Human vtRNA1-1 Levels Modulate Signaling Pathways and Regulate Apoptosis in Human Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MTT Proliferation Assay
2.3. Genome Editing
2.4. Cloning
2.5. Lentiviral Transduction
2.6. Annexin V Staining and Flow Cytometry
2.7. Northern Blot
2.8. Western Blot
2.9. Next Generation mRNA Sequencing
2.10. Bioinformatic Analysis
3. Results
3.1. vtRNA1-1 Knock-Out Stimulates Starvation-Induced Apoptosis
3.2. mRNA Deep Sequencing Reveals a vtRNA1-1 Dependent Increase in Gene Expression of Cellular Signaling Activity
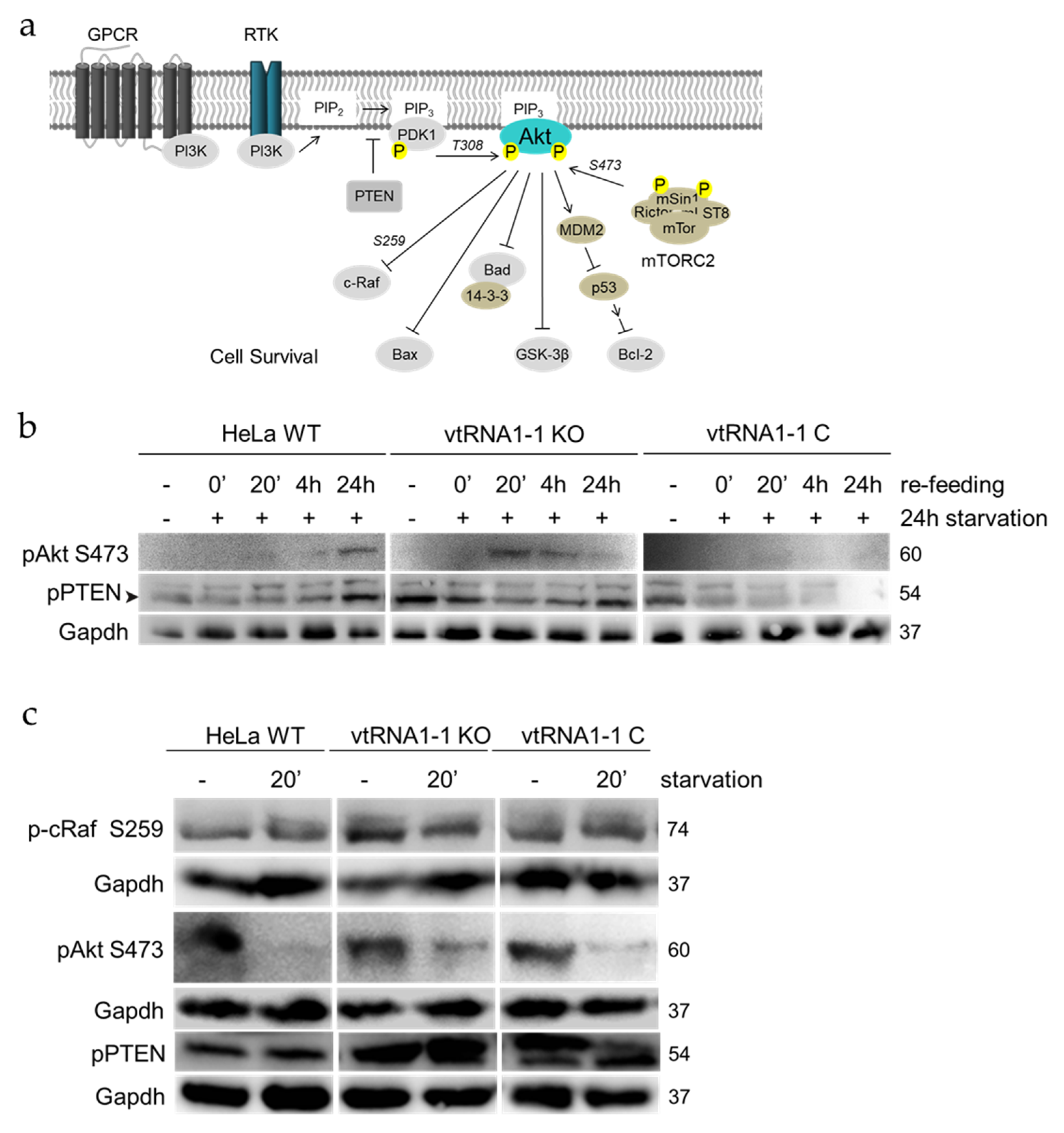
3.3. The Loss of vtRNA1-1 Stimulates PI3K/Akt Pathway Signaling
3.4. The ERK1/2 Cascade Initiation in Starvation-Induced HeLa Cells Is Increased upon vtRNA1-1 Knock-Out
3.5. Only 24 Nucleotides within the vtRNA1-1 Central Domain Are Sufficient to Maintain Apoptosis Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mattick, J.S. RNA regulation: A new genetics? Nat. Rev. Genet. 2004, 5, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.L.; Rome, L.H. Preparative agarose gel electrophoresis for the purification of small organelles and particles. Anal. Biochem. 1986, 156, 161–170. [Google Scholar] [CrossRef]
- Downey, G.P.; Waddell, T.K.; Fukushima, T.; Sue, A.Q.A. Current techniques in cell and molecular biology. J. Critic. Care 1995, 10, 136–149. [Google Scholar] [CrossRef]
- Chugani, D.C.; Rome, L.H.; Kedersha, N.L. Evidence that vault ribonucleoprotein particles localize to the nuclear pore complex. J. Cell Sci. 1993, 106 Pt 1, 23–29. [Google Scholar]
- Kickhoefer, V.A.; Rajavel, K.S.; Scheffer, G.L.; Dalton, W.S.; Scheper, R.J.; Rome, L.H. Vaults are up-regulated in multidrug-resistant cancer cell lines. J. Biol. Chem. 1998, 273, 8971–8974. [Google Scholar] [CrossRef] [Green Version]
- Kitazono, M.; Sumizawa, T.; Takebayashi, Y.; Chen, Z.S.; Furukawa, T.; Nagayama, S.; Tani, A.; Takao, S.; Aikou, T.; Akiyama, S. Multidrug resistance and the lung resistance-related protein in human colon carcinoma SW-620 cells. J. Nat. Cancer Inst. 1999, 91, 1647–1653. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Ginn-Pease, M.E.; Eng, C. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res. 2005, 65, 4108–4116. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Lee, S.; Mian, M.F.; Yun, S.U.; Song, M.; Yi, K.S.; Ryu, S.H.; Suh, P.G. Crosstalk between Src and major vault protein in epidermal growth factor-dependent cell signalling. FEBS J. 2006, 273, 793–804. [Google Scholar] [CrossRef]
- Shimamoto, Y.; Sumizawa, T.; Haraguchi, M.; Gotanda, T.; Jueng, H.C.; Furukawa, T.; Sakata, R.; Akiyama, S. Direct activation of the human major vault protein gene by DNA-damaging agents. Oncol. Rep. 2006, 15, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, M.P.; Dubouix-Bourandy, A.; Bajmoczi, M.; Golan, D.E.; Zaidi, T.; Coutinho-Sledge, Y.S.; Gygi, M.P.; Gygi, S.P.; Wiemer, E.A.; Pier, G.B. Host resistance to lung infection mediated by major vault protein in epithelial cells. Science 2007, 317, 130–132. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.J.; An, H.J.; Oh, Y.S.; Choi, H.R.; Ha, M.K.; Park, S.C. On the role of major vault protein in the resistance of senescent human diploid fibroblasts to apoptosis. Cell Death Differ. 2008, 15, 1673–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmar, F.; Hacker, C.; Zahedi, R.P.; Sickmann, A.; Ewald, A.; Scheer, U.; Dabauvalle, M.C. Assembly of nuclear pore complexes mediated by major vault protein. J. Cell Sci. 2009, 122, 780–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, P.F.; Chen, J.J.; Hackermuller, J.; Hoffmann, S.; Horn, F.; Khaitovich, P.; Kretzschmar, A.K.; Mosig, A.; Prohaska, S.J.; Qi, X.; et al. Evolution of vault RNAs. Mol. Biol. Evol. 2009, 26, 1975–1991. [Google Scholar] [CrossRef] [Green Version]
- Nandy, C.; Mrazek, J.; Stoiber, H.; Grasser, F.A.; Huttenhofer, A.; Polacek, N. Epstein-barr virus-induced expression of a novel human vault RNA. J. Mol. Biol. 2009, 388, 776–784. [Google Scholar] [CrossRef]
- Gopinath, S.C.; Matsugami, A.; Katahira, M.; Kumar, P.K. Human vault-associated non-coding RNAs bind to mitoxantrone, a chemotherapeutic compound. Nucleic Acids Res. 2005, 33, 4874–4881. [Google Scholar] [CrossRef] [Green Version]
- Gopinath, S.C.; Wadhwa, R.; Kumar, P.K. Expression of noncoding vault RNA in human malignant cells and its importance in mitoxantrone resistance. Mol. Cancer Res. MCR 2010, 8, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Persson, H.; Kvist, A.; Vallon-Christersson, J.; Medstrand, P.; Borg, A.; Rovira, C. The non-coding RNA of the multidrug resistance-linked vault particle encodes multiple regulatory small RNAs. Nat. Cell Biol. 2009, 11, 1268–1271. [Google Scholar] [CrossRef]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef]
- Sajini, A.A.; Choudhury, N.R.; Wagner, R.E.; Bornelov, S.; Selmi, T.; Spanos, C.; Dietmann, S.; Rappsilber, J.; Michlewski, G.; Frye, M. Loss of 5-methylcytosine alters the biogenesis of vault-derived small RNAs to coordinate epidermal differentiation. Nat. Commun. 2019, 10, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; OuYang, H.; An, X.; Liu, S. Vault RNAs partially induces drug resistance of human tumor cells MCF-7 by binding to the RNA/DNA-binding protein PSF and inducing oncogene GAGE6. PLoS ONE 2018, 13, e0191325. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Kunkeaw, N.; Jeon, S.H.; Lee, I.; Johnson, B.H.; Kang, G.Y.; Bang, J.Y.; Park, H.S.; Leelayuwat, C.; Lee, Y.S. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA 2011, 17, 1076–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkeaw, N.; Jeon, S.H.; Lee, K.; Johnson, B.H.; Tanasanvimon, S.; Javle, M.; Pairojkul, C.; Chamgramol, Y.; Wongfieng, W.; Gong, B.; et al. Cell death/proliferation roles for nc886, a non-coding RNA, in the protein kinase R pathway in cholangiocarcinoma. Oncogene 2013, 32, 3722–3731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolev, N.G.; Rajan, K.S.; Tycowski, K.T.; Toh, J.Y.; Shi, H.; Lei, Y.; Michaeli, S.; Tschudi, C. The vault RNA of Trypanosoma brucei plays a role in the production of trans-spliced mRNA. J. Biol. Chem. 2019, 294, 15559–15574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kickhoefer, V.A. A new role for vault RNA-TEP1 complexes in mRNA production in trypanosomes. J. Biol. Chem 2019, 294, 15575–15576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horos, R.; Buscher, M.; Kleinendorst, R.; Alleaume, A.M.; Tarafder, A.K.; Schwarzl, T.; Dziuba, D.; Tischer, C.; Zielonka, E.M.; Adak, A.; et al. The small non-coding vault RNA1-1 acts as a riboregulator of autophagy. Cell 2019, 176, 1054–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amort, M.; Nachbauer, B.; Tuzlak, S.; Kieser, A.; Schepers, A.; Villunger, A.; Polacek, N. Expression of the vault RNA protects cells from undergoing apoptosis. Nat. Commun. 2015, 6, 7030. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.; Chauhan, P.; Saha, B.; Kubatzky, K.F. Conceptual evolution of cell signaling. Int. J. Mol. Sci. 2019, 20, 3292. [Google Scholar] [CrossRef] [Green Version]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reber, S.; Mechtersheimer, J.; Nasif, S.; Benitez, J.A.; Colombo, M.; Domanski, M.; Jutzi, D.; Hedlund, E.; Ruepp, M.D. Crispr-trap: A clean approach for the generation of gene knockouts and gene replacements in human cells. M. Biol. Cell 2018, 29, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemr, M.; Buhler, M. Single-step generation of conditional knockout mouse embryonic stem cells. Cell Rep. 2015, 12, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Gebetsberger, J.; Zywicki, M.; Kunzi, A.; Polacek, N. tRNA-derived fragments target the ribosome and function as regulatory non-coding RNA in Haloferax volcanii. Archaea 2012, 2012, 260909. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Erratum: Near-optimal probabilistic RNA-seq quantification. Nat. Biotech. 2016, 34, 888. [Google Scholar] [CrossRef] [Green Version]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
- Wick, M.J.; Dong, L.Q.; Riojas, R.A.; Ramos, F.J.; Liu, F. Mechanism of phosphorylation of protein kinase B/Akt by a constitutively active 3-phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 2000, 275, 40400–40406. [Google Scholar] [CrossRef] [Green Version]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Ross, A.H.; Gericke, A. Phosphorylation keeps PTEN phosphatase closed for business. Proc. Natl. Acad. Sci. USA 2009, 106, 1297–1298. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscher, M.; Horos, R.; Hentze, M.W. ‘High vault-age’: Non-coding RNA control of autophagy. Open Biol. 2020, 10, 190307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitu, V.; Stanley, E.R. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 2006, 18, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Kesavapany, S.; Amin, N.; Zheng, Y.L.; Nijhara, R.; Jaffe, H.; Sihag, R.; Gutkind, J.S.; Takahashi, S.; Kulkarni, A.; Grant, P.; et al. p35/cyclin-dependent kinase 5 phosphorylation of ras guanine nucleotide releasing factor 2 (RasGRF2) mediates rac-dependent extracellular signal-regulated kinase 1/2 activity, altering RasGRF2 and microtubule-associated protein 1b distribution in neurons. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 4421–4431. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Badache, A.; Daly, J.M.; Hynes, N.E. An essential role for Src kinase in ErbB receptor signaling through the MAPK pathway. Exp. Cell Res. 2001, 267, 81–87. [Google Scholar] [CrossRef]
- Moolenaar, W.H.; Kranenburg, O.; Postma, F.R.; Zondag, G.C. Lysophosphatidic acid: G-protein signalling and cellular responses. Curr. Op. Cell Biol. 1997, 9, 168–173. [Google Scholar] [CrossRef]
- Yang, Z.; Xie, C.; Xu, W.; Liu, G.; Cao, X.; Li, W.; Chen, J.; Zhu, Y.; Luo, S.; Luo, Z.; et al. Phosphorylation and inactivation of PTEN at residues Ser380/Thr382/383 induced by Helicobacter pylori promotes gastric epithelial cell survival through PI3K/Akt pathway. Oncotarget 2015, 6, 31916–31926. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Invest. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, S.; Ichikawa, T.; Maneesaay, P.; Saito, Y.; Nagai, K.; Tamura, T.; Manachai, N.; Yamakawa, N.; Hamasaki, M.; Kitabayashi, I.; et al. Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat. Commun. 2014, 5, 3393. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhuo, Y.; Guo, W.; Field, J. P21-activated Kinase 1 (Pak1)-dependent phosphorylation of Raf-1 regulates its mitochondrial localization, phosphorylation of BAD, and Bcl-2 association. J. Biol. Chem. 2005, 280, 24698–24705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molzan, M.; Schumacher, B.; Ottmann, C.; Baljuls, A.; Polzien, L.; Weyand, M.; Thiel, P.; Rose, R.; Rose, M.; Kuhenne, P.; et al. Impaired binding of 14-3-3 to C-RAF in Noonan syndrome suggests new approaches in diseases with increased ras signaling. Mol. Cell. Biol. 2010, 30, 4698–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 1996, 87, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Hogg, J.R.; Collins, K. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 2007, 13, 868–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bracher, L.; Ferro, I.; Pulido-Quetglas, C.; Ruepp, M.-D.; Johnson, R.; Polacek, N. Human vtRNA1-1 Levels Modulate Signaling Pathways and Regulate Apoptosis in Human Cancer Cells. Biomolecules 2020, 10, 614. https://doi.org/10.3390/biom10040614
Bracher L, Ferro I, Pulido-Quetglas C, Ruepp M-D, Johnson R, Polacek N. Human vtRNA1-1 Levels Modulate Signaling Pathways and Regulate Apoptosis in Human Cancer Cells. Biomolecules. 2020; 10(4):614. https://doi.org/10.3390/biom10040614
Chicago/Turabian StyleBracher, Lisamaria, Iolanda Ferro, Carlos Pulido-Quetglas, Marc-David Ruepp, Rory Johnson, and Norbert Polacek. 2020. "Human vtRNA1-1 Levels Modulate Signaling Pathways and Regulate Apoptosis in Human Cancer Cells" Biomolecules 10, no. 4: 614. https://doi.org/10.3390/biom10040614
APA StyleBracher, L., Ferro, I., Pulido-Quetglas, C., Ruepp, M.-D., Johnson, R., & Polacek, N. (2020). Human vtRNA1-1 Levels Modulate Signaling Pathways and Regulate Apoptosis in Human Cancer Cells. Biomolecules, 10(4), 614. https://doi.org/10.3390/biom10040614