An Extra Breath of Fresh Air: Hyperbaric Oxygenation as a Stroke Therapeutic



Abstract
1. Introduction
2. HBOT in Ischemic Stroke: Potential Benefits and Limitations
2.1. Preclinical Functional Outcomes of Post-stroke HBOT
2.2. Clinical Results of HBOT in Stroke
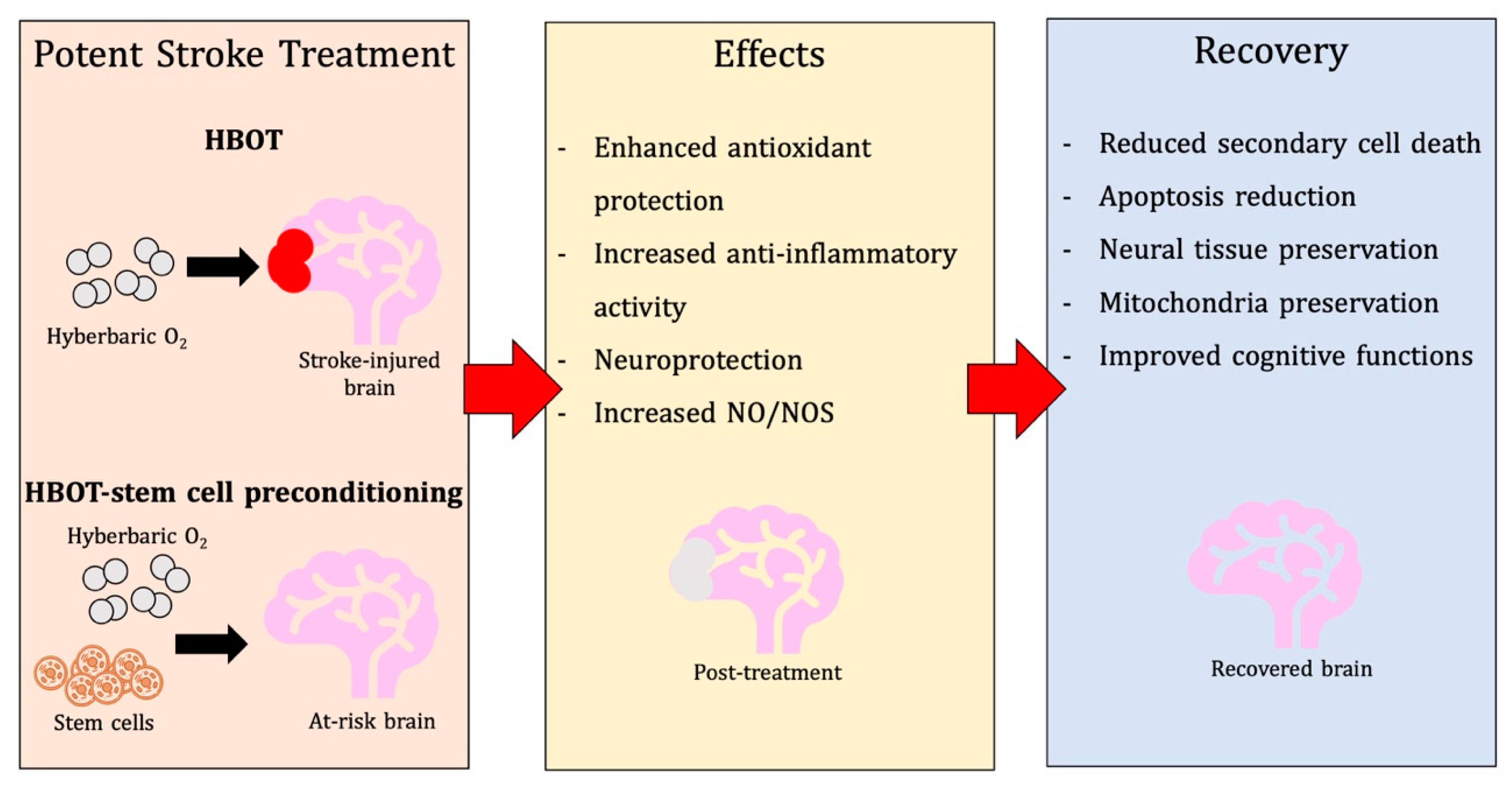
3. Unpacking Mechanisms of Action of HBOT in Stroke
3.1. Physiological and Metabolic Effects
3.2. Antioxidant Effects
3.3. Anti-Inflammatory Effects
3.4. Additional Neuroprotective Mechanisms
4. Implications of HBOT in Other Neurological and Non-Neurological Conditions
4.1. HBOT in Acute and Chronic TBI
4.2. HBOT in Spinal Cord Injury
4.3. HBOT in Other Pathological Contexts
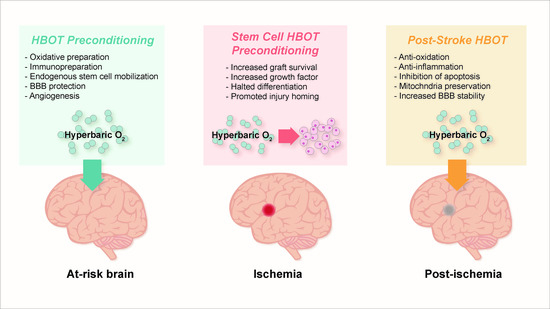
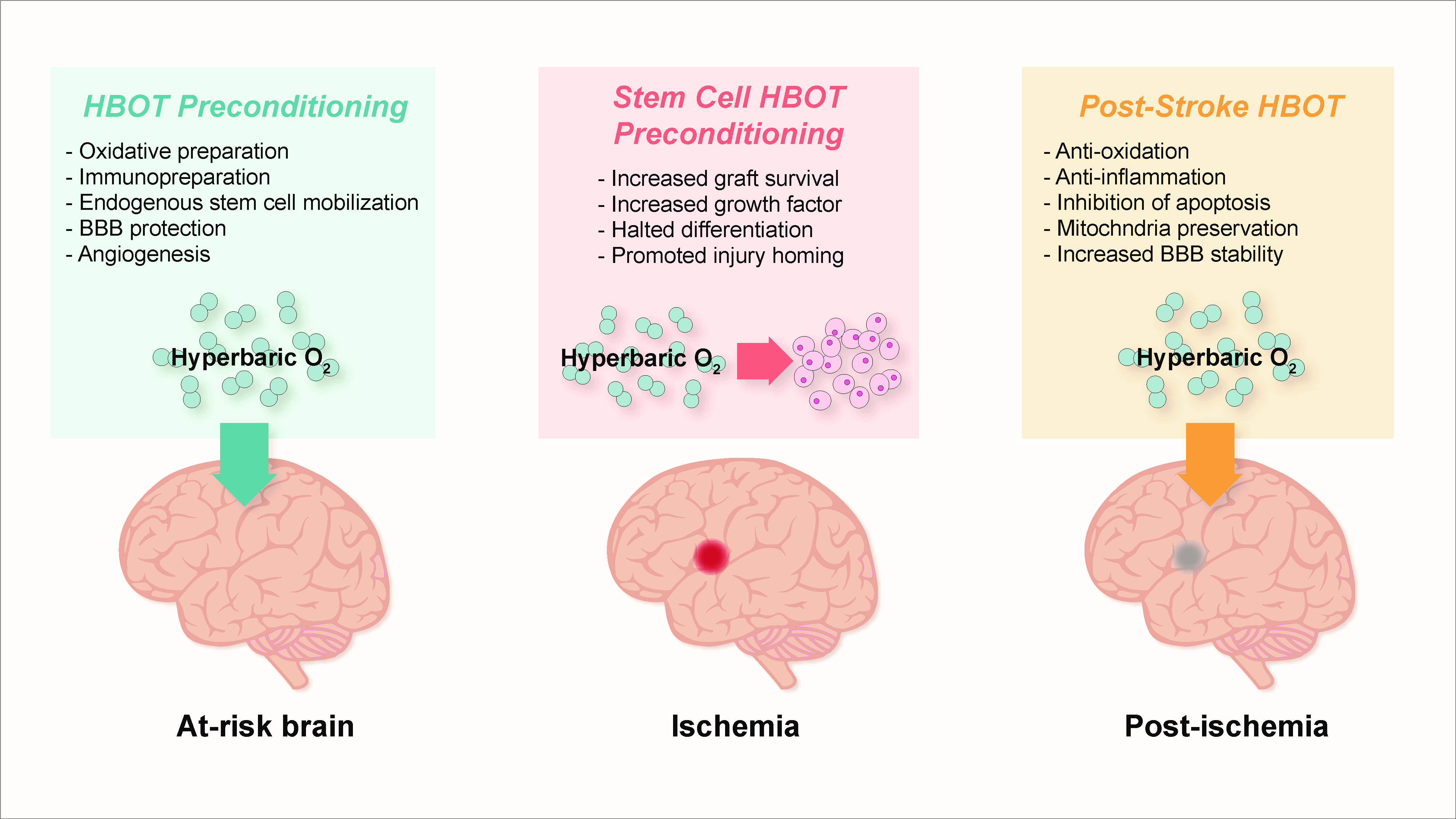
5. Pre-Clinical Findings with HBOT Preconditioning for Stroke
5.1. Preparation for Oxidative Stress
5.2. Reduction of Apoptosis, Activation of Autophagy, and Promotion of Cell Survival
5.3. Immunosuppression and Immunopreparation
5.4. Preservation of Blood-Brain Barrier, Edema Minimization, and Angiogenesis
5.5. Considerations for HBOT Preconditioning Protocols
6. HBOT-Primed Stem Cells as a Promising Therapy
6.1. HBOT Effects on Endogenous Stem Cells
6.2. HBOT and Exogenous Stem Cells
6.3. Effects of HBOT In Vitro: Potential for Stem Cell Priming
6.4. Recent Literature on HBOT and Stroke
6.4.1. Preconditioning
6.4.2. Post-Stroke Treatment
6.4.3. Diseases Resembling Stroke Pathology and HBOT
6.4.4. Optimizing Treatment
7. Future Directions and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tal, S.; Hadanny, A.; Berkovitz, N.; Sasson, E.; Ben-Jacob, E.; Efrati, S. Hyperbaric oxygen may induce angiogenesis in patients suffering from prolonged post-concussion syndrome due to traumatic brain injury. Restor. Neurol. Neurosci. 2015, 33, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Harch, P.G.; Andrews, S.R.; Fogarty, E.F.; Lucarini, J.; Van Meter, K.W. Case control study: Hyperbaric oxygen treatment of mild traumatic brain injury persistent post-concussion syndrome and post- traumatic stress disorder. Med. Gas Res. 2017, 7, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.G.; Lin, G.A.; Xie, H.Q.; Pan, H.T.; Huang, B.Q.; Liu, J.D.; Liu, H.; Zhang, N.; Li, L.; et al. The effects of different hyperbaric oxygen manipulations in rats after traumatic brain injury. Neurosci. Lett. 2014, 563, 38–43. [Google Scholar] [CrossRef]
- Miljkovic-Lolic, M.; Silbergleit, R.; Fiskum, G.; Rosenthal, R.E. Neuroprotective effects of hyperbaric oxygen treatment in experimental focal cerebral ischemia are associated with reduced brain leukocyte myeloperoxidase activity. Brain Res. 2003, 971, 90–94. [Google Scholar] [CrossRef]
- Calvert, J.W.; Zhou, C.; Nanda, A.; Zhang, J.H. Effect of hyperbaric oxygen on apoptosis in neonatal hypoxia-ischemia rat model. J. Appl. Physiol. 2003, 95, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.K.; Cao, H.H.; Ying, X.; Zhang, H.T.; Yu, H.L. The effects of hyperbaric oxygen on macrophage polarization after rat spinal cord injury. Brain Res. 2015, 1606, 68–76. [Google Scholar] [CrossRef]
- Oyinbo, C.A. Secondary injury mechanisms in traumatic spinal cord injury: A nugget of this multiply cascade. Acta Neurobiol. Exp. (Wars) 2011, 71, 281–299. [Google Scholar]
- Wang, L.; Li, W.; Kang, Z.; Liu, Y.; Deng, X.; Tao, H.; Xu, W.; Li, R.; Sun, X.; Zhang, J.H. Hyperbaric oxygen preconditioning attenuates early apoptosis after spinal cord ischemia in rats. J. Neurotrauma 2009, 26, 55–66. [Google Scholar] [CrossRef]
- Shams, Z.; Khalatbary, A.R.; Ahmadvand, H.; Zare, Z.; Kian, K. Neuroprotective effects of hyperbaric oxygen (HBO) therapy on neuronal death induced by sciatic nerve transection in rat. BMC Neurol. 2017, 17, 220. [Google Scholar] [CrossRef]
- Sun, L.; Marti, H.H.; Veltkamp, R. Hyperbaric oxygen reduces tissue hypoxia and hypoxia-inducible factor-1 alpha expression in focal cerebral ischemia. Stroke 2008, 39, 1000–1006. [Google Scholar] [CrossRef]
- Zhang, X.G.; Jiang, Z.L.; Wang, G.H.; Li, Y.C.; Wang, Y.; Li, X.; Shen, H.M. Therapeutic efficacy of hyperbaric oxygen on traumatic brain injury in the rat and the underlying mechanisms. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2012, 28, 42–46. [Google Scholar]
- Lou, M.; Chen, Y.; Ding, M.; Eschenfelder, C.C.; Deuschl, G. Involvement of the mitochondrial ATP- sensitive potassium channel in the neuroprotective effect of hyperbaric oxygenation after cerebral ischemia. Brain Res. Bull. 2006, 69, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, D.; Wang, Q.; Su, P.; Tang, Q. Hyperbaric oxygen treatment of spinal cord injury in rat model. BMC Neurol. 2017, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.W.; Zhang, F.; Liu, H.J.; Li, Z. Hyperbaric oxygen ameliorated the lesion scope and nerve function in acute spinal cord injury patients: A retrospective study. Clin. Biochem. 2018, 53, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Li, F.; Thomas, S.; Yang, Z. Hyperbaric oxygenation alleviates chronic constriction injury (CCI)- induced neuropathic pain and inhibits GABAergic neuron apoptosis in the spinal cord. Scand. J. Pain 2017, 17, 330–338. [Google Scholar] [CrossRef]
- Daruwalla, J.; Christophi, C. Hyperbaric oxygen therapy for malignancy: A review. World J. Surg. 2006, 30, 2112–2131. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Xu, J.; Xu, L.; Wang, S.; Li, R.; Liu, K.; Zheng, J.; Cai, Z.; Zhang, K.; Luo, Y.; et al. Hyperbaric oxygen preconditioning induces tolerance against oxidative injury and oxygen-glucose deprivation by up-regulating heat shock protein 32 in rat spinal neurons. PLoS ONE 2014, 9, e85967. [Google Scholar] [CrossRef]
- Hirata, T.; Cui, Y.J.; Funakoshi, T.; Mizukami, Y.; Ishikawa, Y.; Shibasaki, F.; Matsumoto, M.; Sakabe, T. The temporal profile of genomic responses and protein synthesis in ischemic tolerance of the rat brain induced by repeated hyperbaric oxygen. Brain Res. 2007, 1130, 214–222. [Google Scholar] [CrossRef]
- Gunther, A.; Kuppers-Tiedt, L.; Schneider, P.M.; Kunert, I.; Berrouschot, J.; Schneider, D.; Rossner, S. Reduced infarct volume and differential effects on glial cell activation after hyperbaric oxygen treatment in rat permanent focal cerebral ischaemia. Eur. J. Neurosci. 2005, 21, 3189–3194. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Rossignol, L.W. Hyperbaric oxygen therapy may improve symptoms in autistic children. Med. Hypotheses 2006, 67, 216–228. [Google Scholar] [CrossRef]
- Xiong, L.; Zhu, Z.; Dong, H.; Hu, W.; Hou, L.; Chen, S. Hyperbaric oxygen preconditioning induces neuroprotection against ischemia in transient not permanent middle cerebral artery occlusion rat model. Chin. Med. J. 2000, 113, 836–839. [Google Scholar] [PubMed]
- Mu, J.; Ostrowski, R.P.; Soejima, Y.; Rolland, W.B.; Krafft, P.R.; Tang, J.; Zhang, J.H. Delayed hyperbaric oxygen therapy induces cell proliferation through stabilization of cAMP responsive element binding protein in the rat model of MCAo-induced ischemic brain injury. Neurobiol. Dis. 2013, 51, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, W.; Ding, S.; Xu, W.; Guan, Y.; Zhang, J.H.; Sun, X. Hyperbaric oxygen preconditioning induces tolerance against brain ischemia-reperfusion injury by upregulation of antioxidant enzymes in rats. Brain Res. 2008, 1210, 223–229. [Google Scholar] [CrossRef]
- McDonagh, M.S.; Morgan, D.; Carson, S.; Russman, B.S. Systematic review of hyperbaric oxygen therapy for cerebral palsy: The state of the evidence. Dev. Med. Child Neurol. 2007, 49, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Danesh-Sani, S.A.; Shariati-Sarabi, Z.; Feiz, M.R. Comprehensive review of hyperbaric oxygen therapy. J. Craniofac. Surg. 2012, 23, e483–e491. [Google Scholar] [CrossRef]
- Lim, S.W.; Wang, C.C.; Wang, Y.H.; Chio, C.C.; Niu, K.C.; Kuo, J.R. Microglial activation induced by traumatic brain injury is suppressed by postinjury treatment with hyperbaric oxygen therapy. J. Surg. Res. 2013, 184, 1076–1084. [Google Scholar] [CrossRef]
- Wee, H.Y.; Lim, S.W.; Chio, C.C.; Niu, K.C.; Wang, C.C.; Kuo, J.R. Hyperbaric oxygen effects on neuronal apoptosis associations in a traumatic brain injury rat model. J. Surg. Res. 2015, 197, 382–389. [Google Scholar] [CrossRef]
- Lim, S.W.; Sung, K.C.; Shiue, Y.L.; Wang, C.C.; Chio, C.C.; Kuo, J.R. Hyperbaric oxygen effects on depression-like behavior and neuroinflammation in traumatic brain injury rats. World Neurosurg. 2017, 100, 128–137. [Google Scholar] [CrossRef]
- Vlodavsky, E.; Palzur, E.; Soustiel, J.F. Hyperbaric oxygen therapy reduces neuroinflammation and expression of matrix metalloproteinase-9 in the rat model of traumatic brain injury. Neuropathol. Appl. Neurobiol. 2006, 32, 40–50. [Google Scholar] [CrossRef]
- Harch, P.G.; Andrews, S.R.; Fogarty, E.F.; Amen, D.; Pezzullo, J.C.; Lucarini, J.; Aubrey, C.; Taylor, D.V.; Staab, P.K.; Van Meter, K.W. A phase I study of low-pressure hyperbaric oxygen therapy for blast-induced post-concussion syndrome and post-traumatic stress disorder. J. Neurotrauma 2012, 29, 168–185. [Google Scholar] [CrossRef]
- Borab, Z.; Mirmanesh, M.D.; Gantz, M.; Cusano, A.; Pu, L.L. Systematic review of hyperbaric oxygen therapy for the treatment of radiation-induced skin necrosis. J. Plast. Reconstr. Aesthet. Surg. 2017, 70, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Londahl, M. Hyperbaric oxygen therapy as treatment of diabetic foot ulcers. Diabetes Metab. Res. Rev. 2012, 28 (Suppl. S1), 78–84. [Google Scholar] [CrossRef] [PubMed]
- Londahl, M. Hyperbaric oxygen therapy as adjunctive treatment of diabetic foot ulcers. Med. Clin. N. Am. 2013, 97, 957–980. [Google Scholar] [CrossRef] [PubMed]
- Shuvy, M.; Atar, D.; Gabriel Steg, P.; Halvorsen, S.; Jolly, S.; Yusuf, S.; Lotan, C. Oxygen therapy in acute coronary syndrome: Are the benefits worth the risk? Eur. Heart J. 2013, 34, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.H.; Lehm, J.P.; Jepson, N. Hyperbaric oxygen therapy for acute coronary syndrome. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Bhutani, S.; Vishwanath, G. Hyperbaric oxygen and wound healing. Indian J. Plast. Surg. 2012, 45, 316–324. [Google Scholar] [CrossRef]
- Perez, J.V.; Serrano, A.R.; Andal, M.P.; Aldover, M.C. Delayed hyperbaric intervention in life-threatening decompression illness. Diving Hyperb. Med. 2017, 47, 257–259. [Google Scholar] [CrossRef]
- Harch, P.G.; Fogarty, E.F.; Staab, P.K.; Van Meter, K. Low pressure hyperbaric oxygen therapy and SPECT brain imaging in the treatment of blast-induced chronic traumatic brain injury (post-concussion syndrome) and post traumatic stress disorder: A case report. Cases J. 2009, 2, 6538. [Google Scholar] [CrossRef]
- Xiong, T.; Chen, H.; Luo, R.; Mu, D. Hyperbaric oxygen therapy for people with autism spectrum disorder (ASD). Cochrane Database Syst. Rev. 2016, 10, CD010922. [Google Scholar] [CrossRef]
- Cheng, O.; Ostrowski, R.P.; Wu, B.; Liu, W.; Chen, C.; Zhang, J.H. Cyclooxygenase-2 mediates hyperbaric oxygen preconditioning in the rat model of transient global cerebral Ischemia. Stroke 2011, 42, 484–490. [Google Scholar] [CrossRef]
- Zhang, Y.; Bai, L.; Shi, M.; Lu, H.; Wu, Y.; Tu, J.; Ni, J.; Wang, J.; Cao, L.; Lei, P.; et al. Features and risk factors of carotid atherosclerosis in a population with high stroke incidence in China. Oncotarget 2017, 8, 57477–57488. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Ito, M.; Miyazawa, T.; Katoh, H.; Nawashiro, H.; Shima, K.; Chigasaki, H. Repeated hyperbaric oxygen induces ischemic tolerance in gerbil hippocampus. Brain Res. 1996, 740, 15–20. [Google Scholar] [CrossRef]
- Yamashita, S.; Hirata, T.; Mizukami, Y.; Cui, Y.J.; Fukuda, S.; Ishida, K.; Matsumoto, M.; Sakabe, T. Repeated preconditioning with hyperbaric oxygen induces neuroprotection against forebrain ischemia via suppression of p38 mitogen activated protein kinase. Brain Res. 2009, 1301, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.X.; Rao, J.; Li, Y.H. Hyperbaric oxygen preconditioning improves postoperative cognitive dysfunction by reducing oxidant stress and inflammation. Neural Regen. Res. 2017, 12, 329–336. [Google Scholar] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Xiong, L.; Lao, N.; Chen, S.; Xu, N.; Zhu, Z. Hyperbaric oxygen preconditioning induces tolerance against spinal cord ischemia by upregulation of antioxidant enzymes in rabbits. J. Cereb. Blood Flow Metab. 2006, 26, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Arieli, Y.; Kotler, D.; Eynan, M.; Hochman, A. Hyperbaric oxygen preconditioning protects rats against CNS oxygen toxicity. Respir. Physiol. Neurobiol. 2014, 197, 29–35. [Google Scholar] [CrossRef]
- Ni, X.X.; Ni, M.; Fan, D.F.; Sun, Q.; Kang, Z.M.; Cai, Z.Y.; Liu, Y.; Liu, K.; Li, R.P.; Xu, W.G. Heat-shock protein 70 is involved in hyperbaric oxygen preconditioning on decompression sickness in rats. Exp. Biol. Med. (Maywood) 2013, 238, 12–22. [Google Scholar] [CrossRef]
- Brown, I.R. Heat shock proteins and protection of the nervous system. Ann. N. Y. Acad. Sci. 2007, 1113, 147–158. [Google Scholar] [CrossRef]
- Huang, G.; Diao, J.; Yi, H.; Xu, L.; Xu, J.; Xu, W. Signaling pathways involved in HSP32 induction by hyperbaric oxygen in rat spinal neurons. Redox Biol. 2016, 10, 108–118. [Google Scholar] [CrossRef]
- Vince, R.V.; Oliver, K.; Midgley, A.W.; McNaughton, L.R.; Madden, L.A. In vitro heat shock of human monocytes results in a proportional increase of inducible Hsp70 expression according to the basal content. Amino Acids 2010, 38, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Alfieri, A.; Siow, R.C.; Mann, G.E.; Fraser, P.A. Temporal and spatial distribution of Nrf2 in rat brain following stroke: Quantification of nuclear to cytoplasmic Nrf2 content using a novel immunohistochemical technique. J. Physiol. 2013, 591, 3525–3538. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Huang, G.; Zhang, K.; Sun, J.; Xu, T.; Li, R.; Tao, H.; Xu, W. Nrf2 activation in astrocytes contributes to spinal cord ischemic tolerance induced by hyperbaric oxygen preconditioning. J. Neurotrauma 2014, 31, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Perdrizet, G.A. Preoperative stress conditioning in humans: Is oxygen the drug of choice? Adv. Exp. Med. Biol. 2016, 876, 223–231. [Google Scholar] [PubMed]
- Xue, F.; Huang, J.W.; Ding, P.Y.; Zang, H.G.; Kou, Z.J.; Li, T.; Fan, J.; Peng, Z.W.; Yan, W.J. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning. Behav. Brain Res. 2016, 309, 1–8. [Google Scholar] [CrossRef]
- Zeng, Y.; Xie, K.; Dong, H.; Zhang, H.; Wang, F.; Li, Y.; Xiong, L. Hyperbaric oxygen preconditioning protects cortical neurons against oxygen-glucose deprivation injury: Role of peroxisome proliferator-activated receptor-gamma. Brain Res. 2012, 1452, 140–150. [Google Scholar] [CrossRef]
- Yan, W.; Fang, Z.; Yang, Q.; Dong, H.; Lu, Y.; Lei, C.; Xiong, L. SirT1 mediates hyperbaric oxygen preconditioning-induced ischemic tolerance in rat brain. J. Cereb. Blood Flow Metab. 2013, 33, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Hu, Q.; Liang, X.; Chen, D.; Li, B.; Tang, J.; Zhang, J.H. Hyperbaric oxygen preconditioning attenuates hemorrhagic transformation through increasing PPARgamma in hyperglycemic MCAO rats. Exp. Neurol. 2015, 265, 22–29. [Google Scholar] [CrossRef]
- Goldstein, L.J.; Gallagher, K.A.; Bauer, S.M.; Bauer, R.J.; Baireddy, V.; Liu, Z.J.; Buerk, D.G.; Thom, S.R.; Velazquez, O.C. Endothelial progenitor cell release into circulation is triggered by hyperoxiainduced increases in bone marrow nitric oxide. Stem Cells 2006, 24, 2309–2318. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Sun, X.; Liu, K.; Zhang, J.H.; Xu, W.; Tao, H. Repetitive hyperbaric oxygen exposures enhance sensitivity to convulsion by upregulation of eNOS and nNOS. Brain Res. 2008, 1201, 128–134. [Google Scholar] [CrossRef]
- Chen, C.C.; Hsia, C.W.; Ho, C.W.; Liang, C.M.; Chen, C.M.; Huang, K.L.; Kang, B.H.; Chen, Y.H. Hypoxia and hyperoxia differentially control proliferation of rat neural crest stem cells via distinct regulatory pathways of the HIF1alpha-CXCR4 and TP53-TPM1 proteins. Dev. Dyn. 2017, 246, 162–185. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N.; Liao, J.K. Targeting eNOS and beyond: Emerging heterogeneity of the role of endothelial Rho proteins in stroke protection. Expert Rev. Neurother. 2009, 9, 1171–1186. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, J.; Wullner, U. Inhibition of thioredoxin reductase induces apoptosis in neuronal cell lines: Role of glutathione and the MKK4/JNK pathway. Biochem. Biophys. Res. Commun. 2007, 359, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Feng, S.F.; Wang, Q.; Wang, H.N.; Hou, W.G.; Xiong, L.; Luo, Z.; Tan, Q.R. Hyperbaric oxygen preconditioning ameliorates anxiety-like behavior and cognitive impairments via upregulation of thioredoxin reductases in stressed rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 1018–1025. [Google Scholar] [CrossRef]
- Li, J.S.; Zhang, W.; Kang, Z.M.; Ding, S.J.; Liu, W.W.; Zhang, J.H.; Guan, Y.T.; Sun, X.J. Hyperbaric oxygen preconditioning reduces ischemia-reperfusion injury by inhibition of apoptosis via mitochondrial pathway in rat brain. Neuroscience 2009, 159, 1309–1315. [Google Scholar] [CrossRef]
- Wang, Y.C.; Zhang, S.; Du, T.Y.; Wang, B.; Sun, X.Q. Hyperbaric oxygen preconditioning reduces ischemia- reperfusion injury by stimulating autophagy in neurocyte. Brain Res. 2010, 1323, 149–151. [Google Scholar] [CrossRef]
- Lu, P.G.; Feng, H.; Yuan, S.J.; Zhang, R.W.; Li, M.; Hu, R.; Liu, Z.S.; Yin, J. Effect of preconditioning with hyperbaric oxygen on neural cell apoptosis after spinal cord injury in rats. J. Neurosurg. Sci. 2013, 57, 253–258. [Google Scholar]
- Lu, P.G.; Hu, S.L.; Hu, R.; Wu, N.; Chen, Z.; Meng, H.; Lin, J.K.; Feng, H. Functional recovery in rat spinal cord injury induced by hyperbaric oxygen preconditioning. Neurol. Res. 2012, 34, 944–951. [Google Scholar] [CrossRef]
- Ostrowski, R.P.; Graupner, G.; Titova, E.; Zhang, J.; Chiu, J.; Dach, N.; Corleone, D.; Tang, J.; Zhang, J.H. The hyperbaric oxygen preconditioning-induced brain protection is mediated by a reduction of early apoptosis after transient global cerebral ischemia. Neurobiol. Dis. 2008, 29, 1–13. [Google Scholar] [CrossRef]
- Yang, Y.; Wei, H.; Zhou, X.; Zhang, F.; Wang, C. Hyperbaric oxygen promotes neural stem cell proliferation by activating vascular endothelial growth factor/extracellular signal-regulated kinase signaling after traumatic brain injury. Neuroreport 2017, 28, 1232–1238. [Google Scholar] [CrossRef]
- Gomez, C.R.; Knutson, G.J.; Clifton, K.B.; Schreiber, C.A.; Vuk-Pavlović, S. Age-dependent response of murine female bone marrow cells to hyperbaric oxygen. Biogerontology 2012, 13, 287–297. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, L.; Xie, K.; Zhang, C.; Song, R.; Zhang, H. Hyperbaric oxygen preconditioning attenuates postoperative cognitive impairment in aged rats. Neuroreport 2014, 25, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Song, S.; Xi, G.; Silbergleit, R.; Keep, R.F.; Hoff, J.T.; Hua, Y. Preconditioning with hyperbaric oxygen attenuates brain edema after experimental intracerebral hemorrhage. Neurosurg. Focus 2007, 22, E13. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.N.; Xu, L.; Hu, Q.; Matei, N.; Yang, P.; Tong, L.S.; He, Y.; Guo, Z.; Tang, J.; Yang, Y.; et al. Hyperbaric oxygen preconditioning attenuates hemorrhagic transformation through reactive oxygen species/thioredoxin-interacting protein/nod-like receptor protein 3 pathway in hyperglycemic middle cerebral artery occlusion rats. Crit. Care Med. 2016, 44, e403–e411. [Google Scholar] [CrossRef]
- Jadhav, V.; Ostrowski, R.P.; Tong, W.; Matus, B.; Chang, C.; Zhang, J.H. Hyperbaric oxygen preconditioning reduces postoperative brain edema and improves neurological outcomes after surgical brain injury. Acta Neurochir. Suppl. 2010, 106, 217–220. [Google Scholar]
- Soejima, Y.; Hu, Q.; Krafft, P.R.; Fujii, M.; Tang, J.; Zhang, J.H. Hyperbaric oxygen preconditioning attenuates hyperglycemia-enhanced hemorrhagic transformation by inhibiting matrix metalloproteinases in focal cerebral ischemia in rats. Exp. Neurol. 2013, 247, 737–743. [Google Scholar] [CrossRef]
- Soejima, Y.; Ostrowski, R.P.; Manaenko, A.; Fujii, M.; Tang, J.; Zhang, J.H. Hyperbaric oxygen preconditioning attenuates hyperglycemia enhanced hemorrhagic transformation after transient MCAO in rats. Med. Gas Res. 2012, 2, 9. [Google Scholar] [CrossRef]
- Lin, H.; Chang, C.P.; Lin, H.J.; Lin, M.T.; Tsai, C.C. Attenuating brain edema, hippocampal oxidative stress, and cognitive dysfunction in rats using hyperbaric oxygen preconditioning during simulated high-altitude exposure. J. Trauma Acute Care Surg. 2012, 72, 1220–1227. [Google Scholar] [CrossRef]
- Fang, J.; Li, H.; Li, G.; Wang, L. Effect of hyperbaric oxygen preconditioning on peri-hemorrhagic focal edema and aquaporin-4 expression. Exp. Ther. Med. 2015, 10, 699–704. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Sun, T.; Yu, H.L. Hyperbaric oxygen therapy for the treatment of traumatic brain injury: A meta-analysis. Neurol. Sci. 2016, 37, 693–701. [Google Scholar] [CrossRef]
- Hao, L.; Guo, X.; Zou, C.; Zhou, H.; Tian, H.; Zhang, Y.; Song, C.; Liu, L. Hyperbaric oxygen preconditioning ameliorates blood-brain barrier damage induced by hypoxia through modulation of tight junction proteins in an in vitro model. Croat. Med. J. 2016, 57, 51–57. [Google Scholar] [CrossRef]
- Duan, S.; Shao, G.; Yu, L.; Ren, C. Angiogenesis contributes to the neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Int. J. Neurosci. 2015, 125, 625–634. [Google Scholar] [CrossRef]
- Dong, H.; Xiong, L.; Zhu, Z.; Chen, S.; Hou, L.; Sakabe, T. Preconditioning with hyperbaric oxygen and hyperoxia induces tolerance against spinal cord ischemia in rabbits. Anesthesiology 2002, 96, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Freiberger, J.J.; Suliman, H.B.; Sheng, H.; McAdoo, J.; Piantadosi, C.A.; Warner, D.S. A comparison of hyperbaric oxygen versus hypoxic cerebral preconditioning in neonatal rats. Brain Res. 2006, 1075, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Theodoraki, K.; Tympa, A.; Karmaniolou, I.; Tsaroucha, A.; Arkadopoulos, N.; Smyrniotis, V. Ischemia/reperfusion injury in liver resection: A review of preconditioning methods. Surg. Today 2011, 41, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, K.; Tao, H.; Chen, C.; Zhang, J.H.; Sun, X. Hyperoxia preconditioning: The next frontier in neurology? Neurol. Res. 2012, 34, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Tal, S.; Hadanny, A.; Sasson, E.; Suzin, G.; Efrati, S. Hyperbaric oxygen therapy can induce angiogenesis and regeneration of nerve fibers in traumatic brain injury patients. Front. Hum. Neurosci. 2017, 11, 508. [Google Scholar] [CrossRef]
- Losada, D.M.; Jordani, M.E.; Jordani, M.C.; Piccinato, M.A.; Fina, C.F.; Feres, O.; Chies, A.B.; Evora, P.R.; e Silva, O.D. Should preconditioning hyperbaric oxygenation protect the liver against ischemia-reperfusion injury? An experimental study in a rat model. Transplant. Proc. 2014, 46, 56–62. [Google Scholar] [CrossRef]
- Yokobori, S.; Mazzeo, A.T.; Hosein, K.; Gajavelli, S.; Dietrich, W.D.; Bullock, M.R. Preconditioning for traumatic brain injury. Transl. Stroke Res. 2013, 4, 25–39. [Google Scholar] [CrossRef]
- Sullivan, R.; Duncan, K.; Dailey, T.; Kaneko, Y.; Tajiri, N.; Borlongan, C.V. A possible new focus for stroke treatment—Migrating stem cells. Expert Opin. Biol. Ther. 2015, 15, 949–958. [Google Scholar] [CrossRef]
- Thom, S.R.; Bhopale, V.M.; Velazquez, O.C.; Goldstein, L.J.; Thom, L.H.; Buerk, D.G. Stem cell mobilization by hyperbaric oxygen. Am. J. Physiol. Hear Circ. Physiol. 2006, 290, H1378–H1386. [Google Scholar] [CrossRef]
- Dhar, M.; Neilsen, N.; Beatty, K.; Eaker, S.; Adair, H.; Geiser, D. Equine peripheral blood-derived mesenchymal stem cells: Isolation, identification, trilineage differentiation and effect of hyperbaric oxygen treatment. Equine Vet. J. 2012, 44, 600–605. [Google Scholar] [CrossRef]
- Yang, Y.J.; Wang, X.L.; Yu, X.H.; Wang, X.; Xie, M.; Liu, C.T. Hyperbaric oxygen induces endogenous neural stem cells to proliferate and differentiate in hypoxic-ischemic brain damage in neonatal rats. Undersea Hyperb. Med. 2008, 35, 13–29. [Google Scholar]
- Wei, L.; Wang, J.; Cao, Y.; Ren, Q.; Zhao, L.; Li, X.; Wang, J. Hyperbaric oxygenation promotes neural stem cell proliferation and protects the learning and memory ability in neonatal hypoxic-ischemic brain damage. Int. J. Clin. Exp. Pathol. 2015, 8, 1752–1759. [Google Scholar] [PubMed]
- Heyboer, M., III; Milovanova, T.N.; Wojcik, S.; Grant, W.; Chin, M.; Hardy, K.R.; Lambert, D.S.; Logue, C.; Thom, S.R. CD34+/CD45-dim stem cell mobilization by hyperbaric oxygen—Changes with oxygen dosage. Stem Cell Res. 2014, 12, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Thom, S.R.; Milovanova, T.N.; Yang, M.; Bhopale, V.M.; Sorokina, E.M.; Uzun, G.; Malay, D.S.; Troiano, M.A.; Hardy, K.R.; Lambert, D.S.; et al. Vasculogenic stem cell mobilization and wound recruitment in diabetic patients: Increased cell number and intracellular regulatory protein content associated with hyperbaric oxygen therapy. Wound Repair Regen. 2011, 19, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Krafft, P.R.; Zhang, J.H. Hyperbaric oxygen therapy promotes neurogenesis: Where do we stand? Med. Gas Res. 2011, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, J.; Adler, I.; Manaenko, A.; Schwarz, S.C.; Walkinshaw, G.; Arend, M.; Flippin, L.A.; Storch, A.; Schwarz, J. Non-hypoxic stabilization of hypoxia-inducible factor alpha (HIF-alpha): Relevance in neural progenitor/stem cells. Neurotox Res. 2009, 15, 367–380. [Google Scholar] [CrossRef]
- Qi, C.; Zhang, J.; Chen, X.; Wan, J.; Wang, J.; Zhang, P.; Liu, Y. Hypoxia stimulates neural stem cell proliferation by increasing HIF-1α expression and activating Wnt/β-catenin signaling. Cell. Mol. Biol. (Noisy-le-Grand) 2017, 63, 12–19. [Google Scholar] [CrossRef]
- Belle, J.E.L.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2012, 8, 59–71. [Google Scholar] [CrossRef]
- Lu, K.T.; Sun, C.L.; Wo, P.Y.Y.; Yen, H.H.; Tang, T.H.; Ng, M.C.; Huang, M.L.; Yang, Y.L. Hippocampal neurogenesis after traumatic brain injury is mediated by vascular endothelial growth factor receptor-2 and the Raf/MEK/ERK cascade. J. Neurotrauma 2011, 28, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.J.; Li, Y.P.; Peng, Z.Y.; Xu, J.J.; Kang, Z.M.; Xu, W.G.; Tao, H.Y.; Ostrowski, R.P.; Zhang, J.H.; Sun, X.J. Mechanism of ischemic tolerance induced by hyperbaric oxygen preconditioning involves upregulation of hypoxia-inducible factor-1alpha and erythropoietin in rats. J. Appl. Physiol. 2008, 104, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Ren, P.; Kang, Z.; Du, J.; Lian, Q.; Liu, Y.; Zhang, J.H.; Sun, X. Up-regulated HIF-1alpha is involved in the hypoxic tolerance induced by hyperbaric oxygen preconditioning. Brain Res. 2008, 1212, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Aljitawi, O.S.; Paul, S.; Ganguly, A.; Lin, T.L.; Ganguly, S.; Vielhauer, G.; Capitano, M.L.; Cantilena, A.; Lipe, B.; Mahnken, J.D.; et al. Erythropoietin modulation is associated with improved homing and engraftment after umbilical cord blood transplantation. Blood 2016, 128, 3000–3010. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X.; Liu, Z.G.; Liu, X.J.; Chen, Q.X. Umbilical cord-derived mesenchymal stem cell transplantation combined with hyperbaric oxygen treatment for repair of traumatic brain injury. Neural Regen. Res. 2016, 11, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.K.; Cao, H.H.; Ying, X.; Yu, H.L. Effect of mesenchymal stem cells transplantation combining with hyperbaric oxygen therapy on rehabilitation of rat spinal cord injury. Asian Pac. J. Trop. Med. 2015, 8, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E.J.; Valacchi, F.; Nicora, E.; Brieva, S.; Esteve, C.; Echevarria, L.; Froud, T.; Bernetti, K.; Cayetano, S.M.; Velazquez, O.; et al. Combined treatment of intrapancreatic autologous bone marrow stem cells and hyperbaric oxygen in type 2 diabetes mellitus. Cell Transplant. 2008, 17, 1295–1304. [Google Scholar] [CrossRef]
- Aljitawi, O.S.; Xiao, Y.; Eskew, J.D.; Parelkar, N.K.; Swink, M.; Radel, J.; Lin, T.L.; Kimler, B.F.; Mahnken, J.D.; McGuirk, J.P.; et al. Hyperbaric oxygen improves engraftment of ex-vivo expanded and gene transduced human CD34(+) cells in a murine model of umbilical cord blood transplantation. Blood Cells Mol. Dis. 2014, 52, 59–67. [Google Scholar] [CrossRef]
- Pan, H.C.; Chin, C.S.; Yang, D.Y.; Ho, S.P.; Chen, C.J.; Hwang, S.M.; Chang, M.H.; Cheng, F.C. Human amniotic fluid mesenchymal stem cells in combination with hyperbaric oxygen augment peripheral nerve regeneration. Neurochem. Res. 2009, 34, 1304–1316. [Google Scholar] [CrossRef]
- Schulze, J.; Kaiser, O.; Paasche, G.; Lamm, H.; Pich, A.; Hoffmann, A.; Lenarz, T.; Warnecke, A. Effect of hyperbaric oxygen on BDNF-release and neuroprotection: Investigations with human mesenchymal stem cells and genetically modified NIH3T3 fibroblasts as putative cell therapeutics. PLoS ONE 2017, 12, e0178182. [Google Scholar] [CrossRef]
- Thom, S.R. Oxidative stress is fundamental to hyperbaric oxygen therapy. J. Appl. Physiol. 2009, 106, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.Y.; Berry, A.; Li, D.; Aljitawi, O.S. Hyperbaric oxygen treatment effects on in vitro cultured umbilical cord blood CD34(+) cells. Cytotherapy 2018, 20, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Wakai, T.; Narasimhan, P.; Sakata, H.; Wang, E.; Yoshioka, H.; Kinouchi, H.; Chan, P.H. Hypoxic preconditioning enhances neural stem cell transplantation therapy after intracerebral hemorrhage in mice. J. Cereb. Blood Flow Metab. 2016, 36, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wei, L.; Taylor, T.M.; Wei, J.; Zhou, X.; Wang, J.A.; Yu, S.P. Hypoxic preconditioning enhances bone marrow mesenchymal stem cell migration via Kv2.1 channel and FAK activation. Am. J. Physiol. Cell Physiol. 2011, 301, C362–C372. [Google Scholar] [CrossRef]
- Gross, R.; Golan, H.; Fishlev, G.; Bechor, Y.; Volkov, O.; Bergan, J.; Friedman, M.; Hoofien, D.; Shlamkovitch, N.; Ben-Jacob, E.; et al. Hyperbaric oxygen therapy can improve post concussion syndrome years after mild traumatic brain injury—Randomized prospective trial. PLoS ONE 2013, 8, e79995. [Google Scholar]
- Lee, Y.S.; Chio, C.C.; Chang, C.P.; Wang, L.C.; Chiang, P.M.; Niu, K.C.; Tsai, K.J. Long course hyperbaric oxygen stimulates neurogenesis and attenuates inflammation after ischemic stroke. Mediat. Inflamm. 2013, 2013, 512978. [Google Scholar] [CrossRef]
- Wei, N.; Yu, S.P.; Gu, X.; Taylor, T.M.; Song, D.; Liu, X.F.; Wei, L. Delayed intranasal delivery of hypoxic- preconditioned bone marrow mesenchymal stem cells enhanced cell homing and therapeutic benefits after ischemic stroke in mice. Cell Transplant. 2013, 22, 977–991. [Google Scholar] [CrossRef]
- Wei, L.; Fraser, J.L.; Lu, Z.Y.; Hu, X.; Yu, S.P. Transplantation of hypoxia preconditioned bone marrow mesenchymal stem cells enhances angiogenesis and neurogenesis after cerebral ischemia in rats. Neurobiol. Dis. 2012, 46, 635–645. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Venetsanou, K.; Fildissis, G.; Tokta, R.; Brinias, C.; Baltopoulos, G. The role of nitric oxide in cellular response to hyperbaric conditions. Eur. J. Appl. Physiol. 2012, 112, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Hung, H.F.; Wang, B.W.; Chang, H. Hyperbaric oxygen induces placental growth factor expression in bone marrow-derived mesenchymal stem cells. Life Sci. 2008, 83, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, V.; Ostrowski, R.P.; Tong, W.; Matus, B.; Jesunathadas, R.; Zhang, J.H. Cyclo-xygoenase-2 mediates hyperbaric oxygen preconditioning-induced neuroprotection in the mouse model of surgical brain injury. Stroke 2009, 40, 3139–3142. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Kang, J.; Bai, Y.; Zhang, Y.; Li, H.; Yang, X.; Xiang, X.; Wang, X.; Huang, Y.; Su, J.; et al. Hyperbaric oxygen enlarges the area of brain damage in MCAO rats by blocking autophagy via ERK1/2 activation. Eur. J. Pharmacol. 2014, 728, 93–99. [Google Scholar] [CrossRef]
- He, H.; Li, X.; He, Y. Hyperbaric oxygen therapy attenuates neuronal apoptosis induced by traumatic brain injury via Akt/GSK3β/β-catenin pathway. Neuropsychiatr. Dis. Treat. 2019, 15, 369–374. [Google Scholar] [CrossRef]
- Ying, X.; Tu, W.; Li, S.; Wu, Q.; Chen, X.; Zhou, Y.; Hu, J.; Yang, G.; Jiang, S. Hyperbaric oxygen therapy reduces apoptosis and dendritic/synaptic degeneration via the BDNF/TrkB signaling pathways in SCI rats. Life Sci. 2019, 229, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dong, Q.; Pan, Z.; Song, Y.; Su, P.; Niu, Y.; Sun, Y.; Liu, D. Hyperbaric oxygen improves functional recovery of the injured spinal cord by inhibiting inflammation and glial scar formation. Am. J. Phys. Med. Rehabil. 2019, 98, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Cechin, S.; Alvarez-Cubela, S.; Giraldo, J.A.; Molano, R.D.; Villate, S.; Ricordi, C.; Pileggi, A.; Inverardi, L.; Fraker, C.A.; Dominguez-Bendala, J. Influence of in vitro and in vivo oxygen modulation on beta cell differentiation from human embryonic stem cells. Stem Cells Transl. Med. 2014, 3, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.T.; Fan, G.Y.; Liou, N.H.; Wang, Y.W.; Fu, K.Y.; Ma, K.H.; Liu, J.C.; Chang, S.C.; Huang, K.L.; Dai, L.G.; et al. Histochemical and functional improvement of adipose-derived stem cell-based tissue-engineered cartilage by hyperbaric oxygen/air treatment in a rabbit articular defect model. Ann. Plast. Surg. 2015, 74 (Suppl. S2), S139–S145. [Google Scholar] [CrossRef]
- Lippert, T.; Borlongan, C.V. Prophylactic treatment of hyperbaric oxygen treatment mitigates inflammatory response via mitochondria transfer. CNS Neurosci. Ther. 2019, 25, 815–823. [Google Scholar] [CrossRef]
- Lu, K.; Wang, H.; Ge, X.; Liu, Q.; Chen, M.; Shen, Y.; Liu, X.; Pan, S. Hyperbaric oxygen protects against cerebral damage in permanent middle cerebral artery occlusion rats and inhibits autophagy activity. Neurocrit. Care 2019, 30, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cheng, L.; Chen, Z.L.; Mungur, R.; Xu, S.H.; Wu, J.; Liu, X.L.; Wan, S. Hyperbaric oxygen preconditioning attenuates brain injury after intracerebral hemorrhage by regulating microglia polarization in rats. CNS Neurosci. Ther. 2019, 25, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.C.; Chen, K.H.; Wallace, C.G.; Chen, Y.L.; Ko, S.F.; Lee, M.S.; Yip, H.K. Combined therapy with hyperbaric oxygen and melatonin effectively reduce brain infarct volume and preserve neurological function after acute ischemic infarct in rat. J. Neuropathol. Exp. Neurol. 2019, 78, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Hadanny, A.; Rittblat, M.; Bitterman, M.; May-Raz, I.; Suzin, G.; Boussi-Gross, R.; Zemel, Y.; Bechor, Y.; Catalogna, M.; Efrati, S. Hyperbaric oxygen therapy improves neurocognitive functions of post-stroke patients—A retrospective analysis. Restor. Neurol. Neurosci. 2020, 38, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, S.; Richardson, D.; Santa Mina, D.; Buryk-Iggers, S.; Uehling, J.; Carroll, J.; Clarke, H.; Djaiani, C.; Gershinsky, M.; Katznelson, R. Hyperbaric oxygen and focused rehabilitation program: A feasibility study in improving upper limb motor function after stroke. Appl. Physiol. Nutr. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.X.; Hao, Y.G.; Duan, X.M.; Han, X.L.; Cai, X.X. Hyperbaric oxygen therapy for post-stroke depression: A systematic review and meta-analysis. Clin. Neurol. Neurosurg. 2020, 195, 105910. [Google Scholar] [CrossRef]
- Murphy, R.P.; Donnellan, J. A high-pressure solution for a high-pressure situation: Management of cerebral air embolism with hyperbaric oxygen therapy. Cureus 2019, 11, e5559. [Google Scholar] [CrossRef]
- Sankaran, R.; Radhakrishnan, K.; Sundaram, K.R. Hyperbaric oxygen therapy in patients with hypoxic ischemic encephalopathy. Neurol. India 2019, 67, 728–731. [Google Scholar]
- Golan, H.; Makogon, B.; Volkov, O.; Smolyakov, Y.; Hadanny, A.; Efrati, S. Imaging-based predictors for hyperbaric oxygen therapy outcome in post-stroke patients. Report 1. Med. Hypotheses 2020, 136, 109510. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Wang, Z.; Chen, Y.; Li, R. Intermittent hyperbaric oxygen exposure mobilizing peroxiredoxin 6 to prevent oxygen toxicity. J. Physiol. Sci. 2019, 69, 779–790. [Google Scholar] [CrossRef]
- Guzik, T.J.; Korbut, R.; Adamek-Guzik, T. Nitric oxide and superoxide in inflammation and immune regulation. J. Physiol. Pharmacol. 2003, 54, 469–487. [Google Scholar] [PubMed]
- Go, A.S. Executive summary: Heart disease and stroke statistics—2014 update: A report from the American Heart Association. Circulation 2014, 129, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Knecht, T.; Story, J.; Liu, J.; Davis, W.; Borlongan, C.V.; Pena, I.C.D. Adjunctive therapy approaches for ischemic stroke: Innovations to expand time window of treatment. Int. J. Mol. Sci. 2017, 18, 2756. [Google Scholar] [CrossRef] [PubMed]
- Stonesifer, C.; Corey, S.; Ghanekar, S.; Diamandis, Z.; Acosta, S.A.; Borlongan, C.V. Stem cell therapy for abrogating stroke-induced neuroinflammation and relevant secondary cell death mechanisms. Prog. Neurobiol. 2017, 158, 94–131. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef]
- Wetterling, F.; Chatzikonstantinou, E.; Tritschler, L.; Meairs, S.; Fatar, M.; Schad, L.R.; Ansar, S. Investigating potentially salvageable penumbra tissue in an in vivo model of transient ischemic stroke using sodium, diffusion, and perfusion magnetic resonance imaging. BMC Neurosci. 2016, 17, 82. [Google Scholar] [CrossRef]
- Ostrowski, R.P.; Stepien, K.; Pucko, E.; Matyja, E. The efficacy of hyperbaric oxygen in hemorrhagic stroke: Experimental and clinical implications. Arch. Med. Sci. 2017, 13, 1217–1223. [Google Scholar] [CrossRef]
- Ostrowski, R.P.; Tang, J.; Zhang, J.H. Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke 2006, 37, 1314–1318. [Google Scholar] [CrossRef]
- Cimino, F.; Balestra, C.; Germonpré, P.; De Bels, D.; Tillmans, F.; Saija, A.; Speciale, A.; Virgili, F. Pulsed high oxygen induces a hypoxic-like response in human umbilical endothelial cells and in humans. J. Appl. Physiol. 2012, 113, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yosef, Y.; Miller, A.; Shapiro, S.; Lahat, N. Hypoxia of endothelial cells leads to MMP-2-dependent survival and death. Am. J. Physiol. Cell Physiol. 2005, 289, C1321–C1331. [Google Scholar] [CrossRef] [PubMed]
- Baratz-Goldstein, R.; Toussia-Cohen, S.; Elpaz, A.; Rubovitch, V.; Pick, C.G. Immediate and delayed hyperbaric oxygen therapy as a neuroprotective treatment for traumatic brain injury in mice. Mol. Cell. Neurosci. 2017, 83, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Manaenko, A.; Bian, H.; Guo, Z.; Huang, J.L.; Guo, Z.N.; Yang, P.; Tang, J.; Zhang, J.H. Hyperbaric oxygen reduces infarction volume and hemorrhagic transformation through ATP/NAD(+)/sirt1 pathway in hyperglycemic middle cerebral artery occlusion rats. Stroke 2017, 48, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Niu, K.C.; Hoffer, B.J.; Wang, Y.; Borlongan, C.V. Hyperbaric oxygen therapy for treatment of postischemic stroke in adult rats. Exp. Neurol. 2000, 166, 298–306. [Google Scholar] [CrossRef]
- Singhal, A.B.; Benner, T.; Roccatagliata, L.; Koroshetz, W.J.; Schaefer, P.W.; Lo, E.H.; Buonanno, F.S.; Gonzalez, R.G.; Sorensen, A.G. A pilot study of normobaric oxygen therapy in acute ischemic stroke. Stroke 2005, 36, 797–802. [Google Scholar] [CrossRef]
- Rink, C.; Roy, S.; Khan, M.; Ananth, P.; Kuppusamy, P.; Sen, C.K.; Khanna, S. Oxygen-sensitive outcomes and gene expression in acute ischemic stroke. J. Cereb. Blood Flow Metab. 2010, 30, 1275–1287. [Google Scholar] [CrossRef]
- Matchett, G.A.; Martin, R.D.; Zhang, J.H. Hyperbaric oxygen therapy and cerebral ischemia: Neuroprotective mechanisms. Neurol. Res. 2009, 31, 114–121. [Google Scholar] [CrossRef]
- Nemoto, E.M.; Betterman, K. Neuroprotective effects of hyperbaric oxygen treatment on traumatic brain injury in the rat. Neurol. Res. 2007, 29, 116–126. [Google Scholar] [CrossRef]
- Veltkamp, R.; Siebing, D.A.; Sun, L.; Heiland, S.; Bieber, K.; Marti, H.H.; Nagel, S.; Schwab, S.; Schwaninger, M. Hyperbaric oxygen reduces blood-brain barrier damage and edema after transient focal cerebral ischemia. Stroke 2005, 36, 1679–1683. [Google Scholar] [CrossRef]
- Ploughman, M.; Austin, M.W.; Glynn, L.; Corbett, D. The effects of poststroke aerobic exercise on neuroplasticity: A systematic review of animal and clinical studies. Transl. Stroke Res. 2015, 6, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Hadanny, A.; Golan, H.; Fishlev, G.; Bechor, Y.; Volkov, O.; Suzin, G.; Ben-Jacob, E.; Efrati, S. Hyperbaric oxygen can induce neuroplasticity and improve cognitive functions of patients suffering from anoxic brain damage. Restor. Neurol. Neurosci. 2015, 33, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Tong, W.C.; Lu, X.X.; Peng, H.P. Hyperbaric oxygen therapy in acute ischemic stroke: A review. Interv. Neurol. 2014, 2, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Padma, M.V.; Bhasin, A.; Bhatia, R.; Garg, A.; Singh, M.B.; Tripathi, M.; Prasad, K. Normobaric oxygen therapy in acute ischemic stroke: A pilot study in Indian patients. Ann. Indian Acad. Neurol. 2010, 13, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Qi, Z.; Lou, Y.; Liu, K.J. Normobaric oxygen treatment in acute ischemic stroke: A clinical perspective. Med. Gas Res. 2016, 6, 147–153. [Google Scholar] [PubMed]
- Donati, A.; Damiani, E.; Zuccari, S.; Domizi, R.; Scorcella, C.; Girardis, M.; Giulietti, A.; Vignini, A.; Adrario, E.; Romano, R.; et al. Effects of short-term hyperoxia on erythropoietin levels and microcirculation in critically Ill patients: A prospective observational pilot study. BMC Anesthesiol. 2017, 17, 49. [Google Scholar] [CrossRef]
- Khalifé, M.; Wiams, K.; Ben Aziz, M.; Balestra, C.; Sosnowski, M. Effect of induced relative hypoxia on reticulocyte count in oncological abdominal surgery: A single–centre, controlled, randomized pilot study. Int. J. Sci. Res. 2018, 7, 12–16. [Google Scholar]
- Muralidharan, G.; Rao, G.H.R. Oxygen as a therapeutic drug: Hyperbaric oxygen therapy. Biomed. Pharmacol. J. 2020, 13, 521–528. [Google Scholar] [CrossRef]
- Singhal, A.B.A. Review of oxygen therapy in ischemic stroke. Neurol. Res. 2007, 29, 173–183. [Google Scholar] [CrossRef]
- Zhai, W.W.; Sun, L.; Yu, Z.Q.; Chen, G. Hyperbaric oxygen therapy in experimental and clinical stroke. Med. Gas Res. 2016, 6, 111–118. [Google Scholar]
- Godman, C.A.; Chheda, K.P.; Hightower, L.E.; Perdrizet, G.; Shin, D.G.; Giardina, C. Hyperbaric oxygen induces a cytoprotective and angiogenic response in human microvascular endothelial cells. Cell Stress Chaperones 2010, 15, 431–442. [Google Scholar] [CrossRef]
- Ostrowski, R.P.; Stepien, K.; Pucko, E.; Matyja, E. Hyperbaric oxygen modalities are differentially effective in distinct brain ischemia models. Med. Gas Res. 2016, 6, 39–47. [Google Scholar] [CrossRef]
- Wang, X.L.; Zhao, Y.S.; Yang, Y.J.; Xie, M.; Yu, X.H. Therapeutic window of hyperbaric oxygen therapy for hypoxic-ischemic brain damage in newborn rats. Brain Res. 2008, 1222, 87–94. [Google Scholar] [CrossRef]
- Yin, D.; Zhou, C.; Kusaka, I.; Calvert, J.W.; Parent, A.D.; Nanda, A.; Zhang, J.H. Inhibition of apoptosis by hyperbaric oxygen in a rat focal cerebral ischemic model. J. Cereb. Blood Flow Metab. 2003, 23, 855–864. [Google Scholar] [CrossRef]
- Lou, M.; Eschenfelder, C.C.; Herdegen, T.; Brecht, S.; Deuschl, G. Therapeutic window for use of hyperbaric oxygenation in focal transient ischemia in rats. Stroke 2004, 35, 578–583. [Google Scholar] [CrossRef]
- Yin, D.; Zhang, J.H. Delayed and multiple hyperbaric oxygen treatments expand therapeutic window in rat focal cerebral ischemic model. Neurocrit. Care 2005, 2, 206–211. [Google Scholar] [CrossRef]
- Bennett, M.H.; Weibel, S.; Wasiak, J.; Schnabel, A.; French, C.; Kranke, P. Hyperbaric oxygen therapy for acute ischaemic stroke. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Sanchez, E.C. Mechanisms of action of hyperbaric oxygenation in stroke: A review. Crit. Care Nurs. Q. 2013, 36, 290–298. [Google Scholar] [CrossRef]
- Li, Y.; Dong, H.; Chen, M.; Liu, J.; Yang, L.; Chen, S.; Xiong, L. Preconditioning with repeated hyperbaric oxygen induces myocardial and cerebral protection in patients undergoing coronary artery bypass graft surgery: A prospective, randomized, controlled clinical trial. J. Cardiothorac. Vasc. Anesth. 2011, 25, 908–916. [Google Scholar] [CrossRef]
- Hu, S.L.; Feng, H.; Xi, G.H. Hyperbaric oxygen therapy and preconditioning for ischemic and hemorrhagic stroke. Med. Gas Res. 2016, 6, 232–236. [Google Scholar] [CrossRef]
- Hu, Q.; Liang, X.; Chen, D.; Chen, Y.; Doycheva, D.; Tang, J.; Tang, J.; Zhang, J.H. Delayed hyperbaric oxygen therapy promotes neurogenesis through reactive oxygen species/hypoxia-inducible factor-1alpha/beta-catenin pathway in middle cerebral artery occlusion rats. Stroke 2014, 45, 1807–1814. [Google Scholar] [CrossRef]
- Gao-Yu, C.; Cong-Yina, D.; Li-Jun, Z.; Fei, L.; Hua, F. Effects of hyperbaric oxygen preconditioning on energy metabolism and glutamate level in the peri-infarct area following permanent MCAO. Undersea Hyperb. Med. 2011, 38, 91–99. [Google Scholar]
- Li, Z.; Liu, W.; Kang, Z.; Lv, S.; Han, C.; Yun, L.; Sun, X.; Zhang, J.H. Mechanism of hyperbaric oxygen preconditioning in neonatal hypoxia-ischemia rat model. Brain Res. 2008, 1196, 151–156. [Google Scholar] [CrossRef]
- Yang, Z.J.; Xie, Y.; Bosco, G.M.; Chen, C.; Camporesi, E.M. Hyperbaric oxygenation alleviates MCAO- induced brain injury and reduces hydroxyl radical formation and glutamate release. Eur. J. Appl. Physiol. 2010, 108, 513–522. [Google Scholar] [CrossRef]
- Zhou, J.G.; Fang, Y.Q.; Liu, C.Y.; Zhou, Y.Q.; Ji, Y.F.; Liu, J.C. Effect of hyperbaric oxygen on the expression of nitric oxide synthase mRNA in cortex after acute traumatic cerebral injury. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2012, 28, 38–41. [Google Scholar]
- Yu, M.; Xue, Y.; Liang, W.; Zhang, Y.; Zhang, Z. Protection mechanism of early hyperbaric oxygen therapy in rats with permanent cerebral ischemia. J. Phys. Ther. Sci. 2015, 27, 3271–3274. [Google Scholar] [CrossRef][Green Version]
- Lavrnja, I.; Parabucki, A.; Brkic, P.; Jovanovic, T.; Dacic, S.; Savic, D.; Pantic, I.; Stojiljkovic, M.; Pekovic, S. Repetitive hyperbaric oxygenation attenuates reactive astrogliosis and suppresses expression of inflammatory mediators in the rat model of brain injury. Mediat. Inflamm. 2015, 2015, 498405. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study | Discovery |
|---|---|
| Jadhav et al., 2009 | In surgical brain injury (SBI) mice, HBOT preconditioning ameliorated neurological function and cerebral edema; these neuroprotective effects seemed to be regulated by COX-2 mechanisms, as HBOT attenuated SBI-induced elevation of hypoxia-inducible factor-1alpha and COX-2 activity [170]. |
| Mu et al., 2013 | In permanent MCAO animal models, daily HBOT conditioning at 48 h post-surgery diminished infarct volume and improved neurological function, which correlated with elevated CREB protein expression in the hippocampus and peri-infarct area, boosting cell multiplication. Regarding acute pMCAo models, HBOT increased cerebral PP1-γ expression, alleviating CREB phosphorylation and ubiquitination spurred by ischemia. Moreover, HBOT’s regenerative effects against ischemic stroke can be associated with CREB and PP1-γ mechanisms [37]. |
| Lu et al., 2014 | In transient MCAO rat models, HBOT spurred an increase in ERK1/2 signaling due to higher levels of ROS, leading to the attenuation of autophagy. When U0126, an inhibitor of the ERK1/2 pathway, was applied, infarct size and autophagy were ameliorated [171]. |
| Xue et al., 2016 | MCAO rats subjected to HBOT preconditioning exhibited diminished infarct size, improved neurological behavior, and upregulated Sirt1, Nrf2, HO-1, and SOD1 expression, as well as reduction of MDA. Blocking of Sirt1 or Nrf2 abolished HBOT-induced protective effects, as Nrf2, HO-1, and SOD1 were repressed. Moreover, the protective actions of Sirt1, spurred by HBOT, may consist of the Nrf2/antioxidant defense mechanism [172]. |
| Guo et al., 2016 | Following successive HBOT pre-treatment over five days, rats underwent hyperglycemic MCAO. Preconditioning with HBOT significantly ameliorated hemorrhagic transformation induced by the Nod-like receptor protein 3 signaling and reduced infarct size, altogether rehabilitating neurological performance. HBOT’s neuroprotective effects could be linked to the ROS/thioredoxin-interacting protein/Nod-like receptor protein 3 mechanism [126]. |
| Yang et al., 2017 | HBOT ameliorated neurological impairment in TBI rats via upregulation of VEGF, VEGFR2, Raf-1, MEK1/2, and ERK1/2, stimulating proliferation of neural stem cells (NSC) and homing of these cells to the lesion site. The examination of HBOT’s protective effects in vitro showed similar results, as HBO drastically amplified NSC proliferation and VEGF/ERK signaling [123]. |
| Hu et al., 2017 | In hyperglycemia MCAO rats, exposure to two atmospheres of HBO for an hour immediately after dextrose administration ameliorated depleted ATP and nitcotinamide adenine dinucleotide levels, which in turn elevated silent mating type information regulation 2 homolog 1, alleviating cerebral infarct and neurological dysfunction, along with repressing hemorrhagic transformation [14]. |
| He et al., 2019 | Mice models of acute TBI demonstrated escalated levels of apoptotic neurons and caspase-3 activity, along with attenuation of signaling pathways that regulate apoptosis in neurons (e.g., pAkt/Akt, pGSK3β/GSK3β, and β-catenin). By eliminating the TBI-induced alterations in these pathways, HBOT suppressed neuronal apoptosis [173]. |
| Ying et al., 2019 | BDNF/TrkB signaling has been shown to influence rehabilitation after SCI. In vivo, SCI rat models were exposed to HBOT, and both dendritic/synaptic deterioration and apoptosis were ameliorated, which could be linked to higher levels of BDNF and TrkB activity. When ANA-12, an inhibitor of the BDNF/TrkB pathway, was administered, HBOT’s neuroprotective effects were reversed, indicating that HBOT’s therapeutic benefits are mediated by BDNF/TrkB signaling [174]. |
| Zhou et al., 2019 | Following HBOT, Sprague-Dawley rats with spinal cord injury (SCI) displayed ameliorated motor function and attenuated secondary injuries, such as inflammation and glial scar production. By blocking AKT and NF-kB signaling, HBOT repressed molecules associated with inflammation (iNOS and COX-2) and glial scar generation (GFAP and NG2) [175]. |
| Study | Discovery |
|---|---|
| Yang et al., 2008 | Rats were subject to unilateral carotid artery ligation and then 2 h of hypoxia. HBO2 was then administered following the hypoxic-ischemic event. The HBOT was found to upregulate neural stem cell proliferation in neurogenic environments within the adult brain [143]. |
| Li et al., 2008 | A murine model subjected rats to common carotid artery ligation and hypoxia for 90 min. HBOT was administered 24 h prior to the hypoxic-ischemic injury. Results revealed that rats preconditioned with HBOT had an increased survival rate, and the infarct ratio was decreased. This indicates that HBOT can provide brain protection via the inhibition of neuronal apoptosis pathways [45]. |
| Li et al., 2009 | HBOT preconditioned rats where investigated to determine if apoptotic inhibition through a mitochondrial pathway was correlated with neuroprotection in the ischemic injury in the rat brain. Preconditioning was conducted four times, followed by brain evaluation. Results indicated that HBO-PC significantly reduced brain edema and decreased infarction volume and improved neurological recovery [117]. |
| Rink et al., 2010 | Transient MCAO rodents outline the therapeutic potential of normobaric and hyperbaric oxygen treatments during ischemia and after ischemia. HBOT-treated rodents revealed inhibited leukocyte accumulation in the ischemic area due to a reduction in levels of inflammatory chemokines [50]. |
| Cechin et al., 2014 | This study allowed pancreatic progenitor cells to mature in a perfluorocarbon-based culture device that could adjust the levels of pO2. Enhanced O2 exposure in vitro led to maturation and differentiation of human embryonic stem cell-derived pancreatic progenitor cells [176]. |
| Hadanny et al., 2015 | Patients with cardiac arrest-induced chronic cognitive impairments where treated with sessions of HBOT and analyzed. After administering HBOT five days per week to chronic stroke patients, patients had significant improvements in memory and attention testing [22]. |
| Dai et al., 2015 | A rabbit model seeded human adipose-derived stem cells on a gelatin/polycaprolactone scaffold to determine the functional and histochemical improvement of tissue-engineered cartilage after HBOT. The human adipose-derived stem cells were found to have improved extracellular matrix-secreting abilities after transplantation into a rabbit cartilage defect model when primed with HBOT [177]. |
| Yang et al., 2017 | This study investigated the mechanism of HBOT that promote NSC proliferation and recovery following TBI. The study used 24 rats split into a sham group, a TBI group, and an HBO treated TBI group to determine the neurological differences. Neurological function was evaluated and monitored throughout the week. HBOT was found to promote neural stem cell migration to areas of injury within the brain in rat models of TBI that were preconditioned with HBO [123]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozene, B.; Sadanandan, N.; Gonzales-Portillo, B.; Saft, M.; Cho, J.; Park, Y.J.; Borlongan, C.V. An Extra Breath of Fresh Air: Hyperbaric Oxygenation as a Stroke Therapeutic. Biomolecules 2020, 10, 1279. https://doi.org/10.3390/biom10091279
Cozene B, Sadanandan N, Gonzales-Portillo B, Saft M, Cho J, Park YJ, Borlongan CV. An Extra Breath of Fresh Air: Hyperbaric Oxygenation as a Stroke Therapeutic. Biomolecules. 2020; 10(9):1279. https://doi.org/10.3390/biom10091279
Chicago/Turabian StyleCozene, Blaise, Nadia Sadanandan, Bella Gonzales-Portillo, Madeline Saft, Justin Cho, You Jeong Park, and Cesar V. Borlongan. 2020. "An Extra Breath of Fresh Air: Hyperbaric Oxygenation as a Stroke Therapeutic" Biomolecules 10, no. 9: 1279. https://doi.org/10.3390/biom10091279
APA StyleCozene, B., Sadanandan, N., Gonzales-Portillo, B., Saft, M., Cho, J., Park, Y. J., & Borlongan, C. V. (2020). An Extra Breath of Fresh Air: Hyperbaric Oxygenation as a Stroke Therapeutic. Biomolecules, 10(9), 1279. https://doi.org/10.3390/biom10091279