Resonance in the Rotating Frame. Phys. Rev. 1962, 128, 2042–2053. [Google Scholar]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef]
- Takegoshi, K.; Nakamura, S.; Terao, T. C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001, 344, 631–637. [Google Scholar] [CrossRef]
- Morcombe, C.R.; Gaponenko, V.; Byrd, R.A.; Zilm, K.W. Diluting abundant spins by isotope edited radio frequency field assisted diffusion. J. Am. Chem. Soc. 2004, 126, 7196–7197. [Google Scholar] [CrossRef] [PubMed]
- Morcombe, C.R.; Zilm, K.W. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003, 162, 479–486. [Google Scholar] [CrossRef]
- Burum, D.P.; Ernst, R.R. Net Polarization Transfer via a J-Ordered State for Signal Enhancement of Low-Sensitivity Nuclei. J. Magn. Reson. 1980, 39, 163–168. [Google Scholar] [CrossRef]
- Baldus, M.; Meier, B.H. Total correlation spectroscopy in the solid state. The use of scalar couplings to determine the through-bond connectivity. J. Magn. Reson. Ser. A 1996, 121, 65–69. [Google Scholar] [CrossRef]
- Buck, T.M.; Eledge, J.; Skach, W.R. Evidence for stabilization of aquaporin-2 folding mutants by N-linked glycosylation in endoplasmic reticulum. Am. J. Physiol. Cell Physiol. 2004, 287, C1292–C1299. [Google Scholar] [CrossRef] [Green Version]
- Barth, A.; Zscherp, C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef]
- Tamm, L.K.; Tatulian, S.A. Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 1997, 30, 365–429. [Google Scholar] [CrossRef] [Green Version]
- Vass, E.; Hollosi, M.; Besson, F.; Buchet, R. Vibrational spectroscopic detection of beta- and gamma-turns in synthetic and natural peptides and proteins. Chem. Rev. 2003, 103, 1917–1954. [Google Scholar] [CrossRef]
- Singh, S.; Hedley, D.; Kara, E.; Gras, A.; Iwata, S.; Ruprecht, J.; Strange, P.G.; Byrne, B. A purified C-terminally truncated human adenosine A2A receptor construct is functionally stable and degradation resistant. Protein Expr. Purif. 2010, 74, 80–87. [Google Scholar] [CrossRef]
- Vagenende, V.; Yap, M.G.; Trout, B.L. Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry 2009, 48, 11084–11096. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bhandari, B.; Zhou, P. Insight into the effect of glycerol on stability of globular proteins in high protein model system. Food Chem. 2019, 278, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Pitard, B.; Richard, P.; Duñach, M.; Rigaud, J.L. ATP synthesis by the F0F1 ATP synthase from thermophilic Bacillus PS3 reconstituted into liposomes with bacteriorhodopsin: 2. Relationships between proton motive force and ATP synthesis. Eur. J. Biochem. 1996, 235, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Agre, P.; Mathai, J.C.; Smith, B.L.; Preston, G.M. Functional analyses of aquaporin water channel proteins. Methods Enzym. 1999, 294, 550–572. [Google Scholar] [CrossRef]
- Shinbo, I.; Fushimi, K.; Kasahara, M.; Yamauchi, K.; Sasaki, S.; Marumo, F. Functional analysis of aquaporin-2 mutants associated with nephrogenic diabetes insipidus by yeast expression. Am. J. Physiol. Ren. Physiol. 1999, 277, F734–CF741. [Google Scholar] [CrossRef]
- Shapiro, R.I.; Wen, D.; Levesque, M.; Hronowski, X.; Gill, A.; Garber, E.A.; Galdes, A.; Strauch, K.L.; Taylor, F.R. Expression of Sonic hedgehog-Fc fusion protein in Pichia pastoris. Identification and control of post-translational, chemical, and proteolytic modifications. Protein Expr. Purif. 2003, 29, 272–283. [Google Scholar] [CrossRef]
- Sinha, J.; Plantz, B.A.; Inan, M.; Meagher, M.M. Causes of proteolytic degradation of secreted recombinant proteins produced in methylotrophic yeast Pichia pastoris: Case study with recombinant ovine interferon-τ. Biotechnol. Bioeng. 2005, 89, 102–112. [Google Scholar] [CrossRef]
- Bretthauer, R.K.; Castellino, F.J. Glycosylation of Pichia pastoris-derived proteins. Biotechnol. Appl. Biochem. 1999, 30, 193–200. [Google Scholar]
- Tschopp, J.; Sverlow, G.; Kosson, R.; Craig, W.; Grinna, L. High-level secretion of glycosylated invertase in the methylotrophic yeast, Pichia pastoris. Bio/Technology 1987, 5, 1305. [Google Scholar] [CrossRef]
- Trimble, R.B.; Lubowski, C.; Hauer III, C.R.; Stack, R.; McNaughton, L.; Gemmill, T.R.; Kumar, S.A. Characterization of N-and O-linked glycosylation of recombinant human bile salt-stimulated lipase secreted by Pichia pastoris. Glycobiology 2003, 14, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Azuma, T.; Matsuba, T.; Iida, H.; Suzuki, H.; Yamamoto, K.; Kohli, Y.; Hori, H. Secretion of human intracellular aspartic proteinase cathepsin E expressed in the methylotrophic yeast, Pichia pastoris and characterization of produced recombinant cathepsin E. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1994, 1206, 279–285. [Google Scholar] [CrossRef]
- Grinna, L.S.; Tschopp, J.F. Size distribution and general structural features of N-linked oligosaccharides from the methylotrophic yeast, Pichia pastoris. Yeast 1989, 5, 107–115. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Kempi, N.; Komives, E. Expression of highly disulfide–bonded proteins in Pichia pastoris. Structure 1994, 2, 1003–1005. [Google Scholar] [CrossRef] [Green Version]
- Freivalds, J.; Dislers, A.; Ose, V.; Pumpens, P.; Tars, K.; Kazaks, A. Highly efficient production of phosphorylated hepatitis B core particles in yeast Pichia pastoris. Protein Expr. Purif. 2011, 75, 218–224. [Google Scholar] [CrossRef]
- Harvey, D.J. Proteomic analysis of glycosylation: Structural determination of N-and O-linked glycans by mass spectrometry. Expert Rev. Proteom. 2005, 2, 87–101. [Google Scholar] [CrossRef]
- Brown, D.; Hasler, U.; Nunes, P.; Bouley, R.; Lu, H.A. Phosphorylation events and the modulation of aquaporin 2 cell surface expression. Curr. Opin. Nephrol. Hypertens. 2008, 17, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ing, C.; Emami, S.; Jiang, Y.; Liang, H.; Pomès, R.G.; Brown, L.S.; Ladizhansky, V. Structure and dynamics of extracellular loops in human Aquaporin-1 from solid-state NMR and molecular dynamics. J. Phys. Chem. B 2016, 120, 9887–9902. [Google Scholar] [CrossRef]
- Shi, L.; Lake, E.M.; Ahmed, M.A.; Brown, L.S.; Ladizhansky, V. Solid-state NMR study of proteorhodopsin in the lipid environment: Secondary structure and dynamics. Biochim. Biophys. Acta (Bba)-Biomembr. 2009, 1788, 2563–2574. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Kawamura, I.; Jung, K.H.; Brown, L.S.; Ladizhansky, V. Conformation of a seven-helical transmembrane photosensor in the lipid environment. Angew. Chem. Int. Ed. Engl. 2011, 50, 1302–1305. [Google Scholar] [CrossRef]
- Dingwell, D.A.; Brown, L.S.; Ladizhansky, V. Structure of the Functionally Important Extracellular Loop C of Human Aquaporin 1 Obtained by Solid-State NMR under Nearly Physiological Conditions. J. Phys. Chem. B 2019, 123, 7700–7710. [Google Scholar] [CrossRef]
- Lundborg, M.; Widmalm, G.R. Structural analysis of glycans by NMR chemical shift prediction. Anal. Chem. 2011, 83, 1514–1517. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.-E.; Stenutz, R.; Widmalm, G. Sequence determination of oligosaccharides and regular polysaccharides using NMR spectroscopy and a novel Web-based version of the computer program CASPER. Carbohydr. Res. 2006, 341, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Gellerfors, P.; Axelsson, K.; Helander, A.; Johansson, S.; Kenne, L.; Lindqvist, S.; Pavlu, B.; Skottner, A.; Fryklund, L. Isolation and characterization of a glycosylated form of human insulin-like growth factor I produced in Saccharomyces cerevisiae. J. Biol. Chem. 1989, 264, 11444–11449. [Google Scholar] [PubMed]
- Vervecken, W.; Kaigorodov, V.; Callewaert, N.; Geysens, S.; De Vusser, K.; Contreras, R. In vivo synthesis of mammalian-like, hybrid-type N-glycans in Pichia pastoris. Appl. Environ. Microbiol. 2004, 70, 2639–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukens, B.; De Wachter, C.; Callewaert, N. Engineering the Pichia pastoris N-Glycosylation Pathway Using the GlycoSwitch Technology. Methods Mol. Biol. (Cliftonn. J.) 2015, 1321, 103–122. [Google Scholar] [CrossRef]
- Yang, M.; Yu, X.-W.; Zheng, H.; Sha, C.; Zhao, C.; Qian, M.; Xu, Y. Role of N-linked glycosylation in the secretion and enzymatic properties of Rhizopus chinensis lipase expressed in Pichia pastoris. Microb. Cell Factories 2015, 14, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, G.; Koudijs, M.; van Balkom, B.W.; Oorschot, V.; Klumperman, J.; Deen, P.M.; van der Sluijs, P. Glycosylation is important for cell surface expression of the water channel aquaporin-2 but is not essential for tetramerization in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 2975–2983. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Fushimi, K.; Sasaki, S.; Marumo, F. Structure of aquaporin-2 vasopressin water channel. J. Biol. Chem. 1996, 271, 5171–5176. [Google Scholar]
- Yurugi-Kobayashi, T.; Asada, H.; Shiroishi, M.; Shimamura, T.; Funamoto, S.; Katsuta, N.; Ito, K.; Sugawara, T.; Tokuda, N.; Tsujimoto, H. Comparison of functional non-glycosylated GPCRs expression in Pichia pastoris. Biochem. Biophys. Res. Commun. 2009, 380, 271–276. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Xu, Q.; Du, G.; Hua, Z.; Liu, L.; Li, J.; Chen, J. Lowering induction temperature for enhanced production of polygalacturonate lyase in recombinant Pichia pastoris. Process. Biochem. 2009, 44, 949–954. [Google Scholar] [CrossRef]
- Gao, M.; Dong, S.; Yu, R.; Wu, J.; Zheng, Z.; Shi, Z.; Zhan, X. Improvement of ATP regeneration efficiency and operation stability in porcine interferon-α production by Pichia pastoris under lower induction temperature. Korean J. Chem. Eng. 2011, 28, 1412. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munro, R.; de Vlugt, J.; Ladizhansky, V.; Brown, L.S. Improved Protocol for the Production of the Low-Expression Eukaryotic Membrane Protein Human Aquaporin 2 in Pichia pastoris for Solid-State NMR. Biomolecules 2020, 10, 434. https://doi.org/10.3390/biom10030434
Munro R, de Vlugt J, Ladizhansky V, Brown LS. Improved Protocol for the Production of the Low-Expression Eukaryotic Membrane Protein Human Aquaporin 2 in Pichia pastoris for Solid-State NMR. Biomolecules. 2020; 10(3):434. https://doi.org/10.3390/biom10030434
Chicago/Turabian StyleMunro, Rachel, Jeffrey de Vlugt, Vladimir Ladizhansky, and Leonid S. Brown. 2020. "Improved Protocol for the Production of the Low-Expression Eukaryotic Membrane Protein Human Aquaporin 2 in Pichia pastoris for Solid-State NMR" Biomolecules 10, no. 3: 434. https://doi.org/10.3390/biom10030434
APA StyleMunro, R., de Vlugt, J., Ladizhansky, V., & Brown, L. S. (2020). Improved Protocol for the Production of the Low-Expression Eukaryotic Membrane Protein Human Aquaporin 2 in Pichia pastoris for Solid-State NMR. Biomolecules, 10(3), 434. https://doi.org/10.3390/biom10030434