Different Research Approaches in Unraveling the Venom Proteome of Naja ashei

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Pre-Fractionation and Preparation for LC-MS/MS Analysis
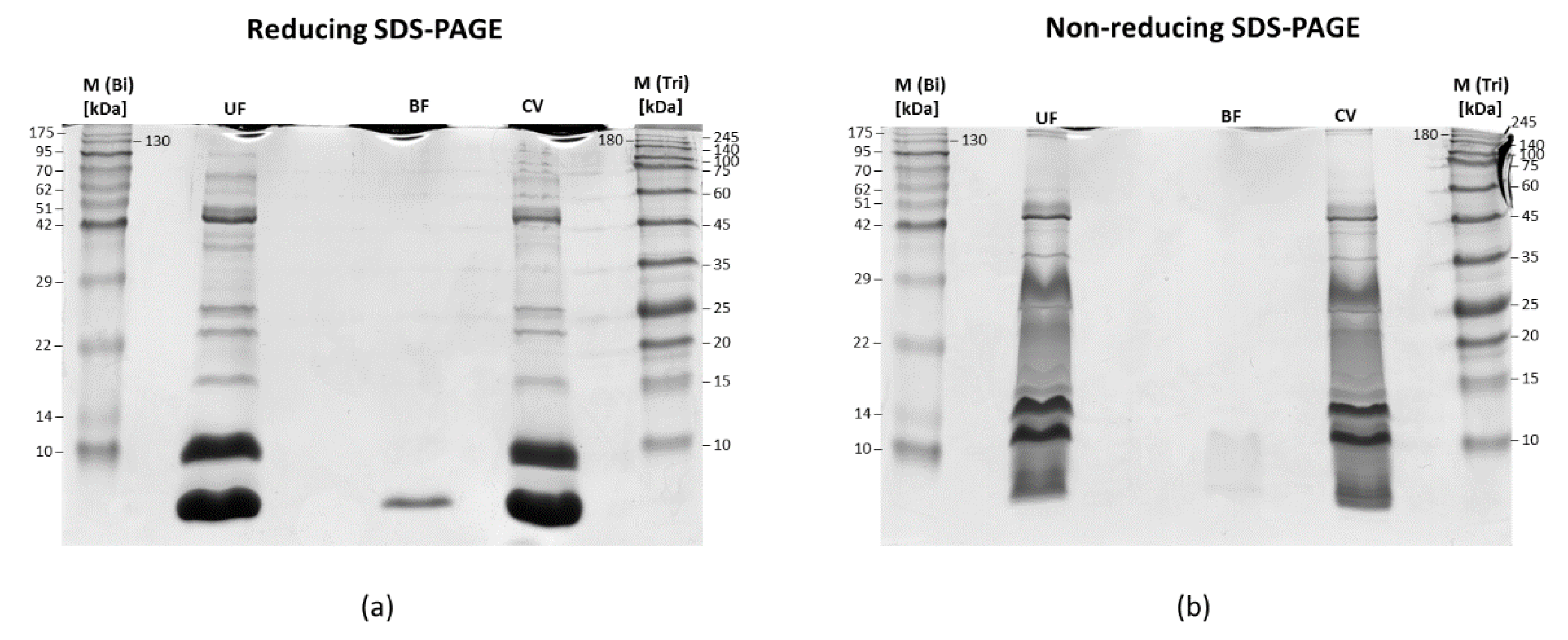
2.2. SDS-PAGE of Fractions Obtained after Membrane Filtration
2.3. Shotgun LC-MS/MS Analysis
2.4. MS Data Processing Protocol
2.4.1. Qualitative and Quantitative Analysis in MaxQuant
2.4.2. Qualitative and Quantitative Analysis in PeptideShaker
3. Results
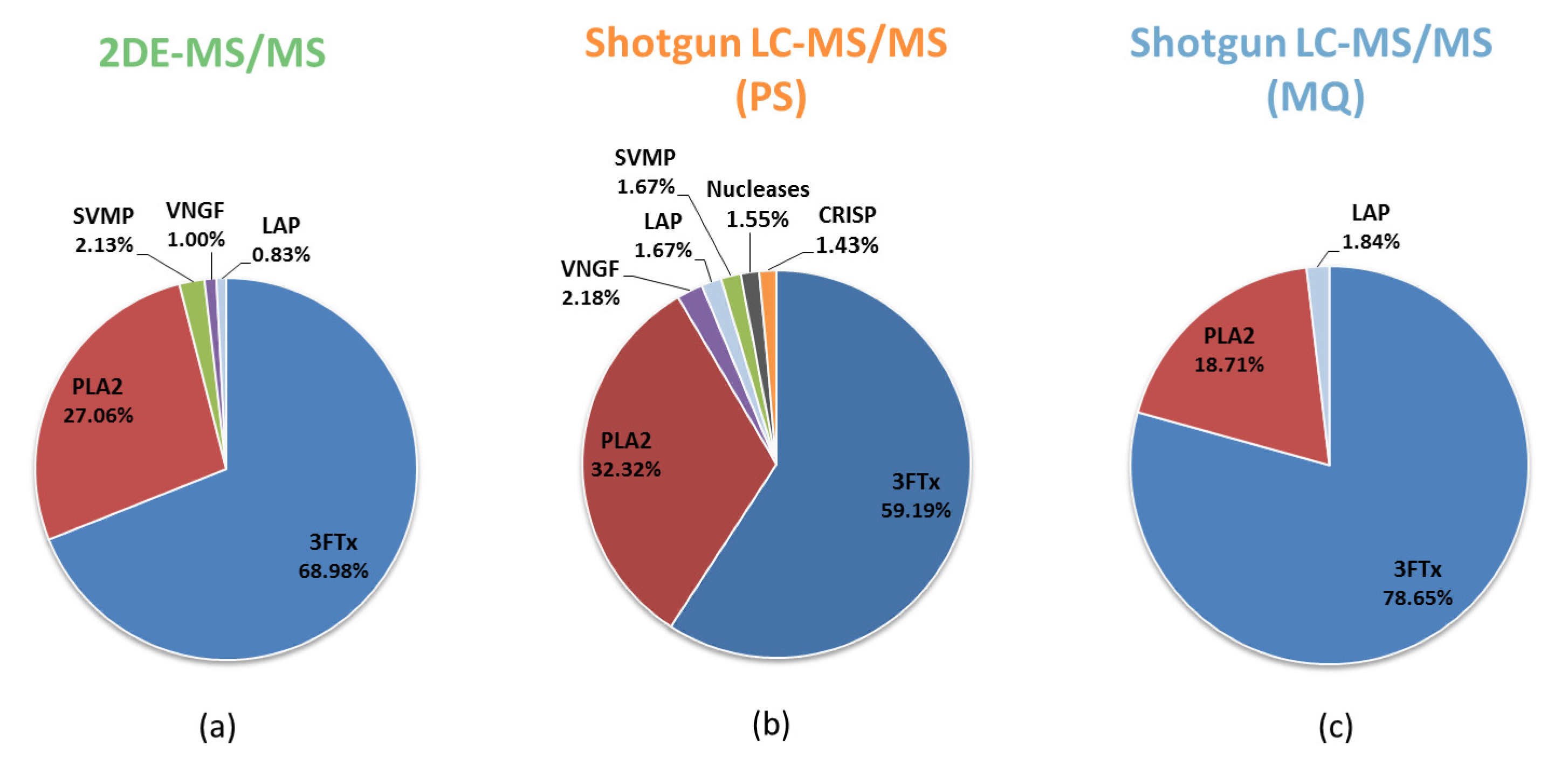
3.1. Comparative Analysis of the Results for the Naja ashei Venom Proteome Obtained Using Different Research Workflows
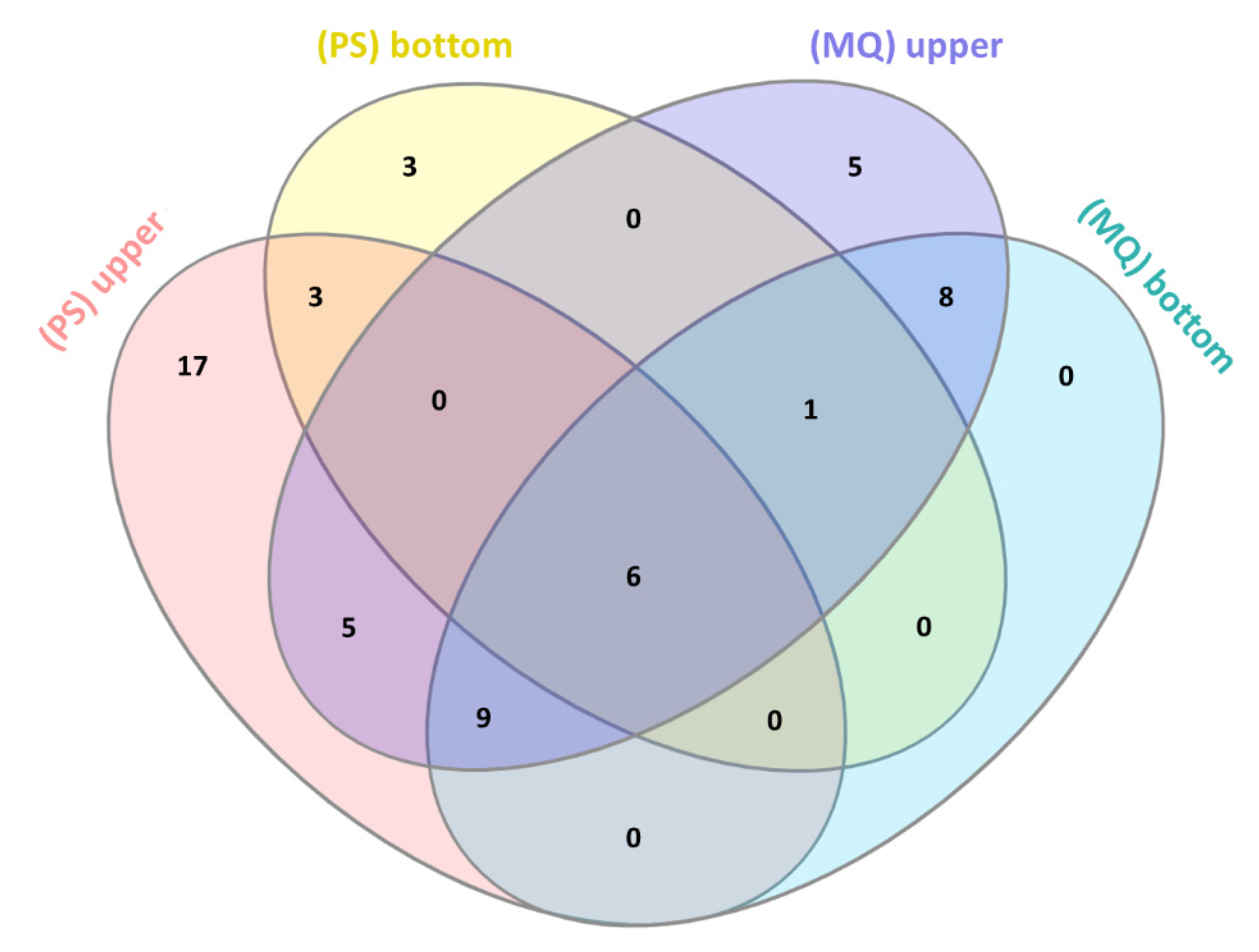
3.2. LC-MS/MS Analysis of Naja ashei Venom after Sample Decomplexation with the Use of Different Data Processing Software and an SDS-PAGE of Obtained Fractions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Calvete, J.J. Snake venomics—From low-resolution toxin-pattern recognition to toxin-resolved venom proteomes with absolute quantification. Expert Rev. Proteom. 2018, 15, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, B.; Calvete, J.J. Strategies in ‘snake venomics’ aiming at an integrative view of compositional, functional, and immunological characteristics of venoms. J. Venom. Anim Toxins 2017, 23, 26. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.W.; Cagney, G. An overview of label-free quantitation methods in proteomics by mass spectrometry. Methods Mol. Biol. 2010, 604, 273–283. [Google Scholar] [PubMed]
- Zelanis, A.; Silva, D.A.; Kitano, E.S.; Liberato, T.; Fukushima, I.; Serrano, S.M.T.; Tashima, A.K. A first step towards building spectral libraries as complementary tools for snake venom proteome/peptidome studies. Comp. Biochem. Phys. D 2019, 31, 100599. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Celis, F.; Encinar, J.R.; Sanz-Medel, A. Standardization approaches in absolute quantitative proteomics with mass spectrometry. Mass Spectrom. Rev. 2017, 37, 715–737. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Shi, J.; Wang, X.; Jiang, H.; Zhu, H.-J. Label-free absolute protein quantification with data-independent acquisition. J. Proteom. 2019, 200, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Sinitcyn, P.; Rudolph, J.D.; Cox, J. Computational Methods for Understanding Mass Spectrometry-Based Shotgun Proteomics Data. Annu. Rev. Biomed. Data Sci. 2018, 1, 207–234. [Google Scholar] [CrossRef]
- Juárez, P.; Sanz, L.; Calvete, J.J. Snake venomics: Characterization of protein families in Sistrurus barbouri venom by cysteine mapping, N-terminal sequencing, and tandem mass spectrometry analysis. Proteomics 2004, 4, 327–338. [Google Scholar]
- Bourmaud, A.; Gallien, S.; Domon, B. A quality control of proteomic experiments based on multiple isotopologous internal standards. EuPA Open Proteom. 2015, 8, 16–21. [Google Scholar] [CrossRef][Green Version]
- Hus, K.K.; Buczkowicz, J.; Petrilla, V.; Petrillová, M.; Łyskowski, A.; Legáth, J.; Bocian, A. First look at the Venom of Naja ashei. Molecules 2018, 23, 609. [Google Scholar] [CrossRef]
- Liu, Y.; Hüttenhain, R.; Collins, B.; Aebersold, R. Mass spectrometric protein maps for biomarker discovery and clinical research. Exp. Rev. Mol. Diagn. 2013, 13, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Ahrné, E.; Molzahn, L.; Glatter, T.; Schmidt, A. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. Proteomics 2013, 13, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Gao, Z.; Ying, W.; Fu, Y.; Zhao, Y.; Wu, S.; Li, M.; Wang, G.; Qian, X.; Zhu, Y.; et al. LFAQ: Toward Unbiased Label-Free Absolute Protein Quantification by Predicting Peptide Quantitative Factors. Anal. Chem. 2019, 91, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Neilson, K.A.; Naveid, A.A.; Muralidharan, S.; Mirzaei, M.; Mariani, M.; Assadourian, G.; Lee, A.; van Sluyter, S.C.; Haynes, P.A. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. [Google Scholar] [PubMed]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Arike, L.; Valgepea, K.; Peil, L.; Nahku, R.; Adamberg, K.; Vilu, R. Comparison and applications of label-free absolute proteome quantification methods on Escherichia coli. J. Proteom. 2012, 75, 5437–5448. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, E.G.; Jung, H.R.; Jung, J.W.; Kim, H.B.; Cho, J.W.; Kim, K.M.; Yi, E.C. Refinements of Lc-MS/MS Spectral Counting Statistics Improve Quantification of Low Abundance Proteins. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platfrom for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Bocian, A.; Sławek, S.; Jaromin, M.; Hus, K.K.; Buczkowicz, J.; Łysiak, D.; Petrílla, V.; Petrillova, M.; Legáth, J. Comparison of Methods for Measuring Protein Concentration in Venom Samples. Animals 2020, 10, 448. [Google Scholar] [CrossRef] [PubMed]
- Bocian, A.; Ciszkowicz, E.; Hus, K.K.; Buczkowicz, J.; Lecka-Szlachta, K.; Pietrowska, M.; Petrilla, V.; Petrillova, M.; Legáth, Ľ.; Legáth, J. Antimicrobial Activity of Protein Fraction from Naja ashei Venom against Staphylococcus epidermidis. Molecules 2020, 25, 293. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research Workflow | |||
|---|---|---|---|
| Protein Family | 2DE-MS/MS | Shotgun LC-MS/MS (PS) | Shotgun LC-MS/MS (MQ) |
| 3FTx | +(68.98%) | +(59.19%) | +(79.28%) |
| PLA2 | +(27.06%) | +(32.32%) | +(18.86%) |
| SVMP | +(2.13%) | +(2.26%) | +(0.43%) |
| VNGF | +(1.00%) | +(2.18%) | +(0.81%) |
| CRISP | +(0.70%) | +(1.43%) | +(0.26%) |
| CVF | +(0.12%) | +(0.14%) | +(0.02%) |
| Nucleases | +(0.01%) | +(1.55%) | +(0.10%) |
| Ig-like | - | +(0.31%) | +(0.08%) |
| GPx | - | +(0.31%) | +(0.05%) |
| LAAO | - | +(0.21%) | +(0.02%) |
| PDE | - | +(0.11%) | +(0.01%) |
| KUN | - | - | +(0.09%) |
| PLB | - | - | +(<0.01%) |
| Venom Fraction (Software) | ||||
|---|---|---|---|---|
| Protein Family | Upper Fraction (PS) | Bottom Fraction (PS) | Upper Fraction (MQ) | Bottom Fraction (MQ) |
| 3FTx | +(77.27%) | +(98.28%) | +(93.36%) | +(99.90%) |
| PLA2 | +(14.18%) | +(1.21%) | +(5.47%) | +(0.08%) |
| SVMP | +(1.74%) | - | +(0.20%) | +(0.001%) |
| VNGF | +(2.63%) | +(0.52%) | +(0.54%) | +(0.02%) |
| CRISP | +(1.92%) | - | +(0.32%) | +(0.005%) |
| CVF | +(0.12%) | - | +(0.005%) | +(0.001%) |
| Nucleases | +(0.90%) | - | +(0.03%) | + |
| Ig-like | +(0.37%) | - | +(0.01%) | + |
| GPx | +(0.45%) | - | +(0.04%) | + |
| LAAO | +(0.16%) | - | +(0.02%) | + |
| PDE | +(0.11%) | - | - | - |
| KUN | - | - | +(0.007%) | +(0.01%) |
| PLB | +(0.09%) | - | - | - |
| TF-like | +(0.07%) | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hus, K.K.; Marczak, Ł.; Petrilla, V.; Petrillová, M.; Legáth, J.; Bocian, A. Different Research Approaches in Unraveling the Venom Proteome of Naja ashei. Biomolecules 2020, 10, 1282. https://doi.org/10.3390/biom10091282
Hus KK, Marczak Ł, Petrilla V, Petrillová M, Legáth J, Bocian A. Different Research Approaches in Unraveling the Venom Proteome of Naja ashei. Biomolecules. 2020; 10(9):1282. https://doi.org/10.3390/biom10091282
Chicago/Turabian StyleHus, Konrad Kamil, Łukasz Marczak, Vladimír Petrilla, Monika Petrillová, Jaroslav Legáth, and Aleksandra Bocian. 2020. "Different Research Approaches in Unraveling the Venom Proteome of Naja ashei" Biomolecules 10, no. 9: 1282. https://doi.org/10.3390/biom10091282
APA StyleHus, K. K., Marczak, Ł., Petrilla, V., Petrillová, M., Legáth, J., & Bocian, A. (2020). Different Research Approaches in Unraveling the Venom Proteome of Naja ashei. Biomolecules, 10(9), 1282. https://doi.org/10.3390/biom10091282