From Cell Culture to Organoids-Model Systems for Investigating Prion Strain Characteristics



Abstract
:1. Introduction
2. Immortalized Cell Lines
2.1. Neuron-Like Cell Lines
2.1.1. Scrapie Mouse Brain (SMB) Cells
2.1.2. PC12 Cells
2.1.3. N2a and Other Neuroblastoma Cells
2.1.4. CAD5 Cells
2.1.5. GT1 Cells
2.1.6. CRBL Cells
2.1.7. 1C11 Cells
2.1.8. Neural Cell Lines from Prnp0/0 Mice
2.2. Microglial Cell Lines
2.3. Astrocyte Cell Line: C8D1A
2.4. Schwann Cell Lines: MovS6 and MovS2
2.5. Fibroblast Cell Lines
2.6. RK13 Cells
2.7. MDBK Cells
2.8. C2C12 Myotubes
2.9. Cells of the Lymphoid Reticular System
3. Primary Culture
4. Stem Cell-Derived Cultures
5. Neurospheres
6. Brain Aggregates
7. Organotypic Slice Culture
8. Organoids
9. Enhancing Prion Propagation in Model Systems
9.1. Temperature
9.2. Culture Media
9.3. PrPC Expression Levels
9.4. Infection Techniques and Mechanisms of Cell-to-Cell Transfer
10. The Influence of Strains on Different Model Systems
10.1. Strain Adaptation
10.2. Strain-Dependent Drug Effects
11. In Conclusion: Choosing the Right Model
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collinge, J. Prion Diseases of Humans and Animals: Their Causes and Molecular Basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulf, M.A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef] [Green Version]
- Oesch, B.; Westaway, D.; Wälchli, M.; McKinley, M.P.; Kent, S.B.H.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Weissmann, C. Molecular genetics of transmissible spongiform encephalopathies. J. Biol. Chem. 1999, 274, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10. [Google Scholar] [CrossRef]
- Diack, A.B.; Head, M.W.; McCutcheon, S.; Boyle, A.; Knight, R.; Ironside, J.W.; Manson, J.C.; Will, R.G. Variant CJD: 18 years of research and surveillance. Prion 2014, 8, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, M.; Dittmar, K.; Llorens, F.; Gelpi, E.; Ferrer, I.; Schulz-Schaeffer, W.J.; Zerr, I. Hereditary Human Prion Diseases: An Update. Mol. Neurobiol. 2017, 54, 4138–4149. [Google Scholar] [CrossRef]
- Mastrianni, J.A. The genetics of prion diseases. Genet. Med. 2010, 12, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, D.M.; Gibbs, C.J.; Sulima, M.P.; Bacote, A.; Amyx, H.; Gajdusek, D.C. Transmission of human spongiform encephalopathies to experimental animals: Comparison of the chimpanzee and squirrel monkey. Dev. Biol. Stand. 1993, 80, 9–13. [Google Scholar] [PubMed]
- Liberski, P.; Gajos, A.; Sikorska, B.; Lindenbaum, S. Kuru, the First Human Prion Disease. Viruses 2019, 11, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorberg, I.; Chiesa, R. Experimental models to study prion disease pathogenesis and identify potential therapeutic compounds. Curr. Opin. Pharmcol. 2019, 44, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A.; Fraser, H. The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J. Gen. Virol. 1989, 70, 2017–2025. [Google Scholar] [CrossRef]
- Telling, G.C.; Scott, M.; Hsiao, K.K.; Foster, D.; Yang, S.L.; Torchia, M.; Sidle, K.C.L.; Collinge, J.; Dearmond, S.J.; Prusiner, S.B. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc. Natl. Acad. Sci. USA 1994, 91, 9936–9940. [Google Scholar] [CrossRef] [Green Version]
- Igel-Egalon, A.; Béringue, V.; Rezaei, H.; Sibille, P. Prion Strains and Transmission Barrier Phenomena. Pathogens 2018, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Parchi, P.; Castellani, R.; Capellari, S.; Ghetti, B.; Young, K.; Chen, S.G.; Farlow, M.; Dickson, D.W.; Sima, A.A.F.; Trojanowski, J.Q.; et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 1996, 39, 767–778. [Google Scholar] [CrossRef]
- Krance, S.H.; Luke, R.; Shenouda, M.; Israwi, A.R.; Colpitts, S.J.; Darwish, L.; Strauss, M.; Watts, J.C. Cellular models for discovering prion disease therapeutics: Progress and challenges. J. Neurochem. 2020, 153, 150–172. [Google Scholar] [CrossRef]
- Kocisko, D.A.; Engel, A.L.; Harbuck, K.; Arnold, K.M.; Olsen, E.A.; Raymond, L.D.; Vilette, D.; Caughey, B. Comparison of protease-resistant prion protein inhibitors in cell cultures infected with two strains of mouse and sheep scrapie. Neurosci. Lett. 2005, 388, 106–111. [Google Scholar] [CrossRef]
- Hannaoui, S.; Gougerot, A.; Privat, N.; Levavasseur, E.; Bizat, N.; Hauw, J.J.; Brandel, J.P.; Haïk, S. Cycline efficacy on the propagation of human prions in primary cultured neurons is strain-specific. J. Infect. Dis. 2014, 209, 1144–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.C.; Haig, D.A. Multiplication of scrapie agent in cell culture. Res. Vet. Sci. 1970, 11, 500–501. [Google Scholar] [CrossRef]
- Birkett, C.R.; Hennion, R.M.; Bembridge, D.A.; Clarke, M.C.; Chree, A.; Bruce, M.E.; Bostock, C.J. Scrapie strains maintain biological phenotypes on propagation in a cell line in culture. EMBO J. 2001, 20, 3351–3358. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.; Carp, R.I.; Callahan, S.M. In vitro replication of scrapie agent in a neuronal model: Infection of PC12 cells. J. Gen. Virol. 1984, 65, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.; Deng, H.; Race, R.E.; Ju, W.; Scalici, C.L.; Papini, M.C.; Kascsak, R.J.; Carp, R.I. Demonstration of scrapie strain diversity in infected PC12 cells. J. Gen. Virol. 1992, 73, 3027–3031. [Google Scholar] [CrossRef] [PubMed]
- Arjona, A.; Simarro, L.; Islinger, F.; Nishida, N.; Manuelidis, L. Two Creutzfeldt-Jakob disease agents reproduce prion protein-independent identities in cell cultures. Proc. Natl. Acad. Sci. USA 2004, 101, 8768–8773. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Harris, D.A.; Vilette, D.; Laude, H.; Frobert, Y.; Grassi, J.; Casanova, D.; Milhavet, O.; Lehmann, S. Successful Transmission of Three Mouse-Adapted Scrapie Strains to Murine Neuroblastoma Cell Lines Overexpressing Wild-Type Mouse Prion Protein. J. Virol. 2000, 74, 320–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klöhn, P.C.; Stoltze, L.; Flechsig, E.; Enari, M.; Weissmann, C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. USA 2003, 100, 11666–11671. [Google Scholar] [CrossRef] [Green Version]
- Mahal, S.P.; Baker, C.A.; Demczyk, C.A.; Smith, E.W.; Julius, C.; Weissmann, C. Prion strain discrimination in cell culture: The cell panel assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20908–20913. [Google Scholar] [CrossRef] [Green Version]
- Oelschlegel, A.M.; Fallahi, M.; Ortiz-Umpierre, S.; Weissmann, C. The Extended Cell Panel Assay Characterizes the Relationship of Prion Strains RML, 79A, and 139A and Reveals Conversion of 139A to 79A-Like Prions in Cell Culture. J. Virol. 2012, 86, 5297–5303. [Google Scholar] [CrossRef] [Green Version]
- Philiastides, A.; Ribes, J.M.; Yip, D.C.-M.; Schmidt, C.; Benilova, I.; Klöhn, P.-C. A New Cell Model for Investigating Prion Strain Selection and Adaptation. Viruses 2019, 11, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourkas, M.E.C.; Arshad, H.; Al-Azzawi, Z.A.M.; Halgas, O.; Shikiya, R.A.; Mehrabian, M.; Schmitt-Ulms, G.; Bartz, J.C.; Watts, J.C. Engineering a Murine Cell Line for the Stable Propagation of Hamster Prions. J. Biol. Chem. 2019, 294, 4911–4923. [Google Scholar] [CrossRef] [PubMed]
- Walia, R.; Ho, C.C.; Lee, C.; Gilch, S.; Schatzl, H.M. Gene-Edited Murine Cell Lines for Propagation of Chronic Wasting Disease Prions. Sci. Rep. 2019, 9, 11151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, G.S.; Magalhães, A.C.; Prado, M.A.M.; Caughey, B. Mouse-Adapted Scrapie Infection of SN56 Cells: Greater Efficiency with Microsome-Associated versus Purified PrP-Res. J. Virol. 2006, 80, 2106–2117. [Google Scholar] [CrossRef] [Green Version]
- Schätzl, H.M.; Laszlo, L.; Holtzman, D.M.; Tatzelt, J.; DeArmond, S.J.; Weiner, R.I.; Mobley, W.C.; Prusiner, S.B. A Hypothalamic Neuronal Cell Line Persistently Infected with Scrapie Prions Exhibits Apoptosis. J. Virol. 1997, 71, 8821–8831. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Katamine, S.; Manuelidis, L. Medicine: Reciprocal Interference between Specific CJD and Scrapie Agents in Neural Cell Cultures. Science 2005, 310, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, K.; Masujin, K.; Okada, H.; Ushiki-Kaku, Y.; Matsuura, Y.; Yokoyama, T. Selective Propagation of Mouse-Passaged Scrapie Prions with Long Incubation Period from a Mixed Prion Population Using GT1-7 Cells. PLoS ONE 2017, 12, e0179317. [Google Scholar] [CrossRef] [Green Version]
- Mays, C.E.; Kang, H.E.; Kim, Y.; Shim, S.H.; Bang, J.E.; Woo, H.J.; Cho, Y.H.; Kim, J.B.; Ryou, C. CRBL Cells: Establishment, Characterization and Susceptibility to Prion Infection. Brain Res. 2008, 1208, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Mouillet-Richard, S.; Nishida, N.; Pradines, E.; Laude, H.; Schneider, B.; Féraudet, C.C.; Grassi, J.; Launay, J.M.; Lehmann, S.; Kellermann, O. Prions Impair Bioaminergic Functions through Serotonin- or Catecholamine-Derived Neurotoxins in Neuronal Cells. J. Biol. Chem. 2008, 283, 23782–23790. [Google Scholar] [CrossRef] [Green Version]
- Maas, E.; Geissen, M.; Groschup, M.H.; Rost, R.; Onodera, T.; Schätzl, H.; Vorberg, I.M. Scrapie Infection of Prion Protein-Deficient Cell Line upon Ectopic Expression of Mutant Prion Proteins. J. Biol. Chem. 2007, 282, 18702–18710. [Google Scholar] [CrossRef] [Green Version]
- McNally, K.L.; Ward, A.E.; Priola, S.A. Cells Expressing Anchorless Prion Protein Are Resistant to Scrapie Infection. J. Virol. 2009, 83, 4469–4475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamaru, Y.; Takenouchi, T.; Ogihara, K.; Hoshino, M.; Takata, M.; Imamura, M.; Tagawa, Y.; Hayashi-Kato, H.; Ushiki-Kaku, Y.; Shimizu, Y.; et al. Microglial Cell Line Established from Prion Protein-Overexpressing Mice Is Susceptible to Various Murine Prion Strains. J. Virol. 2007, 81, 1524–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Gutiérrez, J.F.; Schneider, D.A.; Baszler, T.V.; Greenlee, J.J.; Nicholson, E.M.; Stanton, J.B. HTERT-Immortalized Ovine Microglia Propagate Natural Scrapie Isolates. Virus Res. 2015, 198, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Tahir, W.; Abdulrahman, B.; Abdelaziz, D.H.; Thapa, S.; Walia, R.; Schätzl, H.M. An Astrocyte Cell Line That Differentially Propagates Murine Prions. J. Biol. Chem. 2020, 295, 11572–11583. [Google Scholar] [CrossRef]
- Archer, F.; Bachelin, C.; Andreoletti, O.; Besnard, N.; Perrot, G.; Langevin, C.; le Dur, A.; Vilette, D.; Baron-Van Evercooren, A.; Vilotte, J.-L.; et al. Cultured Peripheral Neuroglial Cells Are Highly Permissive to Sheep Prion Infection. J. Virol. 2004, 78, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Neale, M.H.; Mountjoy, S.J.; Edwards, J.C.; Vilette, D.; Laude, H.; Windl, O.; Saunders, G.C. Infection of Cell Lines with Experimental and Natural Ovine Scrapie Agents. J. Virol. 2010, 84, 2444–2452. [Google Scholar] [CrossRef] [Green Version]
- Follet, J.; Lemaire-Vieille, C.; Blanquet-Grossard, F.; Podevin-Dimster, V.; Lehmann, S.; Chauvin, J.-P.; Decavel, J.-P.; Varea, R.; Grassi, J.; Fontes, M.; et al. PrP Expression and Replication by Schwann Cells: Implications in Prion Spreading. J. Virol. 2002, 76, 2434–2439. [Google Scholar] [CrossRef] [Green Version]
- Vorberg, I.; Raines, A.; Story, B.; Priola, S.A. Susceptibility of Common Fibroblast Cell Lines to Transmissible Spongiform Encephalopathy Agents. J. Infect. Dis. 2004, 189, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Raymond, G.J.; Olsen, E.A.; Lee, K.S.; Raymond, L.D.; Bryant, P.K.; Baron, G.S.; Caughey, W.S.; Kocisko, D.A.; McHolland, L.E.; Favara, C.; et al. Inhibition of Protease-Resistant Prion Protein Formation in a Transformed Deer Cell Line Infected with Chronic Wasting Disease. J. Virol. 2006, 80, 596–604. [Google Scholar] [CrossRef] [Green Version]
- Courageot, M.P.; Daude, N.; Nonno, R.; Paquet, S.; di Bari, M.A.; le Dur, A.; Chapuis, J.; Hill, A.F.; Agrimi, U.; Laude, H.; et al. A Cell Line Infectible by Prion Strains from Different Species. J. Gen. Virol. 2008, 89, 341–347. [Google Scholar] [CrossRef]
- Arellano-Anaya, Z.E.; Huor, A.; Leblanc, P.; Lehmann, S.; Provansal, M.; Raposo, G.; Andréoletti, O.; Vilette, D. Prion Strains Are Differentially Released through the Exosomal Pathway. Cell. Mol. Life Sci. 2015, 72, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Lawson, V.A.; Vella, L.J.; Stewart, J.D.; Sharples, R.A.; Klemm, H.; Machalek, D.M.; Masters, C.L.; Cappai, R.; Collins, S.J.; Hill, A.F. Mouse-Adapted Sporadic Human Creutzfeldt-Jakob Disease Prions Propagate in Cell Culture. Int. J. Biochem. Cell Biol. 2008, 40, 2793–2801. [Google Scholar] [CrossRef] [PubMed]
- Vilette, D.; Andreoletti, O.; Archer, F.; Madelaine, M.F.; Vilotte, J.L.; Lehmann, S.; Laude, H. Ex Vivo Propagation of Infectious Sheep Scrapie Agent in Heterologous Epithelial Cells Expressing Ovine Prion Protein. Proc. Natl. Acad. Sci. USA 2001, 98, 4055–4059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabuncu, E.; Petit, S.; le Dur, A.; Lan Lai, T.; Vilotte, J.-L.; Laude, H.; Vilette, D. PrP Polymorphisms Tightly Control Sheep Prion Replication in Cultured Cells. J. Virol. 2003, 77, 2696–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arellano-Anaya, Z.E.; Savistchenko, J.; Mathey, J.; Huor, A.; Lacroux, C.; Andréoletti, O.; Vilette, D. A Simple, Versatile and Sensitive Cell-Based Assay for Prions from Various Species. PLoS ONE 2011, 6, e20563. [Google Scholar] [CrossRef] [Green Version]
- Dassanayake, R.P.; Zhuang, D.; Truscott, T.C.; Madsen-Bouterse, S.A.; O’Rourke, K.I.; Schneider, D.A. A Transfectant RK13 Cell Line Permissive to Classical Caprine Scrapie Prion Propagation. Prion 2016, 10, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Bian, J.; Napier, D.; Khaychuck, V.; Angers, R.; Graham, C.; Telling, G. Cell-Based Quantification of Chronic Wasting Disease Prions. J. Virol. 2010, 84, 8322–8326. [Google Scholar] [CrossRef] [Green Version]
- Oelschlegel, A.M.; Geissen, M.; Lenk, M.; Riebe, R.; Angermann, M.; Schaetzl, H.; Groschup, M.H. A Bovine Cell Line That Can Be Infected by Natural Sheep Scrapie Prions. PLoS ONE 2015, 10, e0117154. [Google Scholar] [CrossRef] [Green Version]
- Tark, D.; Kim, H.; Neale, M.H.; Kim, M.; Sohn, H.; Lee, Y.; Cho, I.; Joo, Y.; Windl, O. Generation of a Persistently Infected MDBK Cell Line with Natural Bovine Spongiform Encephalopathy (BSE). PLoS ONE 2015, 10, e0115939. [Google Scholar] [CrossRef] [Green Version]
- Herbst, A.; Banser, P.; Velasquez, C.D.; Mays, C.E.; Sim, V.L.; Westaway, D.; Aiken, J.M.; McKenzie, D. Infectious Prions Accumulate to High Levels in Non Proliferative C2C12 Myotubes. PLoS Pathog. 2013, 9, e1003755. [Google Scholar] [CrossRef]
- Akimov, S.; Yakovleva, O.; Vasilyeva, I.; McKenzie, C.; Cervenakova, L. Persistent Propagation of Variant Creutzfeldt-Jakob Disease Agent in Murine Spleen Stromal Cell Culture with Features of Mesenchymal Stem Cells. J. Virol. 2008, 82, 10959–10962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimov, S.; Vasilyeva, I.; Yakovleva, O.; McKenzie, C.; Cervenakova, L. Murine Bone Marrow Stromal Cell Culture with Features of Mesenchymal Stem Cells Susceptible to Mouse-Adapted Human TSE Agent, Fukuoka-1. Folia Neuropathol. 2009, 47, 205–214. [Google Scholar] [PubMed]
- Cervenakova, L.; Akimov, S.; Vasilyeva, I.; Yakovleva, O.; McKenzie, C.; Cervenak, J.; Piccardo, P.; Asher, D.M. Fukuoka-1 Strain of Transmissible Spongiform Encephalopathy Agent Infects Murine Bone Marrow-Derived Cells with Features of Mesenchymal Stem Cells. Transfusion 2011, 51, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Krejciova, Z.; de Sousa, P.; Manson, J.; Ironside, J.W.; Head, M.W. Human Tonsil-Derived Follicular Dendritic-like Cells Are Refractory to Human Prion Infection in Vitro and Traffic Disease-Associated Prion Protein to Lysosomes. Am. J. Pathol. 2014, 184, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Butler, D.A.; Scott, M.R.; Bockman, J.M.; Borchelt, D.R.; Taraboulos, A.; Hsiao, K.K.; Kingsbury, D.T.; Prusiner, S.B. Scrapie-Infected Murine Neuroblastoma Cells Produce Protease-Resistant Prion Proteins. J. Virol. 1988, 62, 1558–1564. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.B.; Lu, D.; Geva, M.; Watts, J.C.; Bhardwaj, S.; Oehler, A.; Renslo, A.R.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Drug Resistance Confounding Prion Therapeutics. Proc. Natl. Acad. Sci. USA 2013, 110, E4160–E4169. [Google Scholar] [CrossRef] [Green Version]
- Race, R.E.; Fadness, L.H.; Chesebro, B. Characterization of scrapie infection in mouse neuroblastoma cells. J. Gen. Virol. 1987, 68, 1391–1399. [Google Scholar] [CrossRef]
- Ladogana, A.; Liu, Q.; Geng Xi, Y.; Pocchiari, M. Proteinase-resistant protein in human neuroblastoma cells infected with brain material from Creutzfeldt-Jakob patient. Lancet 1995, 345, 594–595. [Google Scholar] [CrossRef]
- Arima, K.; Nishida, N.; Sakaguchi, S.; Shigematsu, K.; Atarashi, R.; Yamaguchi, N.; Yoshikawa, D.; Yoon, J.; Watanabe, K.; Kobayashi, N.; et al. Biological and Biochemical Characteristics of Prion Strains Conserved in Persistently Infected Cell Cultures. J. Virol. 2005, 79, 7104–7112. [Google Scholar] [CrossRef] [Green Version]
- Marshall, K.E.; Hughson, A.; Vascellari, S.; Priola, S.A.; Sakudo, A.; Onodera, T.; Baron, G.S. PrP Knockout Cells Expressing Transmembrane PrP Resist Prion Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Zhu, C. Microglia in Prion Diseases. J. Clin. Investig. 2017, 127, 3230–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Jin, M.-Z.; Yang, Z.-Y.; Jin, W.-L. Microglia in Neurodegenerative Diseases. Neural Regen. Res. 2021, 16, 270. [Google Scholar] [CrossRef] [PubMed]
- Fehlinger, A.; Wolf, H.; Hossinger, A.; Duernberger, Y.; Pleschka, C.; Riemschoss, K.; Liu, S.; Bester, R.; Paulsen, L.; Priola, S.A.; et al. Prion Strains Depend on Different Endocytic Routes for Productive Infection. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wüsten, K.A.; Reddy, P.P.; Smiyakin, A.; Bernis, M.E.; Tamgüney, G. A Bioluminescent Cell Assay to Quantify Prion Protein Dimerization. Sci. Rep. 2018, 8, 14178. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Horiuchi, M.; Ishiguro, N.; Muramatsu, Y.; Kai-Uwe, G.D.; Shinagawa, M. Amino Acid Polymorphisms of PrP with Reference to Onset of Scrapie in Suffolk and Corriedale Sheep in Japan. J. Gen. Virol. 1995, 76, 2577–2581. [Google Scholar] [CrossRef]
- Lacroux, C.; Perrin-Chauvineau, C.; Corbiere, F.; Aron, N.; Aguilar-Calvo, P.; Torres, J.M.; Costes, P.; Bremaud, I.; Lugan, S.; Schelcher, F.; et al. Genetic Resistance to Scrapie Infection in Experimentally Challenged Goats. J. Virol. 2014, 88, 2406–2413. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, P.; Alais, S.; Porto-Carreiro, I.; Lehmann, S.; Grassi, J.; Raposo, G.; Darlix, J.L. Retrovirus Infection Strongly Enhances Scrapie Infectivity Release in Cell Culture. EMBO J. 2006, 25, 2674–2685. [Google Scholar] [CrossRef] [Green Version]
- Peden, A.H.; Ritchie, D.L.; Head, M.W.; Ironside, J.W. Detection and Localization of PrPSc in the Skeletal Muscle of Patients with Variant, Iatrogenic, and Sporadic Forms of Creutzfeldt-Jakob Disease. Am. J. Pathol. 2006, 168, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, E.R.; Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Prion Infection of Skeletal Muscle Cells and Papillae in the Tongue. J. Virol. 2004, 78, 6792–6798. [Google Scholar] [CrossRef] [Green Version]
- Dlakic, W.M.; Grigg, E.; Bessen, R.A. Prion Infection of Muscle Cells In Vitro. J. Virol. 2007, 81, 4615–4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadsworth, J.D.F.; Joiner, S.; Hill, A.F.; Campbell, T.A.; Desbruslais, M.; Luthert, P.J.; Collinge, J. Tissue Distribution of Protease Resistant Prion Protein in Variant Creutzfeldt-Jakob Disease Using a Highly Sensitive Immunoblotting Assay. Lancet 2001, 358, 171–180. [Google Scholar] [CrossRef]
- Ponzio, N.M.; Brown, P.H.; Thorbecke, G.J. Host-Tumor Interactions in the SJL Lymphoma Model. Int. Rev. Immunol. 1986, 1, 273–301. [Google Scholar] [CrossRef] [PubMed]
- Cervenakova, L.; Yakovleva, O.; McKenzie, C. Protease-Resistant Prion Protein in Lymphoreticular Tumors of Variant Creutzfeldt-Jakob Disease Mice. Emerg. Infect. Dis. 2006, 12, 511–513. [Google Scholar] [CrossRef]
- Krejciova, Z.; Alibhai, J.; Zhao, C.; Krencik, R.; Rzechorzek, N.M.; Ullian, E.M.; Manson, J.; Ironside, J.W.; Head, M.W.; Chandran, S. Human Stem Cell-Derived Astrocytes Replicate Human Prions in a PRNP Genotype-Dependent Manner. J. Exp. Med. 2017, 214, 3481–3495. [Google Scholar] [CrossRef] [Green Version]
- Cronier, S.; Beringue, V.; Bellon, A.; Peyrin, J.-M.; Laude, H. Prion Strain- and Species-Dependent Effects of Antiprion Molecules in Primary Neuronal Cultures. J. Virol. 2007, 81, 13794–13800. [Google Scholar] [CrossRef] [Green Version]
- Hannaoui, S.; Maatouk, L.; Privat, N.; Levavasseur, E.; Faucheux, B.A.; Haik, S. Prion Propagation and Toxicity Occur In Vitro with Two-Phase Kinetics Specific to Strain and Neuronal Type. J. Virol. 2013, 87, 2535–2548. [Google Scholar] [CrossRef] [Green Version]
- Victoria, G.S.; Arkhipenko, A.; Zhu, S.; Syan, S.; Zurzolo, C. Astrocyte-to-Neuron Intercellular Prion Transfer Is Mediated by Cell-Cell Contact. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cronier, S.; Laude, H.; Peyrin, J.M. Prions Can Infect Primary Cultured Neurons and Astrocytes and Promote Neuronal Cell Death. Proc. Natl. Acad. Sci. USA 2004, 101, 12271–12276. [Google Scholar] [CrossRef] [Green Version]
- Hasebe, R.; Tanaka, M.; Suzuki, A.; Yamasaki, T.; Horiuchi, M. Complement Factors Alter the Amount of PrPSc in Primary-Cultured Mouse Cortical Neurons Associated with Increased Membrane Permeability. Virology 2016, 496, 9–20. [Google Scholar] [CrossRef]
- Fang, C.; Imberdis, T.; Garza, M.C.; Wille, H.; Harris, D.A. A Neuronal Culture System to Detect Prion Synaptotoxicity. PLOS Pathog. 2016, 12, e1005623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milhavet, O.; Casanova, D.; Chevallier, N.; McKay, R.D.G.; Lehmann, S. Neural Stem Cell Model for Prion Propagation. Stem Cells 2006, 24, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Carp, R.I.; Callahan, S.M.; Wisniewski, H.M. Incubation Periods and Survival Times for Mice Injected Stereotaxically with Three Scrapie Strains in Different Brain Regions. J. Gen. Virol. 1987, 68, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Matamoros-Angles, A.; Gayosso, L.M.; Richaud-Patin, Y.; di Domenico, A.; Vergara, C.; Hervera, A.; Sousa, A.; Fernández-Borges, N.; Consiglio, A.; Gavín, R.; et al. IPS Cell Cultures from a Gerstmann-Sträussler-Scheinker Patient with the Y218N PRNP Mutation Recapitulate Tau Pathology. Mol. Neurobiol. 2018, 55, 3033–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, L.S. Neurospheres: Insights into Neural Stem Cell Biology. J. Neurosci. Res. 2004, 78, 761–769. [Google Scholar] [CrossRef]
- Giri, R.K.; Young, R.; Pitstick, R.; DeArmond, S.J.; Prusiner, S.B.; Carlson, G.A. Prion Infection of Mouse Neuropheres. Proc. Natl. Acad. Sci. USA 2006, 103, 3875–3880. [Google Scholar] [CrossRef] [Green Version]
- Herva, M.E.; Relaño-Ginés, A.; Villa, A.; Torres, J.M. Prion Infection of Differentiated Neurospheres. J. Neurosci. Methods 2010, 188, 270–275. [Google Scholar] [CrossRef]
- Iwamaru, Y.; Takenouchi, T.; Imamura, M.; Shimizu, Y.; Miyazawa, K.; Mohri, S.; Yokoyama, T.; Kitani, H. Prion Replication Elicits Cytopathic Changes in Differentiated Neurosphere Cultures. J. Virol. 2013, 87, 8745–8755. [Google Scholar] [CrossRef] [Green Version]
- Iwamaru, Y.; Mathiason, C.K.; Telling, G.C.; Hoover, E.A. Chronic Wasting Disease Prion Infection of Differentiated Neurospheres. Prion 2017, 11, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Bajsarowicz, K.; Ahn, M.; Ackerman, L.; DeArmond, B.N.; Carlson, G.; DeArmond, S.J. A Brain Aggregate Model Gives New Insights Into the Pathobiology and Treatment of Prion Diseases. J. Neuropathol. Exp. Neurol. 2012, 71, 449–466. [Google Scholar] [CrossRef]
- Pineau, H.; Sim, V. POSCAbilities: The Application of the Prion Organotypic Slice Culture Assay to Neurodegenerative Disease Research. Biomolecules 2020, 10, 1079. [Google Scholar] [CrossRef] [PubMed]
- Falsig, J.; Aguzzi, A. The Prion Organotypic Slice Culture Assay-POSCA. Nat. Protoc. 2008, 3, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Croft, C.L.; Cruz, P.E.; Ryu, D.H.; Ceballos-Diaz, C.; Strang, K.H.; Woody, B.M.; Lin, W.L.; Deture, M.; Rodríguez-Lebrón, E.; Dickson, D.W.; et al. RAAV-Based Brain Slice Culture Models of Alzheimer’s and Parkinson’s Disease Inclusion Pathologies. J. Exp. Med. 2019, 216, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, S.; Masuch, A.; Nestel, S.; Katzmarski, N.; Meyer-Luehmann, M.; Biber, K. Forebrain Microglia from Wild-Type but Not Adult 5xFAD Mice Prevent Amyloid-β Plaque Formation in Organotypic Hippocampal Slice Cultures. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Herrmann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A Neuroprotective Role for Microglia in Prion Diseases. J. Exp. Med. 2016, 213, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Falsig, J.; Julius, C.; Margalith, I.; Schwarz, P.; Heppner, F.L.; Aguzzi, A. A Versatile Prion Replication Assay in Organotypic Brain Slices. Nat. Neurosci. 2008, 11, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Falsig, J.; Sonati, T.; Herrmann, U.S.; Saban, D.; Li, B.; Arroyo, K.; Ballmer, B.; Liberski, P.P.; Aguzzi, A. Prion Pathogenesis Is Faithfully Reproduced in Cerebellar Organotypic Slice Cultures. PLoS Pathog. 2012, 8, e1002985. [Google Scholar] [CrossRef] [Green Version]
- Wolf, H.; Hossinger, A.; Fehlinger, A.; Büttner, S.; Sim, V.; McKenzie, D.; Vorberg, I.M. Deposition Pattern and Subcellular Distribution of Disease-Associated Prion Protein in Cerebellar Organotypic Slice Cultures Infected with Scrapie. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [Green Version]
- Campeau, J.L.; Wu, G.; Bell, J.R.; Rasmussen, J.; Sim, V.L. Early Increase and Late Decrease of Purkinje Cell Dendritic Spine Density in Prion-Infected Organotypic Mouse Cerebellar Cultures. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Kondru, N.; Manne, S.; Kokemuller, R.; Greenlee, J.; Greenlee, M.H.W.; Nichols, T.; Kong, Q.; Anantharam, V.; Kanthasamy, A.; Halbur, P.; et al. An Ex Vivo Brain Slice Culture Model of Chronic Wasting Disease: Implications for Disease Pathogenesis and Therapeutic Development. Sci. Rep. 2020, 1–13. [Google Scholar] [CrossRef]
- Cortez, L.M.; Campeau, J.; Norman, G.; Kalayil, M.; van der Merwe, J.; McKenzie, D.; Sim, V.L. Bile Acids Reduce Prion Conversion, Reduce Neuronal Loss, and Prolong Male Survival in Models of Prion Disease. J. Virol. 2015, 89, 7660–7672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Huang, J.; Zhang, L.; Chen, J.; Zeng, Y.; Tang, Y.; Liu, Z. Advances in Cerebral Organoid Systems and Their Application in Disease Modeling. Neuroscience 2019, 399, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Moda, F.; Bolognesi, M.L.; Legname, G. Novel Screening Approaches for Human Prion Diseases Drug Discovery. Expert Opin. Drug Discov. 2019, 14, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Grenier, K.; Kao, J.; Diamandis, P. Three-Dimensional Modeling of Human Neurodegeneration: Brain Organoids Coming of Age. Mol. Psychiatry 2020, 25, 254–274. [Google Scholar] [CrossRef] [PubMed]
- Groveman, B.R.; Foliaki, S.T.; Orru, C.D.; Zanusso, G.; Carroll, J.A.; Race, B.; Haigh, C.L. Sporadic Creutzfeldt-Jakob Disease Prion Infection of Human Cerebral Organoids. Acta Neuropathol. Commun. 2019, 7, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman-Levi, Y.; Meiner, Z.; Canello, T.; Frid, K.; Kovacs, G.G.; Budka, H.; Avrahami, D.; Gabizon, R. Correction: Fatal Prion Disease in a Mouse Model of Genetic E200K Creutzfeldt-Jakob Disease. PLOS Pathog. 2017, 13, e1006294. [Google Scholar] [CrossRef] [Green Version]
- Foliaki, S.; Groveman, B.; Yuan, J.; Walters, R.; Zhang, S.; Tesar, P.; Zou, W.; Haigh, C. Pathogenic Prion Protein Isoforms Are Not Present in Cerebral Organoids Generated from Asymptomatic Donors Carrying the E200K Mutation Associated with Familial Prion Disease. Pathogens 2020, 9, 482. [Google Scholar] [CrossRef]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human IPSC-Based Modeling of Late-Onset Disease via Progerin-Induced Aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Taraboulos, A.; Serban, D.; Prusiner, S.B. Scrapie Prion Proteins Accumulate in the Cytoplasm of Persistently Infected Cultured Cells. J. Cell Biol. 1990, 110, 2117–2132. [Google Scholar] [CrossRef]
- Ghaemmaghami, S.; Phuan, P.W.; Perkins, B.; Ullman, J.; May, B.C.H.; Cohen, F.E.; Prusiner, S.B. Cell Division Modulates Prion Accumulation in Cultured Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 17971–17976. [Google Scholar] [CrossRef] [Green Version]
- Bate, C.; Langeveld, J.; Williams, A. Manipulation of PrPres Production in Scrapie-Infected Neuroblastoma Cells. J. Neurosci. Methods 2004, 138, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions Hijack Tunnelling Nanotubes for Intercellular Spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells Release Prions in Association with Exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collinge, J. Prion Strain Mutation and Selection. Science 2010, 328, 1111–1112. [Google Scholar] [CrossRef] [PubMed]
- Mahal, S.P.; Browning, S.; Li, J.; Suponitsky-Kroyter, I.; Weissmann, C. Transfer of a Prion Strain to Different Hosts Leads to Emergence of Strain Variants. Proc. Natl. Acad. Sci. USA 2010, 107, 22653–22658. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, D.; Homma, T.; Nakagaki, T.; Fuse, T.; Sano, K.; Takatsuki, H.; Atarashi, R.; Nishida, N. Strain-Dependent Effect of Macroautophagy on Abnormally Folded Prion Protein Degradation in Infected Neuronal Cells. PLoS ONE 2015, 10, e0137958. [Google Scholar] [CrossRef] [Green Version]
- Ghaemmaghami, S.; Ahn, M.; Lessard, P.; Giles, K.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Continuous Quinacrine Treatment Results in the Formation of Drug-Resistant Prions. PLoS Pathog. 2009, 5, e1000673. [Google Scholar] [CrossRef]
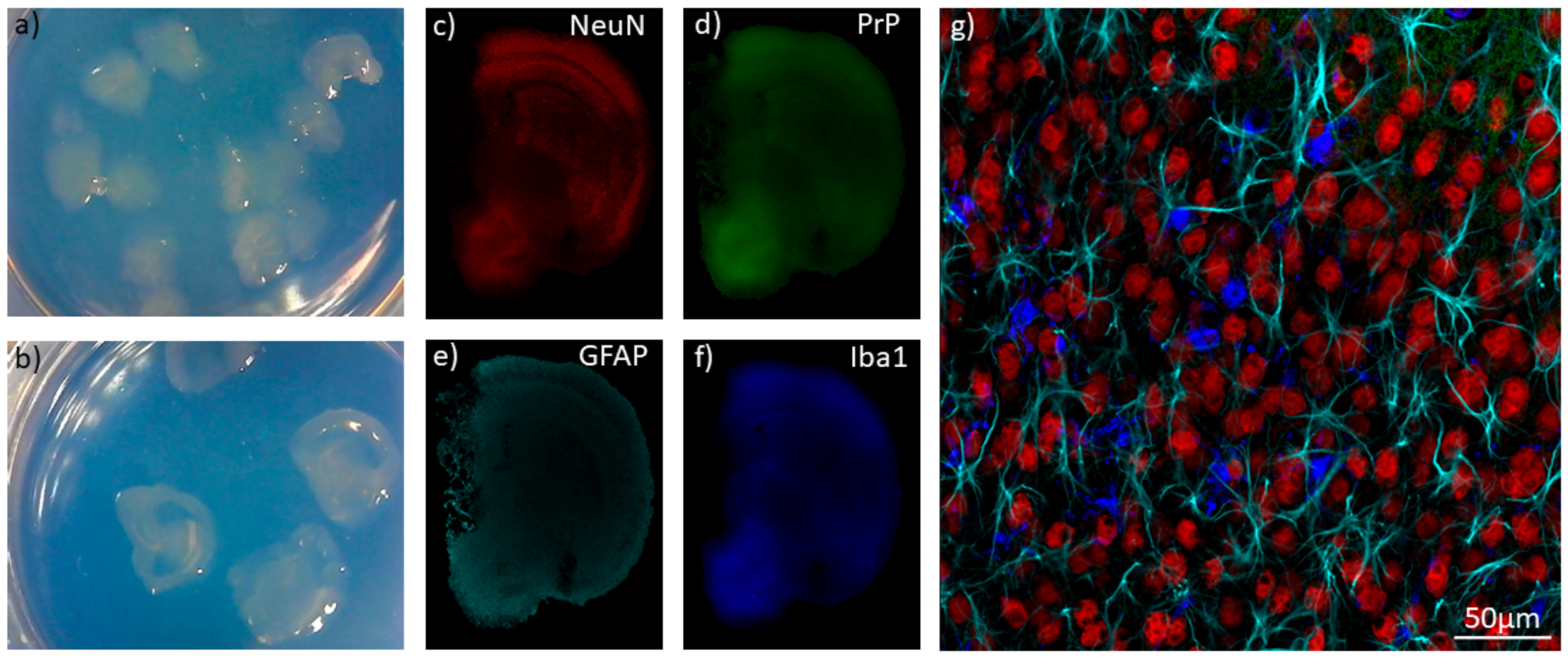

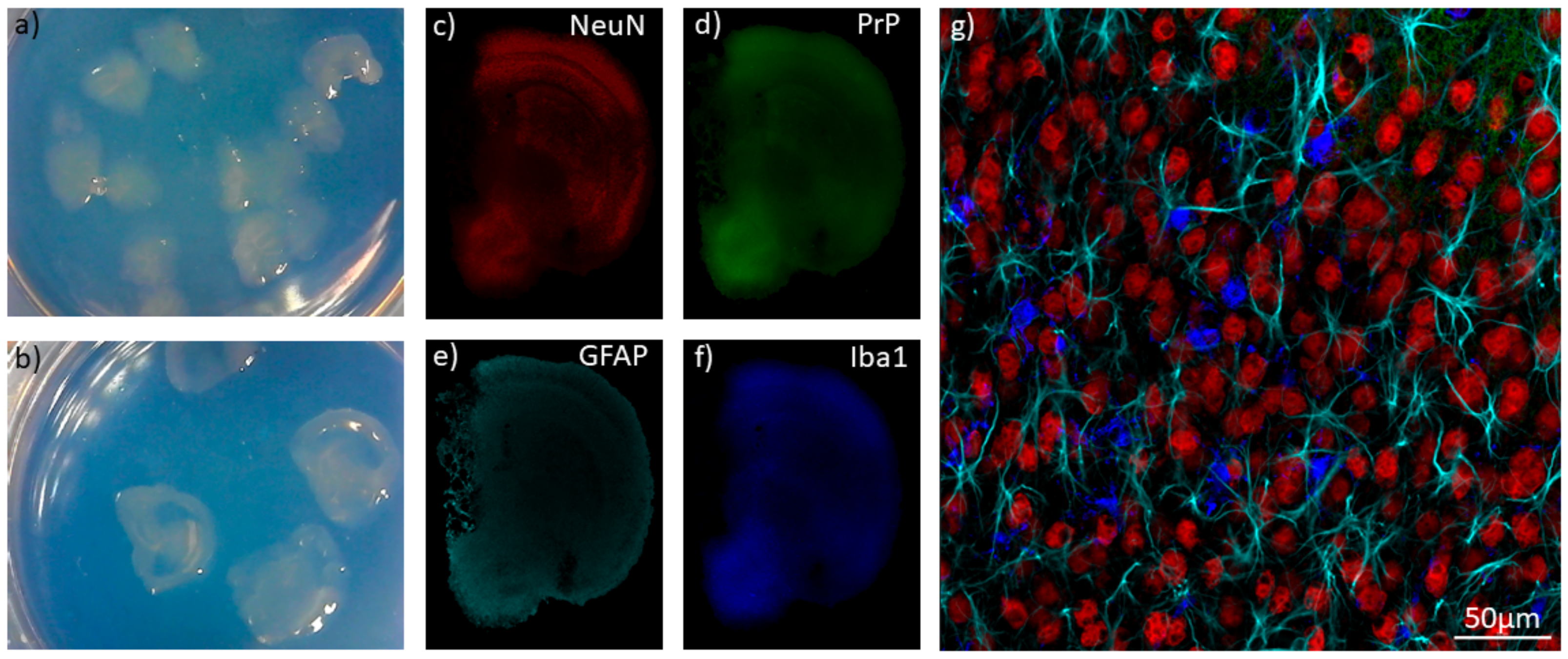{kind=link}
| Prion Disease | Strains |
|---|---|
| Natural sheep scrapie | Kanagawa Scrapie, Obihiro Scrapie, 127S, PG127, LA404 |
| Natural goat scrapie | At least 10 haplotypes |
| Mouse-adapted scrapie | Chandler, 139A, 79A, RML, 22L, 22F, 22A, ME7, 87V |
| Hamster-adapted scrapie | 263K, 139H |
| Rat-adapted scrapie | 139R |
| Bovine spongiform encephalopathy (BSE) | Typical (classic), atypical: H and L strains |
| Mouse-adapted BSE | 301C |
| Transmissible mink encephalopathy (TME) | May have originated as L-type BSE |
| Hamster-adapted TME | Hyper (HY), Drowsy (DY) |
| Cervid chronic wasting disease (CWD) | Species affected: Mule Deer (MD-CWD), White-Tailed Deer (WT-CWD), Elk, Moose |
| Human prion disease | Sporadic Creutzfeldt–Jakob disease (sCJD): subtypes MM1, MM2, MV1, MV2, VV1, VV2 Variant CJD (vCJD) Iatrogenic (iCJD) Genetic: gCJD, Gerstmann–Sträussler–Scheinker (GSS) syndrome, Fatal familial insomnia (FFI) |
| Mouse-adapted CJD | SY, M1000, FU |
| Mouse-adapted GSS | Fukuoka-1 (Fu-1) |
| Cell Line | Cell Type | PrP Species | Strain Propagation |
|---|---|---|---|
| SMB Cells | Scrapie mouse brain cells (from a mouse that was infected with Chandler) | Mouse | Persistently infected with Chandler [22] Pentosan sulfate cured cells can propagate 22F, 139A, 79A [23] CANNOT propagate 263K [23] |
| PC12 Cells | Rat phaeochromocytoma cells | Rat | 139A, ME7 [24,25] CANNOT propagate 263K, 139R [25] |
| N2a | Mouse neuroblastoma | Mouse | FU [26], Chandler, 22L, 139A [27] CANNOT propagate 87V, 22A [27] |
| N2a (PK1 Subclone) | Mouse | RML, 22L, 139A, 79A [28,29,30] CANNOT propagate ME7, 22A, 263K, 301C [28,29] | |
| N2a (R33 Subclone) | Mouse | 22L [28,29] CANNOT propagate ME7, RML, 22A, 263K, 301C [28,29] | |
| N2a (PME Subclones) | Mouse | 22L, RML, ME7 [31] | |
| CAD5 Cells | Mouse catecholaminergic | Mouse | 22L, RML, 139A, 79A, ME7, 301C [29,30] CANNOT propagate 263K [29] |
| Hamster | 263K, HY, 139H [32] CANNOT propagate DY [32] | ||
| Bank vole | 22L, MD-CWD, WT-CWD [33] | ||
| Cervid | MD-CWD, WT-CWD [33] | ||
| SN56 Cells | Mouse cholinergic septal neuronal | Mouse | RML, 22L, ME7 [34] CANNOT propagate 87V, 263K [34] |
| GT1 Cells | Mouse hypothalamic | Mouse | RML [35], 22L, Chandler, FU, SY-CJD [36], Kanagawa scrapie [37] CANNOT propagate 87V, 22A [27] |
| CRBL Cells | Mouse cerebellum | Mouse | 139A [38] CANNOT propagate RML [38] |
| 1C11 Cells | Mouse embryonal carcinoma (neuronal stem cells) | Mouse | Chandler, 22L, Fu-1 [39] CANNOT propagate ME7, 22A [39] |
| HpL3-4 Cells | Mouse hippocampal | Mouse | 22L [40] |
| CF10 Cells | Mouse neuronal | Mouse | 22L [41] |
| MG20 Cells | Mouse microglia | Mouse | Chandler, ME7, Obihiro scrapie, BSE agent [42] |
| MG6 Cells | Mouse microglia | Mouse | CANNOT propagate Chandler, ME7 [42] |
| hTERT Microglia | Mouse microglia | Sheep | natural scrapie [43] |
| C8D1A Cells | Mouse astrocytes | Mouse | 22L, RML [44] CANNOT propagate ME7 [44] |
| MovS6 and MovS2 Cells | Mouse Schwann | Sheep | Natural scrapie (VRQ allele) [45,46] |
| MSC-80 Cells | Mouse Schwann | Mouse | Chandler [47] |
| NIH/3T3 Cells | Mouse fibroblasts | Mouse | 22L [48] |
| L929 Cells | Mouse fibroblasts | Mouse | 22L, RML, 139A, 79A, ME7 [30,48] CANNOT propagate 87V, 301C [29,48] |
| MDB Cells | Mule deer meningeal fibroblasts | Mule deer | MD-CWD [49] |
| MEF Cells | Mouse embryonic fibroblasts | Bank vole | MD-CWD and WT-CWD [33] |
| Cervid | MD-CWD and WT-CWD [33] | ||
| RK13 Cells | Rabbit kidney epithelial | Mouse | Fu-1, Chandler, 22L [50,51] CANNOT propagate ME7 [50], M1000 mouse-adapted CJD, MM2 mouse-adapted CJD [52] |
| Bank Vole | Bank vole-adapted BSE [50] CANNOT propagate Ss3 (bank vole adapted sheep scrapie) [50] | ||
| Sheep (VRQ allele) | natural scrapie (VRQ allele) [46,51,53,54,55] CANNOT propagate Natural scrapie (some VRQ cases and other alleles) [46] | ||
| Goat (ARQ allele) | Goat scrapie (haplotype 1 or 2), Tg338-adapted goat scrapie (haplotype 3 and 4) [56] CANNOT propagate Goat scrapie (haplotype 3 or 4) [56] | ||
| Elk * | Elk CWD [57] CANNOT propagate D10 Deer CWD [57] | ||
| Human (MM) | CANNOT propagate MM2 CJD, M1000 mouse-adapted CJD, mouse-adapted MM2 CJD [52] | ||
| MDBK Cells | Madin–Darby bovine kidney cells | Bovine | Natural scrapie (VRQ and ARQ alleles) [58] CANNOT propagate BSE [58] |
| Bovine ** | BSE [59] | ||
| C2C12 Cells | Mouse myoblasts/myotubes | Mouse | RML, 22L, ME7 [60] CANNOT propagate Hyper strain of hamster-adapted mink encephalopathy [60] |
| tSP-SC Cells | Mouse stromal spleen cells (features of fibroblasts and mesenchymal cells) | Mouse | Fu-1 [61] |
| BMSC/336, O1BM and O2BM Cells | Adipocyte-like cells (derived from mouse bone marrow) | Mouse | Fu-1 [62,63] |
| HK Cells | Follicular dendritic cells. | Human (VV) | CANNOT propagate vCJD, MV sCJD, VV sCJD [64] |
| Cell Type | PrP Species | Strain Propagation |
|---|---|---|
| Cerebellar granular neurons | Mouse | 22L, 139A, ME7, Fu-1 [86,87,88] |
| Hamster (tg7 mice) | Sc237 (Subclone of 263K) [86] | |
| Sheep (tg338 mice) | Natural scrapie, 139A [89] | |
| Human MM PrP (tg650 mice) | MM1 iCJD, MM1 sCDJ, MM vCJD [21] | |
| Human VV PrP (tg152 mice) | VV2 sCJD [21] | |
| Striatal neurons | Mouse | 22L, 139A [87] CANNOT propagate ME7 [87] |
| Cortical neurons | Mouse | 22L, 139A, Chandler [87,90] CANNOT propagate ME7 [87] |
| Hippocampal neurons | Mouse | RML, 22L [91,92] |
| Primary astrocytes | Mouse | 22L [88,92] |
| Sheep (tg338 mice) | Natural scrapie, 139A [89] | |
| Human iPSC-derived astrocytes | Human (MM) | MM vCJD, MM1 sCJD, VV2 sCJD [85] |
| Human (MV) | MM vCJD [85] | |
| Human (VV) | VV2 sCJD [85] CANNOT propagate: MM1 sCJD, MM vCJD, VV1 sCJD [85] |
| Immortalized Cell Culture | Primary Culture | Stem Cell-Derived Cultures | Neuro-Spheres, Brain Aggregates | Organoids | Organotypic Slice Culture | |
|---|---|---|---|---|---|---|
| Difficulty /Cost | Low | Medium | Medium | Medium | High | Medium-High |
| Time | Days to Weeks | Days to Weeks | Days to Weeks | Weeks to Months | Months to Years | Weeks to Months |
| Genetics | Often genetically unstable Human cell lines available | Broad choice of transgenic animals | Human genetics possible | Broad choice of transgenic animals | Human genetics possible | Broad choice of transgenic animals |
| Regenerative capacity | Yes | Depends on cell type | Depends on cell type | Yes | No | No |
| Cell diversity | Low Usually one cell type | Low Usually one cell type | Low Usually one cell type | High Most neural cell types present | Medium Cells derived from neural progenitor cells become more diverse over time | High All neural cell types present |
| Cytoarchitecture | Low monolayer | Low monolayer | Low monolayer | Medium 3D arrangement of cells with some in vivo-like connections | Medium-High Cell populations migrate to resemble in vivo architecture over time Not all regions present | High Mature, in vivo-like cytoarchitecture |
| Strain permissiveness | Low | Medium | Medium | High | More investigation needed | High |
| Pathology | Usually none Infected GT1 cells had abnormal morphology, autophagic vacuoles, DNA fragmentation [40] | Often increased apoptosis [79,81,83] | Increased apoptosis [89] | Increased membrane permeability, astrocyte activation [90] Loss of dendritic spines [88] | Increased neuroinflammation (cytokine release) [109] | Neuronal loss, activation of microglia and astrocytes, spongiform vacuolation [100] Loss of dendritic spines [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pineau, H.; Sim, V.L. From Cell Culture to Organoids-Model Systems for Investigating Prion Strain Characteristics. Biomolecules 2021, 11, 106. https://doi.org/10.3390/biom11010106
Pineau H, Sim VL. From Cell Culture to Organoids-Model Systems for Investigating Prion Strain Characteristics. Biomolecules. 2021; 11(1):106. https://doi.org/10.3390/biom11010106
Chicago/Turabian StylePineau, Hailey, and Valerie L. Sim. 2021. "From Cell Culture to Organoids-Model Systems for Investigating Prion Strain Characteristics" Biomolecules 11, no. 1: 106. https://doi.org/10.3390/biom11010106
APA StylePineau, H., & Sim, V. L. (2021). From Cell Culture to Organoids-Model Systems for Investigating Prion Strain Characteristics. Biomolecules, 11(1), 106. https://doi.org/10.3390/biom11010106