A Novel C1-Esterase Inhibitor Oxygenator Coating Prevents FXII Activation in Human Blood

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Hollow-Fiber Oxygenator Membranes
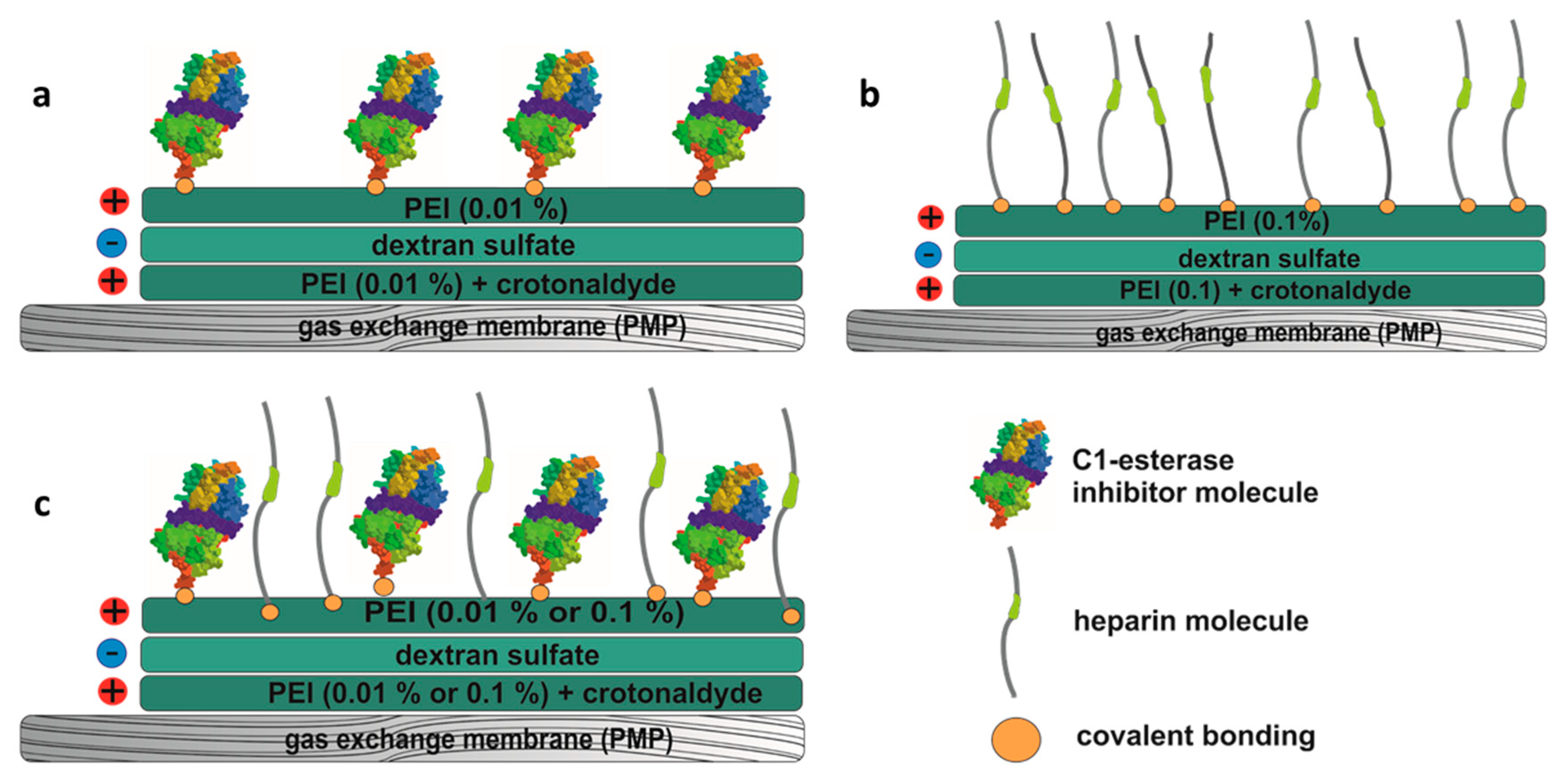
2.2. Coating of the Membranes
2.3. Detection of Bound C1-INH and Heparin
2.4. Blood Sampling
2.5. Blood Incubation
2.6. Soluble Activation Markers
2.7. Scanning Electron Microscopy
2.8. Fluorescence Microscopy
2.9. Statistical Analyses
3. Results
3.1. Quantification of Immobilized C1-INH and Heparin on PMP Membranes
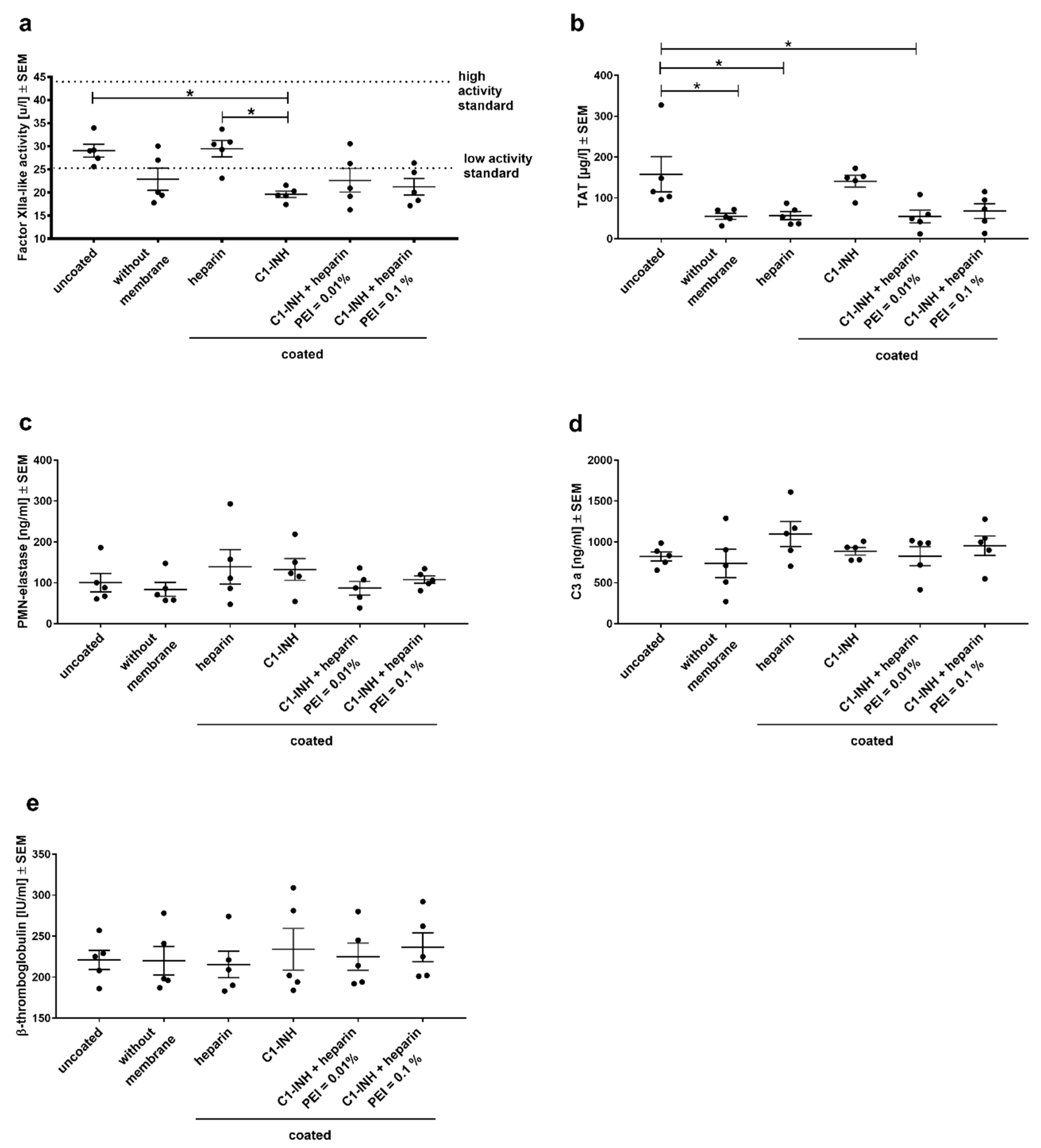
3.2. Analysis of Hemocompatibility
3.3. Membrane Surface Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gulack, B.C.; Hirji, S.A.; Hartwig, M.G. Bridge to lung transplantation and rescue post-transplant: The expanding role of extracorporeal membrane oxygenation. J. Thorac. Dis. 2014, 6, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.M.; Lynch, W.R.; Coopersmith, C.M. Clinical and billing review of extracorporeal membrane oxygenation. Chest 2015, 147, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Marasco, S.F.; Lukas, G.; McDonald, M.; McMillan, J.; Ihle, B. Review of ECMO (Extra Corporeal Membrane Oxygenation) Support in Critically Ill Adult Patients. Hear. Lung Circ. 2008, 17, S41–S47. [Google Scholar] [CrossRef] [PubMed]
- Betit, P. Technical Advances in the Field of ECMO. Respir. Care 2018, 63, 1162–1173. [Google Scholar] [CrossRef]
- MONTOYA, J.P.; SHANLEY, C.J.; MERZ, S.I.; BARTLETT, R.H. Plasma Leakage through Microporous Membranes. ASAIO J. 1992, 38, M399–M405. [Google Scholar] [CrossRef]
- Mottaghy, K.; Oedekoven, B.; Starmans, H.; Müller, B.; Kashefi, A.; Hoffmann, B.; Böhm, S. Technical aspects of plasma leakage prevention in microporous capillary membrane oxygenators. ASAIO Trans. 1989, 35, 640–643. [Google Scholar] [CrossRef]
- Lequier, L.; Horton, S.B.; McMullan, D.M.; Bartlett, R.H. Extracorporeal membrane oxygenation circuitry. Pediatr. Crit. Care Med. 2013, 14, S7–S12. [Google Scholar] [CrossRef]
- Miyashita, T.; Ahmed, A.; Nakanuma, S.; Okamoto, K.; Sakai, S.; Kinoshita, J.; Makino, I.; Nakamura, K.; Hayashi, H.; Oyama, K.; et al. A Three-phase Approach for the Early Identification of Acute Lung Injury Induced by Severe Sepsis. In Vivo (Brooklyn) 2016, 30, 341–350. [Google Scholar]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Sniecinski, R.M.; Chandler, W.L. Activation of the hemostatic system during cardiopulmonary bypass. Anesth. Analg. 2011, 113, 1319–1333. [Google Scholar] [CrossRef]
- Zakkar, M.; Guida, G.; Suleiman, M.-S.; Angelini, G.D. Cardiopulmonary Bypass and Oxidative Stress. Oxid. Med. Cell. Longev. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hein, E.; Munthe-Fog, L.; Thiara, A.S.; Fiane, A.E.; Mollnes, T.E.; Garred, P. Heparin-coated cardiopulmonary bypass circuits selectively deplete the pattern recognition molecule ficolin-2 of the lectin complement pathway in vivo. Clin. Exp. Immunol. 2015, 179, 294–299. [Google Scholar] [CrossRef]
- Kraft, F.; Schmidt, C.; Van Aken, H.; Zarbock, A. Inflammatory response and extracorporeal circulation. Best Pract. Res. Clin. Anaesthesiol. 2015, 29, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Palanzo, D.; Qiu, F.; Baer, L.; Clark, J.B.; Myers, J.L.; Ündar, A. Artificial Organs; John Wiley & Sons, Ltd (10.1111): Hoboken, NJ, USA, 1 November 2010; pp. 869–873. [Google Scholar]
- Weber, M.; Steinle, H.; Golombek, S.; Hann, L.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Blood-Contacting Biomaterials: In Vitro Evaluation of the Hemocompatibility. Front. Bioeng. Biotechnol. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Hogwood, J.; Mulloy, B. The anticoagulant and antithrombotic mechanisms of heparin. Handb. Exp. Pharmacol. 2012, 207, 43–61. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Schmaier, A.H. The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J. Thromb. Haemost. 2016, 14, 28–39. [Google Scholar] [CrossRef]
- Renné, T.; Schmaier, A.H.; Nickel, K.F.; Blombäck, M.; Maas, C. In vivo roles of factor XII. Blood 2012, 120, 4296–4303. [Google Scholar]
- Müller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renné, T. Platelet Polyphosphates Are Proinflammatory and Procoagulant Mediators In Vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef]
- Morrissey, J.H. Polyphosphate: A link between platelets, coagulation and inflammation. Int. J. Hematol. 2012, 95, 346–352. [Google Scholar] [CrossRef]
- Weidmann, H.; Heikaus, L.; Long, A.T.; Naudin, C.; Schlüter, H.; Renné, T. The plasma contact system, a protease cascade at the nexus of inflammation, coagulation and immunity. Biochim. Biophys. Acta - Mol. Cell Res. 2017, 1864, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Renné, T.; Schmaier, A.H.; Nickel, K.F.; Blombäck, M.; Maas, C.; Dc, W.; Schmaier, A.H.; Nickel, K.F.; Blomba, M. In vivo roles of factor XII Review article In vivo roles of factor XII. Blood 2014, 120, 4296–4303. [Google Scholar] [CrossRef]
- Panagiotou, A.; Trendelenburg, M.; Osthoff, M. The Lectin pathway of complement in myocardial ischemia/reperfusion injury-review of its significance and the potential impact of therapeutic interference by C1 esterase inhibitor. Front. Immunol. 2018, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Björkqvist, J.; Nickel, K.F.; Stavrou, E.; Renné, T. In vivo activation and functions of the protease factor XII. Thromb. Haemost. 2014, 112, 868–875. [Google Scholar] [CrossRef]
- Gailani, D.; Bane, C.E.; Gruber, A. Factor XI and contact activation as targets for antithrombotic therapy. J. Thromb. Haemost. 2015, 13, 1383–1395. [Google Scholar] [CrossRef]
- Øvrum, E.; Tangen, G.; Tølløfsrud, S.; Skeie, B.; Ringdal, M.A.L.; Istad, R.; Øystese, R. Heparinized cardiopulmonary bypass circuits and low systemic anticoagulation: An analysis of nearly 6000 patients undergoing coronary artery bypass grafting. J. Thorac. Cardiovasc. Surg. 2011, 141, 1145–1149. [Google Scholar] [CrossRef]
- Ranucci, M.; Balduini, A.; Ditta, A.; Boncilli, A.; Brozzi, S. A Systematic Review of Biocompatible Cardiopulmonary Bypass Circuits and Clinical Outcome. Ann. Thorac. Surg. 2009, 87, 1311–1319. [Google Scholar] [CrossRef]
- Weber, N.; Wendel, H.P.; Ziemer, G. Hemocompatibility of heparin-coated surfaces and the role of selective plasma protein adsorption. Biomaterials 2002, 23, 429–439. [Google Scholar] [CrossRef]
- Begovac, P.C.; Thomson, R.C.; Fisher, J.L.; Hughson, A.; Gällhagen, A. Improvements in GORE-TEX® vascular graft performance by Carmeda® bioactive surface heparin immobilization. Eur. J. Vasc. Endovasc. Surg. 2003, 25, 432–437. [Google Scholar] [CrossRef]
- Biran, R.; Pond, D. Heparin coatings for improving blood compatibility of medical devices. Adv. Drug Deliv. Rev. 2017, 112, 12–23. [Google Scholar] [CrossRef]
- Cai, S.; Dole, V.S.; Bergmeier, W.; Scafidi, J.; Feng, H.; Wagner, D.D.; Davis, A.E. A Direct Role for C1 Inhibitor in Regulation of Leukocyte Adhesion. J. Immunol. 2005, 174, 6462–6466. [Google Scholar] [CrossRef] [PubMed]
- Li, H.(Henry). Self-administered C1 esterase inhibitor concentrates for the management of hereditary angioedema: usability and patient acceptance. Patient Prefer. Adherence 2016, 10, 1727–1737. [Google Scholar] [CrossRef]
- Decher, G.; Hong, J.-D. Buildup of ultrathin multilayer films by a self-assembly process, 1 consecutive adsorption of anionic and cationic bipolar amphiphiles on charged surfaces. Makromol. Chemie. Macromol. Symp. 1991, 46, 321–327. [Google Scholar] [CrossRef]
- Hylton, D.M.; Shalaby, S.W.; Latour, R.A. Direct correlation between adsorption-induced changes in protein structure and platelet adhesion. J. Biomed. Mater. Res. Part A 2005, 73A, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S. The transition of prothrombin to thrombin. J. Thromb. Haemost. 2013, 11, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, M.; Erez, O.; Mastrolia, S.A.; Koifman, A.; Leron, E.; Eshkoli, T.; Mazor, M.; Holcberg, G. The physiologic and therapeutic role of heparin in implantation and placentation. PeerJ 2015, 3, e691. [Google Scholar] [CrossRef]
- Jaffer, I.H.; Fredenburgh, J.C.; Hirsh, J.; Weitz, J.I. Medical device-induced thrombosis: What causes it and how can we prevent it? J. Thromb. Haemost. 2015, 13, S72–S81. [Google Scholar] [CrossRef]
- Visser, M.; Heitmeier, S.; Ten Cate, H.; Spronk, H.M.H. Role of Factor XIa and Plasma Kallikrein in Arterial and Venous Thrombosis. Thromb. Haemost. 2020, 120, 883–993. [Google Scholar] [CrossRef]
- Gailani, D.; Broze, G.J. Factor XII-independent activation of factor XI in plasma: effects of sulfatides on tissue factor-induced coagulation. Blood 1993, 82, 813–819. [Google Scholar] [CrossRef]
- Naudin, C.; Burillo, E.; Blankenberg, S.; Butler, L.; Renné, T. Factor XII Contact Activation. Semin. Thromb. Hemost. 2017, 43, 814–826. [Google Scholar] [CrossRef]
- Zhang, L.; Casey, B.; Galanakis, D.K.; Marmorat, C.; Skoog, S.; Vorvolakos, K.; Simon, M.; Rafailovich, M.H. The influence of surface chemistry on adsorbed fibrinogen conformation, orientation, fiber formation and platelet adhesion. Acta Biomater. 2017, 54, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, C.; Bai, Q.; Zhang, Z. Rare heparin induced thrombocytopenia type I reaction in a hemodialysis patient Case report. Med. (United States) 2018, 97. [Google Scholar] [CrossRef]
- Riedel, R.; Schmieder, A.; Koster, A.; Kim, S.; Baumgarten, G.; Schewe, J.C. Heparin-induced thrombocytopenia type II (HIT II): A medical-economic view. Medizinische Klin. Intensivmed. und Notfallmedizin 2017, 112, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.J.Y.; Parikh, C.R. HEMATOLOGY: ISSUES IN THE DIALYSIS PATIENT: When Heparin Causes Thrombosis: Significance, Recognition, and Management of Heparin-Induced Thrombocytopenia in Dialysis Patients. Semin. Dial. 2006, 19, 297–304. [Google Scholar] [CrossRef]
- Grouzi, E. Update on argatroban for the prophylaxis and treatment of heparin-induced thrombocytopenia type II. J. Blood Med. 2014, 5, 131. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerling, K.; Ölschläger, S.; Avci-Adali, M.; Neumann, B.; Schweizer, E.; Schlensak, C.; Wendel, H.-P.; Stoppelkamp, S. A Novel C1-Esterase Inhibitor Oxygenator Coating Prevents FXII Activation in Human Blood. Biomolecules 2020, 10, 1042. https://doi.org/10.3390/biom10071042
Gerling K, Ölschläger S, Avci-Adali M, Neumann B, Schweizer E, Schlensak C, Wendel H-P, Stoppelkamp S. A Novel C1-Esterase Inhibitor Oxygenator Coating Prevents FXII Activation in Human Blood. Biomolecules. 2020; 10(7):1042. https://doi.org/10.3390/biom10071042
Chicago/Turabian StyleGerling, Katharina, Sabrina Ölschläger, Meltem Avci-Adali, Bernd Neumann, Ernst Schweizer, Christian Schlensak, Hans-Peter Wendel, and Sandra Stoppelkamp. 2020. "A Novel C1-Esterase Inhibitor Oxygenator Coating Prevents FXII Activation in Human Blood" Biomolecules 10, no. 7: 1042. https://doi.org/10.3390/biom10071042
APA StyleGerling, K., Ölschläger, S., Avci-Adali, M., Neumann, B., Schweizer, E., Schlensak, C., Wendel, H.-P., & Stoppelkamp, S. (2020). A Novel C1-Esterase Inhibitor Oxygenator Coating Prevents FXII Activation in Human Blood. Biomolecules, 10(7), 1042. https://doi.org/10.3390/biom10071042