Exomes of Ductal Luminal Breast Cancer Patients from Southwest Colombia: Gene Mutational Profile and Related Expression Alterations

 , ,
, ,  , , and
, , and 
Abstract
:1. Introduction
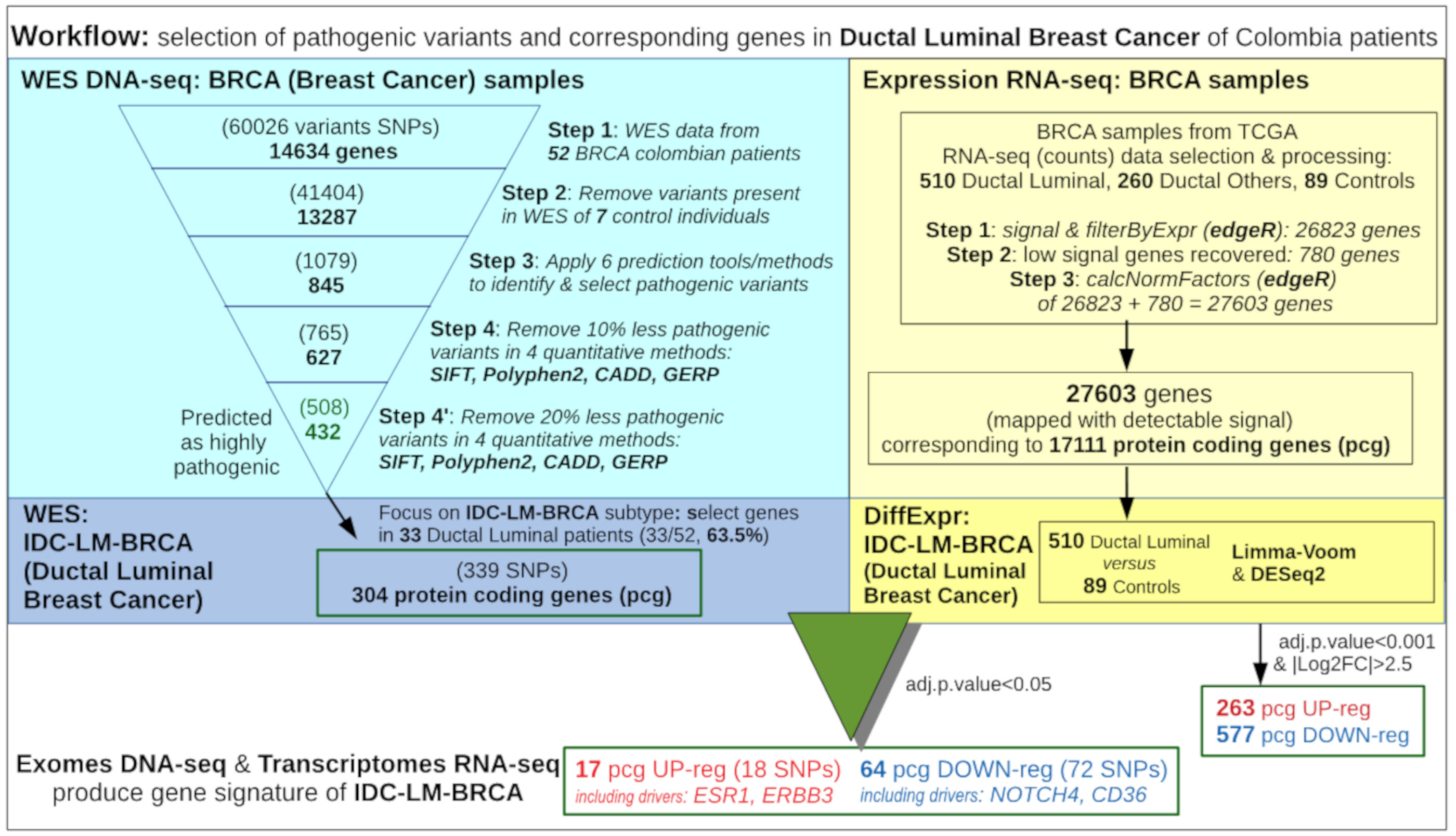
2. Materials and Methods
2.1. Ethical Approval
2.2. Samples Collection and DNA Sequencing
2.3. Exome Mapping and Genetic Variant Calling
2.4. Variant Prioritization Based on Greater Harmful Impact on Protein
2.5. Selection of Samples from TCGA for Comparative Analysis with Colombian Samples
- (i)
- All patients from Colombia and from TCGA selection were women of similar age, presenting an average of 61.6 years old at diagnosis (standard deviation ± 12.6) for the Colombian cohort and an average of 57.3 years (SD ± 13.2) for the TCGA patients.
- (ii)
- Both cohorts of patients were mostly White. A recent genetic study by Norris et al. (2017) [17] stated that the population from Antioquia, a close Colombian state culturally very similar to the patient’s region (Valle del Cauca), shows averages of 64% European ancestry, 29% Native American ancestry and 7% African ancestry. The majority of the selected TCGA patients were also White of European ancestry (496/770, 64%). Therefore, to a large extent, the Colombian and the TCGA patients have a similar genetic background. The remaining TCGA patients were: Black or African American (148/770, 19.2%), Asian (47/770, 6%), American Indian or Alaska Native (1/770, 0.01%), and of unreported race (78/770, 10%).
- (iii)
- With respect to the cellular subtypes, all the breast cancer patients selected from TCGA were invasive ductal carcinoma. In this way, we matched with the main cellular subtype of the WES samples from Colombia: 42/52 (81%) invasive ductal carcinoma (IDC).
- (iv)
- With respect to the breast cancer intrinsic subtypes, the whole set of 770 tumor samples from TCGA were: luminal A (339), luminal B (171), basal (165), Her2 (73) and normal (22). For the comparison with the Colombian cohort, we only used the luminal samples (339 + 171 = 510), because the majority of the Colombian samples (within the ductal) were of luminal subtype.
- (v)
- With respect to tumor stage, in both groups of patients, the majority of the samples corresponded to stage I and II tumors: 81% of the Colombian patients and 76% of the patients selected from TCGA. Furthermore, none of the in-house patients from Colombia or TCGA patients had metastases.
2.6. Selection of Samples from TCGA for Expression Calculation
2.7. Recovery of Some Genes Expressed Only in Some Groups
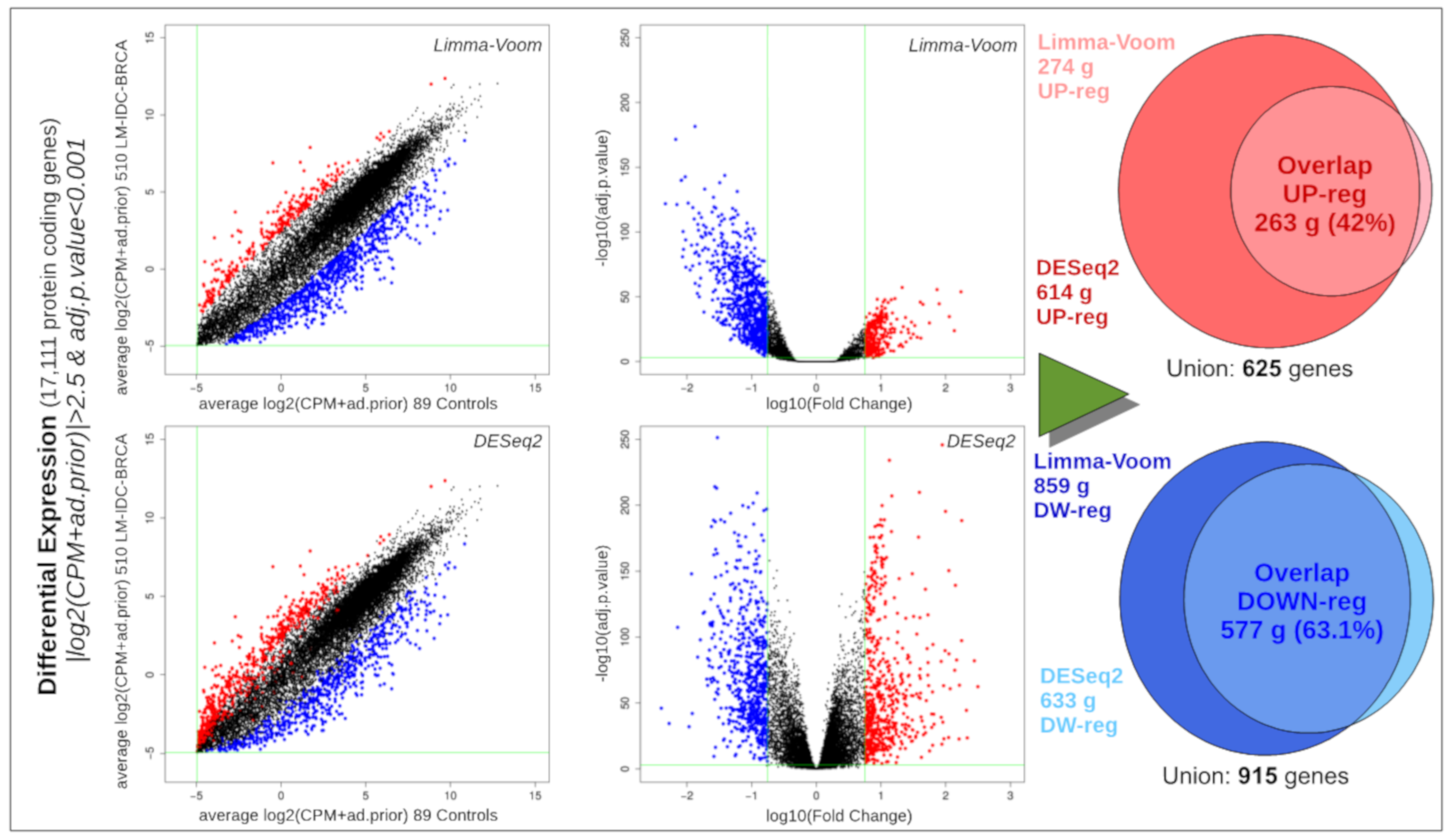
2.8. Differential Expression Analysis of Ductal Luminal Breast Cancer (Idc-Lm-Brca) Subtype
2.9. Functional Analysis and Annotation of the Variants
2.10. Combined Analysis of Wes Data From the Colombian and Tcga Cohorts
3. Results and Discussion
3.1. Analysis of the Whole Exome Sequencing Data to Identify Relevant Genetic Variants
3.2. Genes Including Genetic Variants Considered Driver Mutations
3.3. Functional Involvement in Cancer of Genes Found with Driver Mutations
3.4. Global Differential Expression of Ductal Luminal Breast Cancer Samples
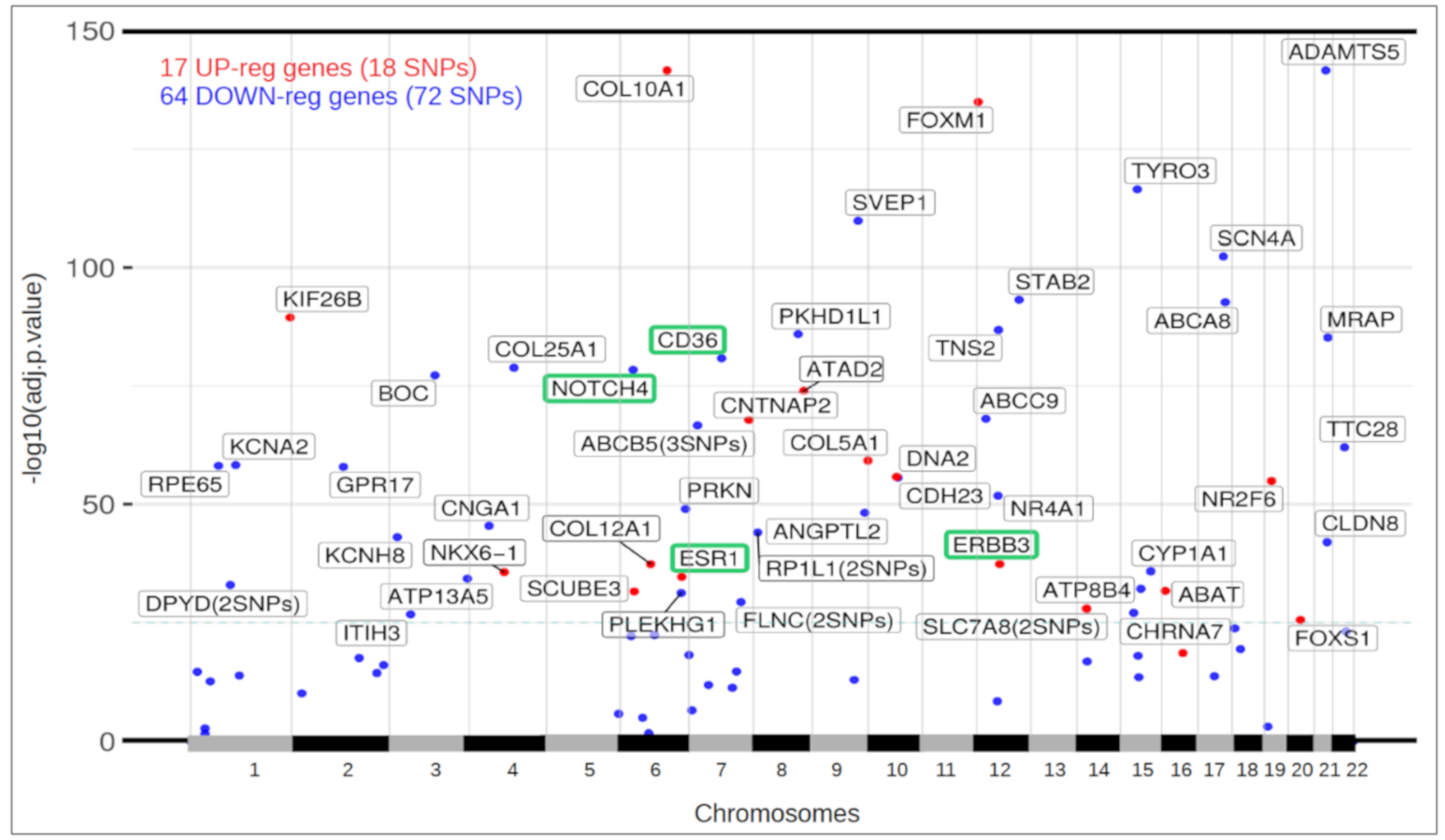
3.5. Differential Expression of Ductal Luminal Breast Cancer Samples in Genes That Suffer Mutations
3.6. Functional View of the Genes Altered in Ductal Luminal Breast Cancer
3.7. Mutations Found in CBLB, a Gene That Inhibits the TGF-β Pathway
3.8. Common Mutated Genes in Ductal Luminal Breast Cancer Patients from Colombia and TCGA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zaidi, Z.; Dib, H.A. Abstract 4191: The worldwide female breast cancer incidence and survival, 2018. Cancer Res. 2019, 79, 4191. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, C.; Rivera, A.L.; Trochez, D.; Solarte, M.; Gómez, D.; Cifuentes, L.; Barreto, G. Mutational analysis of BRCA1 and BRCA2 genes in women with familial breast cancer from different regions of Colombia. Hered. Cancer Clin. Pract. 2019, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Pardo, C.; de Vries, E. Breast and cervical cancer survival at Instituto Nacional de Cancerología, Colombia. Colomb. Med. 2018, 49, 102–108. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.N.; Dave, R.; Sanadya, J.; Sharma, P.; Sharma, K.K. Various types and management of breast cancer: An overview. J. Adv. Pharm. Technol. Res. 2010, 1, 109–126. [Google Scholar]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Urbach, D.; Lupien, M.; Karagas, M.R.; Moore, J.H. Cancer heterogeneity: Origins and implications for genetic association studies. Trends Genet. 2012, 28, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Hosen, M.I.; Ahmed, M.; Shekhar, H.U. Onco-Multi-OMICS Approach: A New Frontier in Cancer Research. Biomed. Res. Int. 2018, 2018, 9836256. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, M.D.; Cabanski, C.R.; Sun, W.; Hoadley, K.A.; Walter, V.; Mose, L.E.; Troester, M.A.; Hammerman, P.S.; Parker, J.S.; Perou, C.M.; et al. Integrated RNA and DNA sequencing improves mutation detection in low purity tumors. Nucleic Acids Res. 2014, 42, e107. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [Green Version]
- Norris, E.T.; Wang, L.; Conley, A.B.; Rishishwar, L.; Mariño-Ramírez, L.; Valderrama-Aguirre, A.; Jordan, I.K. Genetic ancestry, admixture and health determinants in Latin America. BMC Genom. 2018, 19, 861. [Google Scholar] [CrossRef] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Res 2016, 5, 1542. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res 2016, 5, 1438. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Schroeder, M.P.; Rubio-Perez, C.; Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveROLE classifies cancer driver genes in loss of function and activating mode of action. Bioinformatics 2014, 30, i549–i555. [Google Scholar] [CrossRef] [Green Version]
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; López-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef] [Green Version]
- Lever, J.; Zhao, E.Y.; Grewal, J.; Jones, M.R.; Jones, S.J.M. CancerMine: A literature-mined resource for drivers, oncogenes and tumor suppressors in cancer. Nat. Methods 2019, 16, 505–507. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ainscough, B.J.; Griffith, M.; Coffman, A.C.; Wagner, A.H.; Kunisaki, J.; Choudhary, M.N.; McMichael, J.F.; Fulton, R.S.; Wilson, R.K.; Griffith, O.L.; et al. DoCM: A database of curated mutations in cancer. Nat. Methods 2016, 13, 806–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.; Lee, J.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Alonso-López, D.; Gutiérrez, M.A.; Lopes, K.P.; Prieto, C.; Santamaría, R.; De Las Rivas, J. APID interactomes: Providing proteome-based interactomes with controlled quality for multiple species and derived networks. Nucleic Acids Res. 2016, 44, W529–W535. [Google Scholar] [CrossRef] [Green Version]
- Fontanillo, C.; Nogales-Cadenas, R.; Pascual-Montano, A.; De Las Rivas, J. Functional Analysis beyond Enrichment: Non-Redundant Reciprocal Linkage of Genes and Biological Terms. PLoS ONE 2011, 6, e24289. [Google Scholar] [CrossRef] [Green Version]
- Conley, A.B.; Rishishwar, L.; Norris, E.T.; Valderrama-Aguirre, A.; Mariño-Ramírez, L.; Medina-Rivas, M.A.; Jordan, I.K. A Comparative Analysis of Genetic Ancestry and Admixture in the Colombian Populations of Chocó and Medellín. G3 Genes Genomes Genet. 2017, 7, 3435–3447. [Google Scholar] [CrossRef] [Green Version]
- Neelamraju, Y.; Gonzalez-Perez, A.; Bhat-Nakshatri, P.; Nakshatri, H.; Janga, S.C. Mutational landscape of RNA-binding proteins in human cancers. RNA Biol. 2018, 15, 115–129. [Google Scholar] [CrossRef]
- Kang, J.M.; Park, S.; Kim, S.J.; Hong, H.Y.; Jeong, J.; Kim, H.S.; Kim, S.J. CBL enhances breast tumor formation by inhibiting tumor suppressive activity of TGF-β signaling. Oncogene 2012, 50, 5123–5131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, M.W.; Maquat, L.E. Nonsense-mediated mRNA Decay and Cancer. Curr. Opin. Genet. Dev. 2018, 48, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Vidoz, L.; Sinilnikova, O.M.; Stoppa-Lyonnet, D.; Lenoir, G.M.; Mazoyer, S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum. Mol. Genet. 2002, 11, 2805–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, A.E.; Smith, I.E.; Ashley, S.; Fulford, L.G.; Lakhani, S.R. Oestrogen receptor status, pathological complete response and prognosis in patients receiving neoadjuvant chemotherapy for early breast cancer. Br. J. Cancer 2004, 91, 2012–2017. [Google Scholar] [CrossRef]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast Cancer Molecular Subtypes Respond Differently to Preoperative Chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef] [Green Version]
- Boon, K.-L.; Norman, C.M.; Grainger, R.J.; Newman, A.J.; Beggs, J.D. Prp8p dissection reveals domain structure and protein interaction sites. RNA 2006, 12, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Kurtovic-Kozaric, A.; Przychodzen, B.; Singh, J.; Konarska, M.M.; Clemente, M.J.; Otrock, Z.K.; Nakashima, M.; Hsi, E.D.; Yoshida, K.; Shiraishi, Y.; et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia 2015, 29, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Henry, N.L.; Hayes, D.F. Cancer biomarkers. Mol. Oncol. 2012, 6, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gómez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.-M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.T.; Gou, X.; Seker, S.; Ellis, M.J. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J. Cancer Metastasis Treat. 2019, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018, 33, 173–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, S.; Yamamoto-Ibusuki, M.; Yamamoto, S.; Yamamoto, Y.; Iwase, H. Association of ErbB1-4 expression in invasive breast cancer with clinicopathological characteristics and prognosis. Breast Cancer 2014, 21, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, X.; Lee, C.K.; Liu, B. Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene 2010, 29, 4225–4236. [Google Scholar] [CrossRef]
- Balko, J.M.; Miller, T.W.; Morrison, M.M.; Hutchinson, K.; Young, C.; Rinehart, C.; Sánchez, V.; Jee, D.; Polyak, K.; Prat, A.; et al. The receptor tyrosine kinase ErbB3 maintains the balance between luminal and basal breast epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Chambard, J.-C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the PI3K-Akt pathway in human cancer: Rationale and promise. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Barnes, D.M.; Millis, R.R.; Gillett, C.E.; Ryder, K.; Skilton, D.; Fentiman, I.S.; Rubens, R.D. The interaction of oestrogen receptor status and pathological features with adjuvant treatment in relation to survival in patients with operable breast cancer: A retrospective study of 2660 patients. Endocr. Relat. Cancer 2004, 11, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Nadal, A.; Ropero, A.B.; Laribi, O.; Maillet, M.; Fuentes, E.; Soria, B. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2000, 97, 11603–11608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhana, P.; Trivelato Junior, R.R.; Beirão, P.S.L.; Cruz, J.S.; Rodrigues, A.L.P. Is there a role for voltage-gated Na+ channels in the aggressiveness of breast cancer? Braz. J. Med. Biol. Res. 2017, 50, e6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.R.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-gated ion channels in cancer cell proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ye, C.; Guo, X.; Wen, W.; Long, J.; Gao, Y.T.; Shu, X.O.; Zheng, W.; Cai, Q. Evaluation of potential regulatory function of breast cancer risk locus at 6q25.1. Carcinogenesis 2016, 37, 163–168. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene HGNC Symbol | Nucleotide Change | Protein AA Change | dbSNP_ID(rs) | Frequency in IDC-LM-BRCA | Cancer-Genome Interpreter Prediction | SNPs (Known, Reported, New) | Human Population with More Frequency |
|---|---|---|---|---|---|---|---|
| ABCB4 | c.G2363A | p.R788Q | rs8187801 | 3/33 | Driver_mutation | reported | ExAC_AFR |
| ATM | c.C7375G | p.R2459G | rs730881383 | 1/33 | Driver_mutation | reported | ExAC_OTH |
| ATM | c.C7468T | p.L2490F | rs753262623 | 1/33 | Driver_mutation | reported | ExAC_SAS |
| CD36 | c.G1016T | p.G339V | rs146027667 | 1/33 | Driver_mutation | known | ExAC_OTH |
| CHD8 | c.C871T | p.L291F | rs192989929 | 1/33 | Driver_mutation | reported | ExAC_OTH/ExAC_AMR |
| DPYD | c.A2846T | p.D949V | rs67376798 | 1/33 | known in cancer | reported | ExAC_NFE |
| EPHA1 | c.C2371T | p.R791C | rs766301333 | 1/33 | Driver_mutation | reported | ExAC_NFE |
| ERBB3 | c.G2167C | p.V723L | rs189789018 | 1/33 | Driver_mutation | known | ExAC_AMR |
| ESR1 | c.G1138C | p.E380Q # | rs1057519827 | 1/33 | Driver_mutation | known | all populations similar |
| MLH1 | c.A1129G | p.K377E | rs35001569 | 1/33 | Driver_mutation | reported | ExAC_NFE |
| MSH3 | c.T2732G | p.L911W | rs41545019 | 2/33 | Driver_mutation | reported | ExAC_NFE |
| NOTCH1 | c.G2983A | p.G995S ## | rs868369610 | 1/33 | Driver_mutation | reported | all populations similar |
| NOTCH4 | c.G2504T | p.G835V | rs9267835 | 2/33 | Driver_mutation | known | ExAC_AFR/ExAC_AMR |
| STAT6 | c.C1069T | p.R357W | rs776930978 | 1/33 | Driver_mutation | reported | all populations similar |
| TP53 | c.G338T | p.G113V | rs121912656 | 1/33 | Driver_mutation | reported | ExAC_EAS |
| TP53 | c.T215A | p.L72Q | rs1057519997 | 1/33 | Driver_mutation | reported | all populations similar |
| UPF3B | c.G1082A | p.R361H | rs143538947 | 1/33 | Driver_mutation | reported | ExAC_AFR |
| CBLB | c.G1972A | p.G658S | locus (chr:3q13.11;exon:13) | 1/33 | Driver_mutation | new | NA |
| PRPF8 | c.G4153T | p.V1385F | locus (chr:17p13.3;exon:25) | 1/33 | Driver_mutation | new | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes-Urrea, C.; Bueno-Gutiérrez, F.; Solarte, M.; Guevara-Burbano, M.; Tobar-Tosse, F.; Vélez-Varela, P.E.; Bonilla, J.C.; Barreto, G.; Velasco-Medina, J.; Moreno, P.A.; et al. Exomes of Ductal Luminal Breast Cancer Patients from Southwest Colombia: Gene Mutational Profile and Related Expression Alterations. Biomolecules 2020, 10, 698. https://doi.org/10.3390/biom10050698
Cortes-Urrea C, Bueno-Gutiérrez F, Solarte M, Guevara-Burbano M, Tobar-Tosse F, Vélez-Varela PE, Bonilla JC, Barreto G, Velasco-Medina J, Moreno PA, et al. Exomes of Ductal Luminal Breast Cancer Patients from Southwest Colombia: Gene Mutational Profile and Related Expression Alterations. Biomolecules. 2020; 10(5):698. https://doi.org/10.3390/biom10050698
Chicago/Turabian StyleCortes-Urrea, Carolina, Fernando Bueno-Gutiérrez, Melissa Solarte, Miguel Guevara-Burbano, Fabian Tobar-Tosse, Patricia E. Vélez-Varela, Juan Carlos Bonilla, Guillermo Barreto, Jaime Velasco-Medina, Pedro A. Moreno, and et al. 2020. "Exomes of Ductal Luminal Breast Cancer Patients from Southwest Colombia: Gene Mutational Profile and Related Expression Alterations" Biomolecules 10, no. 5: 698. https://doi.org/10.3390/biom10050698
APA StyleCortes-Urrea, C., Bueno-Gutiérrez, F., Solarte, M., Guevara-Burbano, M., Tobar-Tosse, F., Vélez-Varela, P. E., Bonilla, J. C., Barreto, G., Velasco-Medina, J., Moreno, P. A., & De Las Rivas, J. (2020). Exomes of Ductal Luminal Breast Cancer Patients from Southwest Colombia: Gene Mutational Profile and Related Expression Alterations. Biomolecules, 10(5), 698. https://doi.org/10.3390/biom10050698