Post-Translational Regulation of ARF: Perspective in Cancer

Abstract
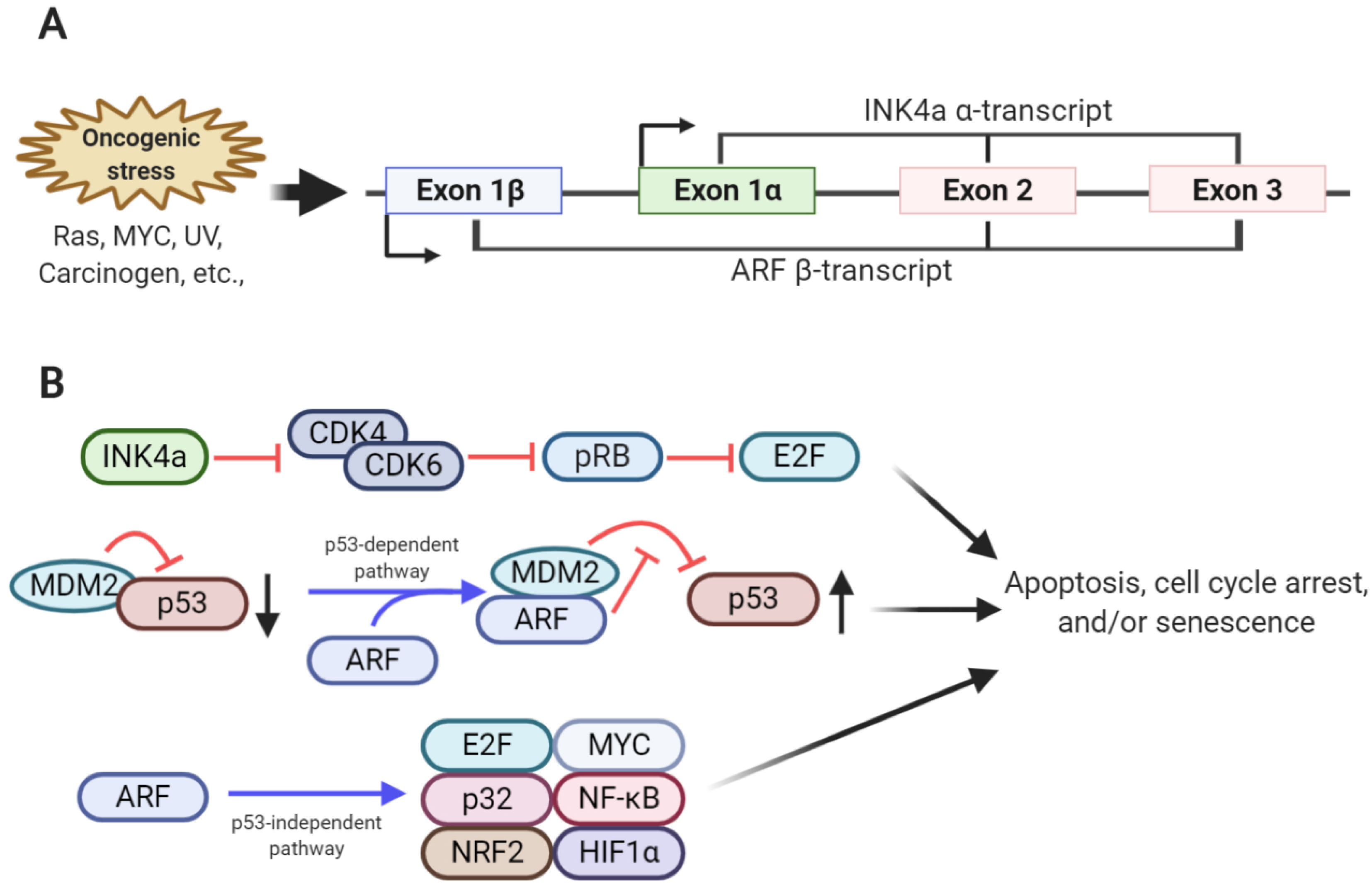
:1. Introduction
2. Transcriptional Regulation of ARF
2.1. Activators of ARF Transcription
2.2. Suppressors of ARF Transcription
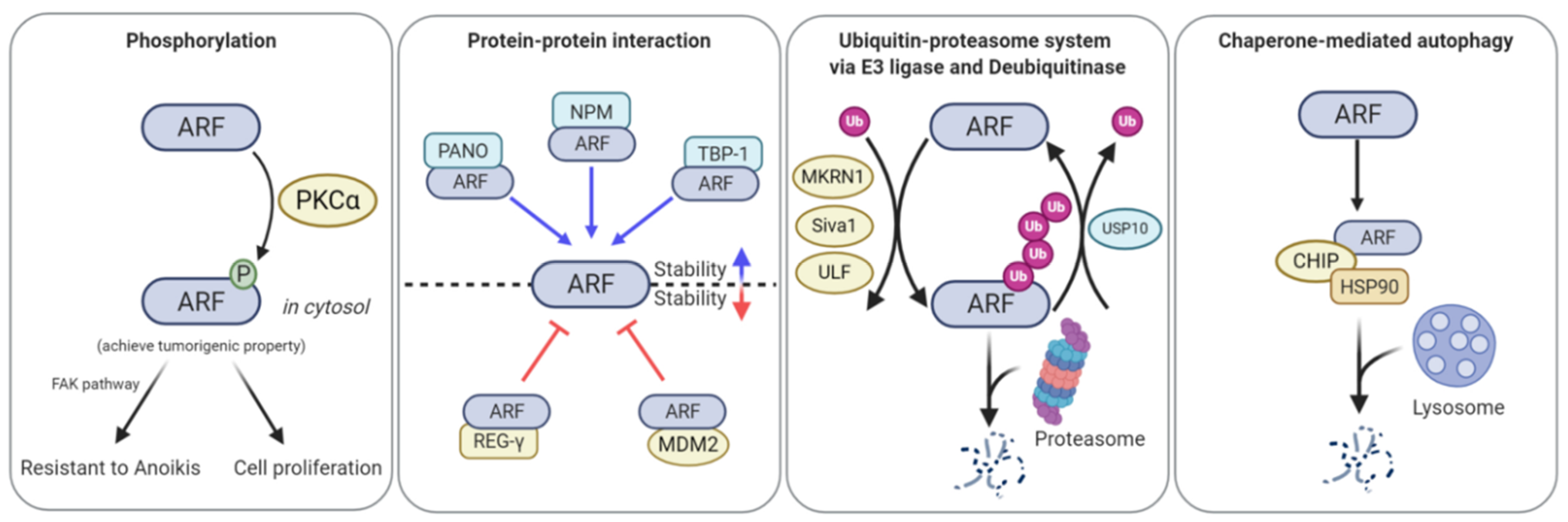
3. Post-Translational Regulation of ARF Regulates Its Functional Roles in Cellular Physiology
3.1. ARF Phosphorylation
3.2. ARF Regulation via Degradation
3.2.1. The Ubiquitin-Proteasome System (UPS)
3.2.2. Chaperone-Mediated Autophagy (CMA)
3.3. Protein–Protein Interaction (PPI)
4. Post-Translational Regulation of ARF in Human Cancer
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hall, M.; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and cdk inhibitors in human cancer. Adv. Cancer Res. 1996, 68, 67–108. [Google Scholar] [PubMed]
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual role of the alternative reading frame arf protein in cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Han, S.Y.; Song, J. Dynamics of arf regulation that control senescence and cancer. BMB Rep. 2016, 49, 598–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.; Han, S.Y.; Song, J. Regulatory network of arf in cancer development. Mol. Cells 2018, 41, 381–389. [Google Scholar] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16ink4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the arf tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin d/cdk4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Russo, A.A.; Tong, L.; Lee, J.O.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the cyclin-dependent kinase cdk6 by the tumour suppressor p16ink4a. Nature 1998, 395, 237–243. [Google Scholar] [CrossRef]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the arf tumor suppressor with p53 and mdm2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The ink4a tumor suppressor gene product, p19arf, interacts with mdm2 and neutralizes mdm2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Eymin, B.; Karayan, L.; Seite, P.; Brambilla, C.; Brambilla, E.; Larsen, C.J.; Gazzeri, S. Human arf binds e2f1 and inhibits its transcriptional activity. Oncogene 2001, 20, 1033–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, F.; Hamilton, T.; Silver, D.P.; Sharpless, N.E.; Bardeesy, N.; Rokas, M.; DePinho, R.A.; Livingston, D.M.; Grossman, S.R. P19arf targets certain e2f species for degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 4455–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Nag, A.; Raychaudhuri, P. Differential regulation of e2f1, dp1, and the e2f1/dp1 complex by arf. Mol. Cell. Biol. 2002, 22, 8398–8408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Sen, J.; Hagen, J.; Korgaonkar, C.K.; Caffrey, M.; Quelle, D.E.; Hughes, D.E.; Ackerson, T.J.; Costa, R.H.; Raychaudhuri, P. Arf directly binds dp1: Interaction with dp1 coincides with the g1 arrest function of arf. Mol. Cell. Biol. 2005, 25, 8024–8036. [Google Scholar] [CrossRef] [Green Version]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of arf-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Nag, A.; Pan, W.; Hay, N.; Gartel, A.L.; Colamonici, O.; Mori, Y.; Raychaudhuri, P. Myc-arf (alternate reading frame) interaction inhibits the functions of myc. J. Biol. Chem. 2004, 279, 36698–36707. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Gregory, M.A.; Li, Z.; Brousal, J.P.; West, K.; Hann, S.R. P19arf directly and differentially controls the functions of c-myc independently of p53. Nature 2004, 431, 712–717. [Google Scholar] [CrossRef]
- Rocha, S.; Campbell, K.J.; Perkins, N.D. P53- and mdm2-independent repression of nf-kappa b transactivation by the arf tumor suppressor. Mol. Cell 2003, 12, 15–25. [Google Scholar] [CrossRef]
- Fatyol, K.; Szalay, A.A. The p14arf tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1alpha (hif-1alpha ) and inhibits hif-1-mediated transcription. J. Biol. Chem. 2001, 276, 28421–28429. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. Nrf2 is a major target of arf in p53-independent tumor suppression. Mol. Cell 2017, 68, 224–232. [Google Scholar] [CrossRef]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19arf induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef] [PubMed]
- van Oosterwijk, J.G.; Li, C.; Yang, X.; Opferman, J.T.; Sherr, C.J. Small mitochondrial arf (smarf) protein corrects p53-independent developmental defects of arf tumor suppressor-deficient mice. Proc. Natl. Acad. Sci. USA 2017, 114, 7420–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiGiammarino, E.L.; Filippov, I.; Weber, J.D.; Bothner, B.; Kriwacki, R.W. Solution structure of the p53 regulatory domain of the p19arf tumor suppressor protein. Biochemistry 2001, 40, 2379–2386. [Google Scholar] [PubMed]
- Bothner, B.; Lewis, W.S.; DiGiammarino, E.L.; Weber, J.D.; Bothner, S.J.; Kriwacki, R.W. Defining the molecular basis of arf and hdm2 interactions. J. Mol. Biol. 2001, 314, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The arf tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef]
- Müller-Tidow, C.; Metzelder, S.; Buerger, H.; Packeisen, J.; Ganser, A.; Heil, G.; Kügler, K.; Adigüzel, G.; Schwäble, J.; Steffen, B. Expression of the p14 arf tumor suppressor predicts survival in acute myeloid leukemia. Leukemia 2004, 18, 720–726. [Google Scholar] [CrossRef]
- Elliott, M.J.; Dong, Y.B.; Yang, H.; McMasters, K.M. E2f-1 up-regulates c-myc and p14arf and induces apoptosis in colon cancer cells. Clin. Cancer Res. 2001, 7, 3590–3597. [Google Scholar]
- Bouchard, C.; Lee, S.; Paulus-Hock, V.; Loddenkemper, C.; Eilers, M.; Schmitt, C.A. Foxo transcription factors suppress myc-driven lymphomagenesis via direct activation of arf. Genes Dev. 2007, 21, 2775–2787. [Google Scholar] [CrossRef] [Green Version]
- Freeman-Anderson, N.E.; Zheng, Y.; McCalla-Martin, A.C.; Treanor, L.M.; Zhao, Y.D.; Garfin, P.M.; He, T.C.; Mary, M.N.; Thornton, J.D.; Anderson, C.; et al. Expression of the arf tumor suppressor gene is controlled by tgfbeta2 during development. Development 2009, 136, 2081–2089. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhao, Y.D.; Gibbons, M.; Abramova, T.; Chu, P.Y.; Ash, J.D.; Cunningham, J.M.; Skapek, S.X. Tgfbeta signaling directly induces arf promoter remodeling by a mechanism involving smads 2/3 and p38 mapk. J. Biol. Chem. 2010, 285, 35654–35664. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Roussel, M.F.; Sherr, C.J. Induction of arf tumor suppressor gene expression and cell cycle arrest by transcription factor dmp1. Proc. Natl. Acad. Sci. USA 1999, 96, 3993–3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglic, D.; Stovall, D.B.; Cline, J.M.; Fry, E.A.; Mallakin, A.; Taneja, P.; Caudell, D.L.; Willingham, M.C.; Sui, G.; Inoue, K. Dmp1beta, a splice isoform of the tumour suppressor dmp1 locus, induces proliferation and progression of breast cancer. J. Pathol. 2015, 236, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Fry, E.A. Aberrant splicing of the dmp1-arf-mdm2-p53 pathway in cancer. Int. J. Cancer 2016, 139, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Ozenne, P.; Dayde, D.; Brambilla, E.; Eymin, B.; Gazzeri, S. P14(arf) inhibits the growth of lung adenocarcinoma cells harbouring an egfr l858r mutation by activating a stat3-dependent pro-apoptotic signalling pathway. Oncogene 2013, 32, 1050–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayde, D.; Guerard, M.; Perron, P.; Hatat, A.S.; Barrial, C.; Eymin, B.; Gazzeri, S. Nuclear trafficking of egfr by vps34 represses arf expression to promote lung tumor cell survival. Oncogene 2016, 35, 3986–3994. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, W. Functional characterization of e2f3b in human hepg2 liver cancer cell line. J. Cell. Biochem. 2018, 119, 3429–3439. [Google Scholar] [CrossRef]
- Jacobs, J.J.; Keblusek, P.; Robanus-Maandag, E.; Kristel, P.; Lingbeek, M.; Nederlof, P.M.; van Welsem, T.; van de Vijver, M.J.; Koh, E.Y.; Daley, G.Q.; et al. Senescence bypass screen identifies tbx2, which represses cdkn2a (p19(arf)) and is amplified in a subset of human breast cancers. Nat. Genet. 2000, 26, 291–299. [Google Scholar] [CrossRef]
- Pietersen, A.M.; Horlings, H.M.; Hauptmann, M.; Langerød, A.; Ajouaou, A.; Cornelissen-Steijger, P.; Wessels, L.F.; Jonkers, J.; Van De Vijver, M.J.; van Lohuizen, M. Ezh2 and bmi1 inversely correlate with prognosis and tp53 mutation in breast cancer. Breast Cancer Res. 2008, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; He, L.; Kapoor, A.; Gillis, A.; Rybak, A.P.; Cutz, J.-C.; Tang, D. Bmi1 promotes prostate tumorigenesis via inhibiting p16ink4a and p14arf expression. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Bernard, D.; Martinez-Leal, J.F.; Rizzo, S.; Martinez, D.; Hudson, D.; Visakorpi, T.; Peters, G.; Carnero, A.; Beach, D.; Gil, J. Cbx7 controls the growth of normal and tumor-derived prostate cells by repressing the ink4a/arf locus. Oncogene 2005, 24, 5543–5551. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Ghosh, P.; O’Farrell, T.; Munk, R.; Rezanka, L.J.; Sasaki, C.Y.; Longo, D.L. Transforming growth factor beta1 (tgf-beta1) suppresses growth of b-cell lymphoma cells by p14(arf)-dependent regulation of mutant p53. J. Biol. Chem. 2012, 287, 23184–23195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakouros, D.; Isenmann, S.; Cooper, L.; Zannettino, A.; Anderson, P.; Glackin, C.; Gronthos, S. Twist-1 induces ezh2 recruitment regulating histone methylation along the ink4a/arf locus in mesenchymal stem cells. Mol. Cell. Biol. 2012, 32, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of myc and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, 1–24. [Google Scholar] [CrossRef] [PubMed]
- DeGregori, J.; Leone, G.; Miron, A.; Jakoi, L.; Nevins, J.R. Distinct roles for e2f proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7245–7250. [Google Scholar] [CrossRef] [Green Version]
- Aslanian, A.; Iaquinta, P.J.; Verona, R.; Lees, J.A. Repression of the arf tumor suppressor by e2f3 is required for normal cell cycle kinetics. Genes Dev. 2004, 18, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Komori, H.; Enomoto, M.; Nakamura, M.; Iwanaga, R.; Ohtani, K. Distinct e2f-mediated transcriptional program regulates p14arf gene expression. EMBO J. 2005, 24, 3724–3736. [Google Scholar] [CrossRef] [Green Version]
- Humbert, P.O.; Verona, R.; Trimarchi, J.M.; Rogers, C.; Dandapani, S.; Lees, J.A. E2f3 is critical for normal cellular proliferation. Genes Dev. 2000, 14, 690–703. [Google Scholar]
- Tschan, M.P.; Federzoni, E.A.; Haimovici, A.; Britschgi, C.; Moser, B.A.; Jin, J.; Reddy, V.A.; Sheeter, D.A.; Fischer, K.M.; Sun, P.; et al. Human dmtf1beta antagonizes dmtf1alpha regulation of the p14(arf) tumor suppressor and promotes cellular proliferation. Biochim. Biophys. Acta 2015, 1849, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Wen, R.; Rehg, J.E.; Adachi, M.; Cleveland, J.L.; Roussel, M.F.; Sherr, C.J. Disruption of the arf transcriptional activator dmp1 facilitates cell immortalization, ras transformation, and tumorigenesis. Genes Dev. 2000, 14, 1797–1809. [Google Scholar]
- McKeller, R.N.; Fowler, J.L.; Cunningham, J.J.; Warner, N.; Smeyne, R.J.; Zindy, F.; Skapek, S.X. The arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc. Natl Acad. Sci. USA 2002, 99, 3848–3853. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.L.; Thornton, J.D.; Martin, A.C.; Rehg, J.E.; Bertwistle, D.; Zindy, F.; Skapek, S.X. Arf-dependent regulation of pdgf signaling in perivascular cells in the developing mouse eye. EMBO J. 2005, 24, 2803–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromley, A.; Churchman, M.L.; Zindy, F.; Sherr, C.J. Transient expression of the arf tumor suppressor during male germ cell and eye development in arf-cre reporter mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6285–6290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Lohuizen, M.; Verbeek, S.; Scheijen, B.; Wientjens, E.; van der Gulden, H.; Berns, A. Identification of cooperating oncogenes in e mu-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [Google Scholar] [CrossRef]
- Haupt, Y.; Alexander, W.S.; Barri, G.; Klinken, S.P.; Adams, J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in e mu-myc transgenic mice. Cell 1991, 65, 753–763. [Google Scholar] [CrossRef]
- van Lohuizen, M.; Frasch, M.; Wientjens, E.; Berns, A. Sequence similarity between the mammalian bmi-1 proto-oncogene and the drosophila regulatory genes psc and su(z)2. Nature 1991, 353, 353–355. [Google Scholar] [CrossRef]
- Alkema, M.J.; Jacobs, J.; Voncken, J.W.; Jenkins, N.A.; Copeland, N.G.; Satijn, D.P.; Otte, A.P.; Berns, A.; van Lohuizen, M. Mpc2, a new murine homolog of the drosophila polycomb protein is a member of the mouse polycomb transcriptional repressor complex. J. Mol. Biol. 1997, 273, 993–1003. [Google Scholar] [CrossRef]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef]
- Gil, J.; Bernard, D.; Martinez, D.; Beach, D. Polycomb cbx7 has a unifying role in cellular lifespan. Nat. Cell Biol. 2004, 6, 67–72. [Google Scholar] [CrossRef]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The egfr odyssey—From activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Inoue, R.; Shiraishi, T. Pkcalpha is involved in phorbol ester tpa-mediated stabilization of p14arf. Biochem. Biophys. Res. Commun. 2005, 330, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Ranieri, M.; Sansone, F.; Santoriello, C.; Calogero, R.A.; Calabro, V.; Pollice, A.; La Mantia, G. Mimicking p14arf phosphorylation influences its ability to restrain cell proliferation. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef]
- Fontana, R.; Guidone, D.; Sangermano, F.; Calabro, V.; Pollice, A.; La Mantia, G.; Vivo, M. Pkc dependent p14arf phosphorylation on threonine 8 drives cell proliferation. Sci. Rep. 2018, 8, 7056–7065. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, M.W.; Bae, K.H.; Lee, S.C.; Song, J.; Lee, E.W. The roles of ubiquitination in extrinsic cell death pathways and its implications for therapeutics. Biochem. Pharmacol. 2019, 162, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of e3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.L.; den Besten, W.; Bertwistle, D.; Roussel, M.F.; Sherr, C.J. N-terminal polyubiquitination and degradation of the arf tumor suppressor. Genes Dev. 2004, 18, 1862–1874. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Shan, J.; Zhu, W.G.; Qin, J.; Gu, W. Transcription-independent arf regulation in oncogenic stress-mediated p53 responses. Nature 2010, 464, 624–627. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Kon, N.; Zhong, J.; Zhang, P.; Yu, L.; Gu, W. Differential effects on arf stability by normal versus oncogenic levels of c-myc expression. Mol. Cell 2013, 51, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Chio, I.I.C.; Sasaki, M.; Ghazarian, D.; Moreno, J.; Done, S.; Ueda, T.; Inoue, S.; Chang, Y.L.; Chen, N.J.; Mak, T.W. Tradd contributes to tumour suppression by regulating ulf-dependent p19arf ubiquitylation. Nat. Cell Biol 2012, 14, 625–633. [Google Scholar] [CrossRef]
- Lo, D.; Zhang, Y.; Dai, M.S.; Sun, X.X.; Zeng, S.X.; Lu, H. Nucleostemin stabilizes arf by inhibiting the ubiquitin ligase ulf. Oncogene 2015, 34, 1688–1697. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Cho, Y.E.; Kim, S.H.; Kim, Y.J.; Park, J.H. Gltscr2 promotes the nucleoplasmic translocation and subsequent degradation of nucleolar arf. Oncotarget 2017, 8, 16293–16302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.; Shin, J.Y.; Seo, J.; Lee, K.D.; Lee, E.W.; Lee, M.S.; Lee, H.W.; Choi, I.J.; Jeong, J.S.; Chun, K.H.; et al. Acceleration of gastric tumorigenesis through mkrn1-mediated posttranslational regulation of p14arf. J. Natl. Cancer Inst. 2012, 104, 1660–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zha, M.; Zhao, X.; Jiang, P.; Du, W.; Tam, A.Y.; Mei, Y.; Wu, M. Siva1 inhibits p53 function by acting as an arf e3 ubiquitin ligase. Nat. Commun. 2013, 4, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Araki, T.; Nakagawa, M.; Hirao, A.; Unno, M.; Nakayama, K. S6 kinase- and beta-trcp2-dependent degradation of p19arf is required for cell proliferation. Mol. Cell Biol. 2015, 35, 3517–3527. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Han, S.Y.; Choi, C.H.; Cho, H.; Lee, M.S.; Kim, S.Y.; Song, J.S.; Hong, K.M.; Lee, H.W.; Hewitt, S.M.; et al. Oncogene-induced senescence mediated by c-myc requires usp10 dependent deubiquitination and stabilization of p14arf. Cell Death Differ. 2018, 25, 1050–1062. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.B.; Shi, G.M.; Dong, Z.R.; Ke, A.W.; Ma, H.H.; Gao, Q.; Shen, Z.Z.; Huang, X.Y.; Chen, H.; Yu, D.D.; et al. Ubiquitin-specific protease 7 accelerates p14(arf) degradation by deubiquitinating thyroid hormone receptor-interacting protein 12 and promotes hepatocellular carcinoma progression. Hepatology 2015, 61, 1603–1614. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Han, S.Y.; Ko, A.; Kitano, H.; Choi, C.H.; Lee, M.S.; Seo, J.; Fukuoka, J.; Kim, S.Y.; Hewitt, S.M.; Chung, J.Y.; et al. Molecular chaperone hsp90 is necessary to prevent cellular senescence via lysosomal degradation of p14arf. Cancer Res. 2017, 77, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Han, S.Y.; Seong, D.; Han, H.J.; Song, J. Multifaceted c-terminus of hsp70-interacting protein regulates tumorigenesis via protein quality control. Arch. Pharm. Res. 2019, 42, 63–75. [Google Scholar] [CrossRef]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef]
- Korgaonkar, C.; Hagen, J.; Tompkins, V.; Frazier, A.A.; Allamargot, C.; Quelle, F.W.; Quelle, D.E. Nucleophosmin (b23) targets arf to nucleoli and inhibits its function. Mol. Cell. Biol. 2005, 25, 1258–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Bonetti, P.; Lazzerini Denchi, E.; Martinelli, P.; Zamponi, R.; Marine, J.C.; Helin, K.; Falini, B.; Pelicci, P.G. Nucleophosmin is required for DNA integrity and p19arf protein stability. Mol. Cell. Biol. 2005, 25, 8874–8886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Besten, W.; Kuo, M.L.; Williams, R.T.; Sherr, C.J. Myeloid leukemia-associated nucleophosmin mutants perturb p53-dependent and independent activities of the arf tumor suppressor protein. Cell Cycle 2005, 4, 1593–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogev, O.; Saadon, K.; Anzi, S.; Inoue, K.; Shaulian, E. DNA damage-dependent translocation of b23 and p19 arf is regulated by the jun n-terminal kinase pathway. Cancer Res. 2008, 68, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase atm and arf tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef]
- Hamilton, G.; Abraham, A.G.; Morton, J.; Sampson, O.; Pefani, D.E.; Khoronenkova, S.; Grawenda, A.; Papaspyropoulos, A.; Jamieson, N.; McKay, C.; et al. Akt regulates npm dependent arf localization and p53mut stability in tumors. Oncotarget 2014, 5, 6142–6167. [Google Scholar] [CrossRef]
- Pollice, A.; Nasti, V.; Ronca, R.; Vivo, M.; Lo Iacono, M.; Calogero, R.; Calabro, V.; La Mantia, G. Functional and physical interaction of the human arf tumor suppressor with tat-binding protein-1. J. Biol Chem 2004, 279, 6345–6353. [Google Scholar] [CrossRef] [Green Version]
- Pollice, A.; Sepe, M.; Villella, V.R.; Tolino, F.; Vivo, M.; Calabro, V.; La Mantia, G. Tbp-1 protects the human oncosuppressor p14arf from proteasomal degradation. Oncogene 2007, 26, 5154–5162. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Barton, L.F.; Chi, Y.; Clurman, B.E.; Roberts, J.M. Ubiquitin-independent degradation of cell-cycle inhibitors by the reggamma proteasome. Mol. Cell 2007, 26, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Vivo, M.; Matarese, M.; Sepe, M.; Di Martino, R.; Festa, L.; Calabro, V.; La Mantia, G.; Pollice, A. Mdm2-mediated degradation of p14arf: A novel mechanism to control arf levels in cancer cells. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Watari, A.; Li, Y.; Higashiyama, S.; Yutsudo, M. A novel proapoptotic gene pano encodes a post-translational modulator of the tumor suppressor p14arf. Exp. Cell Res. 2012, 318, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse ink4a locus mediated by the alternative reading frame product p19arf. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, T.; Bodner, S.; van de Kamp, E.; Randle, D.H.; Sherr, C.J. Tumor spectrum in arf-deficient mice. Cancer Res. 1999, 59, 2217–2222. [Google Scholar] [PubMed]
- Sharpless, N.E.; Ramsey, M.R.; Balasubramanian, P.; Castrillon, D.H.; DePinho, R.A. The differential impact of p16(ink4a) or p19(arf) deficiency on cell growth and tumorigenesis. Oncogene 2004, 23, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.; Dominguez, G.; Silva, J.M.; Garcia, J.M.; Gallego, I.; Corbacho, C.; Provencio, M.; Espana, P.; Bonilla, F. Analysis of genetic and epigenetic processes that influence p14arf expression in breast cancer. Oncogene 2001, 20, 4586–4590. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, G.; Carballido, J.; Silva, J.; Silva, J.M.; Garcia, J.M.; Menendez, J.; Provencio, M.; Espana, P.; Bonilla, F. P14arf promoter hypermethylation in plasma DNA as an indicator of disease recurrence in bladder cancer patients. Clin. Cancer Res. 2002, 8, 980–985. [Google Scholar] [PubMed]
- Silva, J.; Silva, J.M.; Dominguez, G.; Garcia, J.M.; Cantos, B.; Rodriguez, R.; Larrondo, F.J.; Provencio, M.; Espana, P.; Bonilla, F. Concomitant expression of p16ink4a and p14arf in primary breast cancer and analysis of inactivation mechanisms. J. Pathol. 2003, 199, 289–297. [Google Scholar] [CrossRef]
- Dominguez, G.; Silva, J.; Garcia, J.M.; Silva, J.M.; Rodriguez, R.; Munoz, C.; Chacon, I.; Sanchez, R.; Carballido, J.; Colas, A.; et al. Prevalence of aberrant methylation of p14arf over p16ink4a in some human primary tumors. Mutat. Res. 2003, 530, 9–17. [Google Scholar] [CrossRef]
- Lee, M.; Sup Han, W.; Kyoung Kim, O.; Hee Sung, S.; Sun Cho, M.; Lee, S.N.; Koo, H. Prognostic value of p16ink4a and p14arf gene hypermethylation in human colon cancer. Pathol. Res. Pract. 2006, 202, 415–424. [Google Scholar] [CrossRef]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14(arf) is independent of p16(ink4a) methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar]
- Tannapfel, A.; Busse, C.; Geissler, F.; Witzigmann, H.; Hauss, J.; Wittekind, C. Ink4a-arf alterations in liver cell adenoma. Gut 2002, 51, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannapfel, A.; Sommerer, F.; Benicke, M.; Weinans, L.; Katalinic, A.; Geissler, F.; Uhlmann, D.; Hauss, J.; Wittekind, C. Genetic and epigenetic alterations of the ink4a-arf pathway in cholangiocarcinoma. J. Pathol. 2002, 197, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Akiyama, Y.; Nakajima, T.; Ichikawa, W.; Nihei, Z.; Sugihara, K.; Yuasa, Y. Alterations and hypermethylation of the p14(arf) gene in gastric cancer. Int. J. Cancer 2000, 87, 654–658. [Google Scholar] [CrossRef]
- Zochbauer-Muller, S.; Fong, K.M.; Virmani, A.K.; Geradts, J.; Gazdar, A.F.; Minna, J.D. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001, 61, 249–255. [Google Scholar]
- Chaar, I.; Amara, S.; Elamine, O.E.; Khiari, M.; Ounissi, D.; Khalfallah, T.; Ben Hmida, A.; Mzabi, S.; Bouraoui, S. Biological significance of promoter hypermethylation of p14/arf gene: Relationships to p53 mutational status in tunisian population with colorectal carcinoma. Tumour Biol. 2014, 35, 1439–1449. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.S.; Wang, Y.C.; Tseng, R.C.; Chang, J.W.; Chen, J.T.; Shih, C.M.; Chen, C.Y.; Wang, Y.C. 5’ cytosine-phospho-guanine island methylation is responsible for p14arf inactivation and inversely correlates with p53 overexpression in resected non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 4734–4741. [Google Scholar] [CrossRef] [Green Version]
- Berggren, P.; Kumar, R.; Sakano, S.; Hemminki, L.; Wada, T.; Steineck, G.; Adolfsson, J.; Larsson, P.; Norming, U.; Wijkstrom, H.; et al. Detecting homozygous deletions in the cdkn2a(p16(ink4a))/arf(p14(arf)) gene in urinary bladder cancer using real-time quantitative pcr. Clin. Cancer Res. 2003, 9, 235–242. [Google Scholar]
- Shintani, S.; Nakahara, Y.; Mihara, M.; Ueyama, Y.; Matsumura, T. Inactivation of the p14(arf), p15(ink4b) and p16(ink4a) genes is a frequent event in human oral squamous cell carcinomas. Oral Oncol. 2001, 37, 498–504. [Google Scholar] [CrossRef]
- Konishi, N.; Nakamura, M.; Kishi, M.; Nishimine, M.; Ishida, E.; Shimada, K. Heterogeneous methylation and deletion patterns of the ink4a/arf locus within prostate carcinomas. Am. J. Pathol. 2002, 160, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Sailasree, R.; Abhilash, A.; Sathyan, K.M.; Nalinakumari, K.R.; Thomas, S.; Kannan, S. Differential roles of p16ink4a and p14arf genes in prognosis of oral carcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Nishida, N.; Fukuda, Y.; Nishimura, T.; Komeda, T.; Nakao, K. Alteration of the p14(arf) gene and p53 status in human hepatocellular carcinomas. J. Gastroenterol. 2004, 39, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Bilim, V.; Hara, N.; Takahashi, K.; Tomita, Y. Homozygous deletions of the ink4a/arf locus in renal cell cancer. Anticancer Res. 2006, 26, 4299–4305. [Google Scholar] [PubMed]
- Rizos, H.; Puig, S.; Badenas, C.; Malvehy, J.; Darmanian, A.P.; Jimenez, L.; Mila, M.; Kefford, R.F. A melanoma-associated germline mutation in exon 1beta inactivates p14arf. Oncogene 2001, 20, 5543–5547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randerson-Moor, J.A.; Harland, M.; Williams, S.; Cuthbert-Heavens, D.; Sheridan, E.; Aveyard, J.; Sibley, K.; Whitaker, L.; Knowles, M.; Bishop, J.N.; et al. A germline deletion of p14(arf) but not cdkn2a in a melanoma-neural system tumour syndrome family. Hum. Mol. Genet. 2001, 10, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizos, H.; Darmanian, A.P.; Holland, E.A.; Mann, G.J.; Kefford, R.F. Mutations in the ink4a/arf melanoma susceptibility locus functionally impair p14arf. J. Biol. Chem. 2001, 276, 41424–41434. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, C.; Lee Wu, C.; Evans, G.; Howell, A.; Elles, R.G.; Jordan, R.; Sloan, P.; Read, A.P.; Thakker, N. Germline mutation of arf in a melanoma kindred. Hum. Mol. Genet. 2002, 11, 1273–1279. [Google Scholar] [CrossRef] [Green Version]
- Vonlanthen, S.; Heighway, J.; Tschan, M.P.; Borner, M.M.; Altermatt, H.J.; Kappeler, A.; Tobler, A.; Fey, M.F.; Thatcher, N.; Yarbrough, W.G.; et al. Expression of p16ink4a/p16alpha and p19arf/p16beta is frequently altered in non-small cell lung cancer and correlates with p53 overexpression. Oncogene 1998, 17, 2779–2785. [Google Scholar] [CrossRef] [Green Version]
- Gazzeri, S.; Della Valle, V.; Chaussade, L.; Brambilla, C.; Larsen, C.J.; Brambilla, E. The human p19arf protein encoded by the beta transcript of the p16ink4a gene is frequently lost in small cell lung cancer. Cancer Res. 1998, 58, 3926–3931. [Google Scholar]
- Song, J.S.; Yi, J.M.; Cho, H.; Choi, C.H.; Park, Y.; Chung, E.J.; Song, J.; Chung, J.Y.; Hong, S.M. Dual loss of usp10 and p14arf protein expression is associated with poor prognosis in patients with small intestinal adenocarcinoma. Tumour Biol. 2018, 40, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Han, G.H.; Chay, D.B.; Yi, J.M.; Cho, H.; Chung, J.Y.; Kim, J.H. Loss of both usp10 and p14arf protein expression is an independent prognostic biomarker for poor prognosis in patients with epithelial ovarian cancer. Cancer Genom. Proteom. 2019, 16, 553–562. [Google Scholar] [CrossRef]
- Vivo, M.; Fontana, R.; Ranieri, M.; Capasso, G.; Angrisano, T.; Pollice, A.; Calabro, V.; La Mantia, G. P14arf interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 2017, 36, 4913–4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates e-cadherin repression via p19arf in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Lu, W.; Liu, S.; Yang, Q.; Goodwin, J.S.; Sathyanarayana, S.A.; Pratap, S.; Chen, Z. Mmp7 interacts with arf in nucleus to potentiate tumor microenvironments for prostate cancer progression in vivo. Oncotarget 2016, 7, 47609–47619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Aguilera, A.; Sanchez-Beato, M.; Garcia, J.F.; Prieto, I.; Pollan, M.; Piris, M.A. P14(arf) nuclear overexpression in aggressive b-cell lymphomas is a sensor of malfunction of the common tumor suppressor pathways. Blood 2002, 99, 1411–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.K.; Park, J.Y.; Kang, H.J.; Cho, H.C. Overexpression of p16ink4a and p14arf in haematological malignancies. Clin. Lab. Haematol. 2003, 25, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Ferru, A.; Fromont, G.; Gibelin, H.; Guilhot, J.; Savagner, F.; Tourani, J.M.; Kraimps, J.L.; Larsen, C.J.; Karayan-Tapon, L. The status of cdkn2a alpha (p16ink4a) and beta (p14arf) transcripts in thyroid tumour progression. Br. J. Cancer 2006, 95, 1670–1677. [Google Scholar] [CrossRef] [Green Version]
- Humbey, O.; Pimkina, J.; Zilfou, J.T.; Jarnik, M.; Dominguez-Brauer, C.; Burgess, D.J.; Eischen, C.M.; Murphy, M.E. The arf tumor suppressor can promote the progression of some tumors. Cancer Res. 2008, 68, 9608–9613. [Google Scholar] [CrossRef] [Green Version]
- Owczarek, T.B.; Kobayashi, T.; Ramirez, R.; Rong, L.; Puzio-Kuter, A.M.; Iyer, G.; Teo, M.Y.; Sanchez-Vega, F.; Wang, J.; Schultz, N.; et al. Arf confers a context-dependent response to chemotherapy in muscle-invasive bladder cancer. Cancer Res. 2017, 77, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. Arf induces autophagy by virtue of interaction with bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef] [Green Version]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine arf tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M., Jr.; Zaika, A.I. Helicobacter pylori pathogen regulates p14arf tumor suppressor and autophagy in gastric epithelial cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Transcription Factor | Cancer Type | Correlation with ARF Expression | Molecular Mechanism | Ref. |
|---|---|---|---|---|
| MYC | Acute myeloid leukemia | Positive correlation with ARF The combined expression of high MYC and ARF in AML Patients with low ARF expression worsen overall survival rates | MYC overexpression increases ARF mRNA transcription. ARF null mice exhibit resistance to MYC-driven apoptosis. | [26] |
| E2F1/E2F2 | Colon cancer | Positive correlation with ARF The combined expression of high E2Fs and ARF in colon cancer | E2Fs bind to the conserved sequence of ARF promoter, increasing ARF transcription. Overexpression of E2F1 leads to G2/M arrest with increase in ARF protein levels. | [27] |
| FoxO | Primary lymphoma | Positive correlation with ARF FoxO proteins have an instructive role in regulating ARF expression during MYC-induced lymphomagenesis | FoxO increases ARF transcription via interacting with FoxO-binding site region in the first intron of ARF. Lymphomas expressing a dominant-negative mutant of FoxO (dnFoxO) have low levels of ARF mRNA regardless of the p53 status. | [28] |
| TGF-β2/ SMAD2/3 | Unknown | Positive correlation with ARF TGFβ2-deficient embryos show hyperplasia phenotype in the eyes at embryonic day 13.5 with low ARF expression | SMAD2/3 bind to a proximal region of the ARF locus in a TGFβ2-dependent manner. | [29,30] |
| DMP1α | Unknown | Positive correlation with ARF | DMP1α binds to the consensus sequence of the ARF promoter, leading to an increase in ARF transcription. | [31] |
| DMP1β | Breast cancer | Inverse correlation with ARF The correlation between high DMP1β expression and shorter survival of breast cancer patients | DMP1β binds to DMP1α, which inhibit its transcriptional activity, thereby leading to a decrease in ARF transcription. High DMP1β and low DMP1α expression due to alternative splicing is frequently observed in breast cancer patients. | [32,33] |
| EGFR/VPS34 | Lung cancer | Inverse correlation with ARF The expression of low ARF in lung tumors harbouring constitutive active mutant EGFR | Active EGFR interacts with VPS34, which moves to the nucleus, thus inhibiting ARF expression via binding to the AT-rich sequence of the ARF promoter. | [34,35] |
| E2F3b | Hepatocarcinoma | Inverse correlation with ARF The expression of high E2F3 in hepatocellular carcinoma (HCC) | E2F3b represses ARF mRNA expression via binding to ARF promoter. E2F3b induces G1/S phase transition and markedly increases cell proliferation, but has a minor effect on apoptosis. | [36] |
| TBX2 | Breast cancer | Inverse correlation with ARF TBX2 amplification in human breast cancer | ARF expression in BMI-1 deficient cells is suppressed by TBX2 without any change in INK4a level. | [37] |
| BMI-1 | Breast cancer | Inverse correlation with ARF | Overexpression of BMI-1 results in the elevation of expression of polycomb group (PcG)-target genes followed by the inhibition of ARF expression. | [38] |
| Prostate cancer | Inverse correlation with ARF The combined expression of high BMI-1 and low ARF in prostate cancer | BMI-1-expressing DU145 cells form drastic large tumors in NOD/SCID mice. | [39] | |
| CBX7 | Prostate cancer | Inverse correlation with ARF | CBX7 ablation retards cell proliferation via the ARF/p53 and INK4a/Rb pathways. | [40] |
| TGF-β1 | B-cell lymphoma | Inverse correlation with ARF | In B-cell lymphoma expressing mutant p53, activation of TGFβ1 leads to a decrease in E2F1 expression, leading to the reduction in ARF transcription. The low expression of ARF induces the destabilization of mutant p53. | [41] |
| Twist/Ezh2 | Unknown | Inverse correlation with ARF | Twist-1 recognizes H3K27me3 on the ARF locus followed by interaction with Ezh2, which leads to suppression of ARF transcription via PRC2 complex. | [42] |
| Post-Translational Regulator | Cancer Type | Correlation with ARF Expression | Molecular Mechanism | Ref. |
|---|---|---|---|---|
| MKRN1 | Gastric adenocarcinoma | Inverse correlation with ARF The combined expression of high MKRN1 and low ARF in well-differentiated adenocarcinoma | MKRN1 promotes ARF ubiquitination, which leads to the proteasome-dependent degradation of ARF | [72] |
| TRADD | Invasive breast cancer | Positive correlation with ARF Low TRADD expression correlates with poor prognosis. | TRADD competes with ULF for interaction with ARF, protecting ARF from ULF-mediated ubiquitination. | [69] |
| ATM | Lung carcinoma | Inverse correlation with ARF | ATM-PP1 axis inhibits Nek2 kinase activity, which induces the de-phosphorylation of NPM, thus leading to the nucleoplasm localization and degradation of ARF. | [85] |
| USP7/ULF | Hepatocarcinoma | Inverse correlation with ARF The combined expression of low USP7 and ULF worsen overall survival rates. | USP7 forms a complex with ULF that protects ULF protein from proteasome-mediated degradation via removal of ubiquitin. | [76] |
| HSP90/CHIP | NSCLC | Inverse correlation with ARF The combined expression of high HSP90, CHIP, and low ARF worsen overall survival rates. | HSP90 and CHIP complex form an interaction with ARF, which induces lysosomal degradation of ARF through binding to LAMP2A. The E3 ligase activity of CHIP is not required for formation of a tertiary complex and lysosomal degradation of ARF. | [78,79] |
| USP10 | NSCLC | Positive correlation with ARF The combined expression of low USP10 and ARF worsen overall survival rates. | MYC increases the stability of ARF protein via induction of USP10, which is a deubiquitinase of ARF. | [75] |
| Small intestine cancer | Positive correlation with ARF The combined expression of high USP10 and ARF are negatively correlated with vascular and lymphatic invasion. The combined expression of low USP10 and ARF worsen overall survival rates. | Several patients with intestinal adenocarcinoma contain aberrant hyper-methylations in the USP10 and ARF promoter regions with low expression of both proteins. | [119] | |
| Ovarian Cancer | Positive correlation with ARF The combined expression of low USP10 and ARF is displayed in cancer. The combined expression of low USP10 and ARF worsen overall survival rates. | High degree of methylation in USP10 and ARF CpG islands detected by methylation specific PCR analysis in ovarian cancer patients | [120] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, J.; Seong, D.; Lee, S.R.; Oh, D.-B.; Song, J. Post-Translational Regulation of ARF: Perspective in Cancer. Biomolecules 2020, 10, 1143. https://doi.org/10.3390/biom10081143
Seo J, Seong D, Lee SR, Oh D-B, Song J. Post-Translational Regulation of ARF: Perspective in Cancer. Biomolecules. 2020; 10(8):1143. https://doi.org/10.3390/biom10081143
Chicago/Turabian StyleSeo, Jinho, Daehyeon Seong, Seung Ri Lee, Doo-Byoung Oh, and Jaewhan Song. 2020. "Post-Translational Regulation of ARF: Perspective in Cancer" Biomolecules 10, no. 8: 1143. https://doi.org/10.3390/biom10081143
APA StyleSeo, J., Seong, D., Lee, S. R., Oh, D.-B., & Song, J. (2020). Post-Translational Regulation of ARF: Perspective in Cancer. Biomolecules, 10(8), 1143. https://doi.org/10.3390/biom10081143