Brain Disorders and Chemical Pollutants: A Gap Junction Link? †




Abstract
:
1. Introduction
2. Gap Junction and Connexin Roles in Brain
2.1. Brain Development and Connexin Expression
2.2. Adult Brain Organization and Connexin Involvement
2.2.1. Gap-Junction Compartments in Brain
The Neuronal Gap-Junction Compartment
The Panglial Syncytium
The Vascular Gap-Junction Compartment
Other Putative Gap-Junction Compartments in Brain
- Stem Cell Niches
- ii.
- Microglia
2.2.2. Connexin Paracrine/Autocrine Functions in Brain
Connexin Paracrine/Autocrine Communication and Neuronal Activity
Connexin Paracrine/Autocrine Communication and Microglia Activation
2.3. Possible Consequences of Connexin Dysfunction in Brain
3. Inhibition of Gap-Junctional Intercellular Communication by Environmental Chemical Pollutants
3.1. Pesticides
3.1.1. Insecticides
Organochlorine Insecticides
Toxaphene
3.1.2. Herbicides
3.1.3. Fungicides
3.2. Bisphenol A
3.3. Phthalates
3.4. Others
4. Pathologies
4.1. Neurodevelopmental Disorders
4.1.1. Autism Spectrum Disorders
4.1.2. Attention Deficit Hyperactivity Disorders
4.1.3. Epilepsy
4.2. Neurodegenerative Disorders
4.2.1. Parkinson’s Disease
4.2.2. Alzheimer’s Disease
4.3. Neurobehavioral Disorders
4.3.1. Migraines
4.3.2. Major Depressive Disorders
4.4. Glioma
5. Discussion
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef] [Green Version]
- Kleihues, P.; Barnholtz-Sloan, J.; Ohgaki, H. Tumours of the nervous system. In World Cancer Report 2014; Stewart, B.W., Wild, C.P., Eds.; International Agency for Research on Cancer: Lyon, France, 2014; pp. 511–521. [Google Scholar]
- Gage, F.H. Adult neurogenesis in mammals. Science 2019, 364, 827–828. [Google Scholar] [CrossRef]
- Luo, Y.; Weibman, D.; Halperin, J.M.; Li, X. A Review of Heterogeneity in Attention Deficit/Hyperactivity Disorder (ADHD). Front Hum Neurosci. 2019, 13, 42. [Google Scholar] [CrossRef] [Green Version]
- Polanczyk, G.V.; Salum, G.A.; Sugaya, L.S.; Caye, A.; Rohde, L.A. Annual research review: A meta-analysis of the worldwide prevalence of mental disorders in children and adolescents. J. Child Psychol. Psychiatry 2015, 56, 345–365. [Google Scholar] [CrossRef]
- Bhandari, R.; Paliwal, J.K.; Kuhad, A. Neuropsychopathology of Autism Spectrum Disorder: Complex Interplay of Genetic, Epigenetic, and Environmental Factors. Adv. Neurobiol. 2020, 24, 97–141. [Google Scholar]
- Lyall, K.; Croen, L.; Daniels, J.; Fallin, M.D.; Ladd-Acosta, C.; Lee, B.K.; Park, B.Y.; Snyder, N.W.; Schendel, D.; Volk, H.; et al. The Changing Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2017, 20, 81–102. [Google Scholar] [CrossRef] [Green Version]
- Elsabbagh, M.; Divan, G.; Koh, Y.J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.; Wang, C.; et al. Global Prevalence of Autism and Other Pervasive Developmental Disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [Green Version]
- Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 20 June 2019).
- Hurd, M.D.; Martorell, P.; Delavande, A.; Mullen, K.J.; Langa, K.M. Monetary costs of dementia in the United States. N. Engl. J. Med. 2013, 368, 1326–1334. [Google Scholar] [CrossRef] [Green Version]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Beitz, J.M. Parkinson’s disease: A review. Front. Biosci. 2014, 6, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Vandeleur, C.L.; Fassassi, S.; Castelao, E.; Glaus, J.; Strippoli, M.F.; Lasserre, A.M.; Rudaz, D.; Gebreab, S.; Pistis, G.; Aubry, J.M.; et al. Prevalence and correlates of DSM-5 major depressive and related disorders in the community. Psychiatry Res. 2017, 250, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, E.R.; Druss, B.G. Rate and Predictors of Persistent Major Depressive Disorder in a Nationally Representative Sample. Community Ment. Health J. 2015, 51, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Leso, V.; Gervetti, P.; Mauro, S.; Macrini, M.C.; Ercolano, M.L.; Iavicoli, I. Shift work and migraine: A systematic review. J. Occup. Health 2020, 62, e12116. [Google Scholar] [CrossRef] [Green Version]
- Willecke, K.; Eiberger, J.; Degen, J.; Eckardt, D.; Romualdi, A.; Güldenagel, M.; Deutsch, U.; Söhl, G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 2002, 383, 725–737. [Google Scholar] [CrossRef]
- Nakase, T.; Naus, C.C. Gap junctions and neurological disorders of the central nervous system. Biochim. Biophys. Acta 2004, 1662, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Bruzzone, R. Learning the language of cell-cell communication through connexin channels. Genome Biol. 2001, 2, REPORTS4027. [Google Scholar] [CrossRef]
- Thompson, R.J.; Macvicar, B.A. Connexin and pannexin hemichannels of neurons and astrocytes. Channels 2008, 2, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Kreuzberg, M.M.; Deuchars, J.; Weiss, E.; Schober, A.; Sonntag, S.; Wellershaus, K.; Draguhn, A.; Willecke, K. Expression of connexin30.2 in interneurons of the central nervous system in the mouse. Mol. Cell. Neurosci. 2008, 37, 119–134. [Google Scholar] [CrossRef]
- Dere, E.; Zheng-Fischhöfer, Q.; Viggiano, D.; Gironi Carnevale, U.A.; Ruocco, L.A.; Zlomuzica, A.; Schnichels, M.; Willecke, K.; Huston, J.P.; Sadile, A.G. Connexin31.1 deficiency in the mouse impairs object memory and modulates open-field exploration, acetylcholine esterase levels in the striatum, and cAMP response element-binding protein levels in the striatum and piriform cortex. Neuroscience 2008, 153, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Swayne, L.A.; Bennett, S.A.L. Connexins and pannexins in neuronal development and adult neurogenesis. BMC Cell Biol. 2016, 17 (Suppl. 1), 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belluardo, N.; Mudò, G.; Trovato-Salinaro, A.; Le Gurun, S.; Charollais, A.; Serre-Beinier, V.; Amato, G.; Haefliger, J.A.; Meda, P.; Condorelli, D.F. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000, 865, 121–138. [Google Scholar] [CrossRef]
- Condorelli, D.F.; Belluardo, N.; Trovato-Salinaro, A.; Mudò, G. Expression of Cx36 in mammalian neurons. Brain Res. Rev. 2000, 32, 72–85. [Google Scholar] [CrossRef]
- Nagy, J.I.; Pereda, A.E.; Rash, J.E. Electrical synapses in mammalian CNS: Past eras, present focus and future directions. Biochim. Biophys. Acta Biomembr. 2018, 1860, 102–123. [Google Scholar] [CrossRef] [PubMed]
- Schock, S.C.; Leblanc, D.; Hakim, A.M.; Thompson, C.S. ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro. Biochem. Biophys. Res. Commun. 2008, 368, 138–144. [Google Scholar] [CrossRef]
- Venance, L.; Glowinski, J.; Giaume, C. Electrical and chemical transmission between striatal GABAergic output neurones in rat brain slices. J. Physiol. 2004, 559, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Dedek, K.; Schultz, K.; Pieper, M.; Dirks, P.; Maxeiner, S.; Willecke, K.; Weiler, R.; Janssen-Bienhold, U. Localization of heterotypic gap junctions composed of connexin45 and connexin36 in the rod pathway of the mouse retina. Eur. J. Neurosci. 2006, 24, 1675–1686. [Google Scholar] [CrossRef]
- Schubert, T.; Maxeiner, S.; Krüger, O.; Willecke, K.; Weiler, R. Connexin45 mediates gap junctional coupling of bistratified ganglion cells in the mouse retina. J. Comp. Neurol. 2005, 490, 29–39. [Google Scholar] [CrossRef]
- Van Der Giessen, R.S.; Maxeiner, S.; French, P.J.; Willecke, K.; De Zeeuw, C.I. Spatiotemporal distribution of Connexin45 in the olivocerebellar system. J. Comp. Neurol. 2006, 495, 173–184. [Google Scholar] [CrossRef]
- Hombach, S.; Janssen-Bienhold, U.; Söhl, G.; Schubert, T.; Büssow, H.; Ott, T.; Weiler, R.; Willecke, K. Functional expression of connexin57 in horizontal cells of the mouse retina. Eur. J. Neurosci. 2004, 19, 2633–2640. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Prado, N.; Sonntag, S.; Skeberdis, V.A.; Willecke, K.; Bukauskas, F.F. Gating, permselectivity and pH-dependent modulation of channels formed by connexin57, a major connexin of horizontal cells in the mouse retina. J. Physiol. 2009, 587, 3251–3269. [Google Scholar] [CrossRef] [PubMed]
- Ciolofan, C.; Lynn, B.; Wellershaus, K.; Willecke, K.; Nagy, J.I. Spatial relationships of connexin36, connexin57 and zonula occludens-1 in the outer plexiform layer of mouse retina. Neuroscience 2007, 148, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Frisch, C.; De Souza-Silva, M.A.; Söhl, G.; Güldenagel, M.; Willecke, K.; Huston, J.P.; Dere, E. Stimulus complexity dependent memory impairment and changes in motor performance after deletion of the neuronal gap junction protein connexin36 in mice. Behav. Brain Res. 2005, 157, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Belousov, A.B. Deletion of neuronal gap junction protein connexin 36 impairs hippocampal LTP. Neurosci. Lett. 2011, 502, 30–32. [Google Scholar] [CrossRef] [Green Version]
- Hempelmann, A.; Heils, A.; Sander, T. Confirmatory evidence for an association of the connexin-36 gene with juvenile myoclonic epilepsy. Epilepsy Res. 2006, 71, 223–228. [Google Scholar] [CrossRef]
- Mas, C.; Taske, N.; Deutsch, S.; Guipponi, M.; Thomas, P.; Covanis, A.; Friis, M.; Kjeldsen, M.J.; Pizzolato, G.P.; Villemure, J.G.; et al. Association of the connexin36 gene with juvenile myoclonic epilepsy. J. Med. Genet. 2004, 41, e93. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.I.; Patel, D.; Ochalski, P.A.; Stelmack, J.L. Connexin30 in rodent, cat and human brain: Selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience 1999, 88, 447–468. [Google Scholar] [CrossRef]
- Nagy, J.I.; Li, X.; Rempel, J.; Stelmack, G.; Patel, D.; Staines, W.A.; Yasumura, T.; Rash, J.E. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J. Neurosci. 2001, 21, 1983–2000. [Google Scholar]
- Houades, V.; Koulakoff, A.; Ezan, P.; Seif, I.; Giaume, C. Gap junction-mediated astrocytic networks in the mouse barrel cortex. J. Neurosci. 2008, 28, 5207–5217. [Google Scholar] [CrossRef]
- Filippov, M.A.; Hormuzdi, S.G.; Fuchs, E.C.; Monyer, H. A reporter allele for investigating connexin 26 gene expression in the mouse brain. Eur. J. Neurosci. 2003, 18, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Montero, T.D.; Orellana, J.A. Hemichannels: New pathways for gliotransmitter release. Neuroscience 2015, 286, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow. Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neurosci. 2004, 129, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallraff, A.; Köhling, R.; Heinemann, U.; Theis, M.; Willecke, K.; Steinhäuser, C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 2006, 26, 5438–5447. [Google Scholar] [CrossRef] [Green Version]
- Pannasch, U.; Vargová, L.; Reingruber, J.; Ezan, P.; Holcman, D.; Giaume, C.; Syková, E.; Rouach, N. Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 8467–8472. [Google Scholar] [CrossRef] [Green Version]
- Ezan, P.; André, P.; Cisternino, S.; Saubaméa, B.; Boulay, A.C.; Doutremer, S.; Thomas, M.A.; Quenech’du, N.; Giaume, C.; Cohen-Salmon, M. Deletion of astroglial connexins weakens the blood-brain barrier. J. Cereb. Blood Flow. Metab. 2012, 32, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Sáez, P.J.; Sáez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Abudara, V.; Roux, L.; Dallérac, G.; Matias, I.; Dulong, J.; Mothet, J.P.; Rouach, N.; Giaume, C. Activated microglia impairs neuroglial interaction by opening Cx43 hemichannels in hippocampal astrocytes. Glia 2015, 63, 795–811. [Google Scholar] [CrossRef]
- Rusakov, D.A. Depletion of extracellular Ca2+ prompts astroglia to moderate synaptic network activity. Sci. Signal. 2012, 5, pe4. [Google Scholar] [CrossRef] [Green Version]
- Contreras, J.E.; Sáez, J.C.; Bukauskas, F.F.; Bennett, M.V. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad. Sci. USA 2003, 100, 11388–11393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovegno, M.; Sáez, J.C. Role of astrocyte connexin hemichannels in cortical spreading depression. Biochim. Biophys. Acta Biomembr. 2018, 1860, 216–223. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Decrock, E.; Wang, N.; Bol, M.; Vinken, M.; Bultynck, G.; Leybaert, L. The dual face of connexin-based astroglial Ca(2+) communication: A key player in brain physiology and a prime target in pathology. Biochim. Biophys. Acta 2014, 1843, 2211–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chever, O.; Pannasch, U.; Ezan, P.; Rouach, N. Astroglial connexin 43 sustains glutamatergic synaptic efficacy. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theis, M.; Jauch, R.; Zhuo, L.; Speidel, D.; Wallraff, A.; Doring, B.; Frisch, C.; Sohl, G.; Teubner, B.; Euwens, C.; et al. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J. Neurosci. 2003, 23, 766–776. [Google Scholar] [CrossRef]
- Frisch, C.; Theis, M.; De Souza Silva, M.A.; Dere, E.; Söhl, G.; Teubner, B.; Namestkova, K.; Willecke, K.; Huston, J.P. Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur. J. Neurosci. 2003, 18, 2313–2318. [Google Scholar] [CrossRef]
- Dere, E.; De Souza-Silva, M.A.; Frisch, C.; Teubner, B.; Söhl, G.; Willecke, K.; Huston, J.P. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur. J. Neurosci. 2003, 18, 629–638. [Google Scholar] [CrossRef]
- Lutz, S.E.; Zhao, Y.; Gulinello, M.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci. 2009, 29, 7743–7752. [Google Scholar] [CrossRef] [Green Version]
- De Bock, M.; Kerrebrouck, M.; Wang, N.; Leybaert, L. Neurological manifestations of oculodentodigital dysplasia: A Cx43 channelopathy of the central nervous system? Front. Pharm. 2013, 4, 120. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ionescu, A.V.; Lynn, B.D.; Lu, S.; Kamasawa, N.; Morita, M.; Davidson, K.G.; Yasumura, T.; Rash, J.E.; Nagy, J.I. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience 2004, 126, 611–630. [Google Scholar] [CrossRef] [Green Version]
- Kamasawa, N.; Sik, A.; Morita, M.; Yasumura, T.; Davidson, K.G.; Nagy, J.I.; Rash, J.E. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: Implications for ionic homeostasis and potassium siphoning. Neuroscience 2005, 136, 65–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagiava, A.; Theophilidis, G.; Sargiannidou, I.; Kyriacou, K.; Kleopa, K.A. Oxaliplatin-induced neurotoxicity is mediated through gap junction channels and hemichannels and can be prevented by octanol. Neuropharmacology 2015, 97, 289–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, J.I.; Ionescu, A.V.; Lynn, B.D.; Rash, J.E. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia 2003, 44, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odermatt, B.; Wellershaus, K.; Wallraff, A.; Seifert, G.; Degen, J.; Euwens, C.; Fuss, B.; Büssow, H.; Schilling, K.; Steinhäuser, C.; et al. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 2003, 23, 4549–4559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglione, M.; Tress, O.; Haas, B.; Karram, K.; Trotter, J.; Willecke, K.; Kettenmann, H. Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32. Glia 2010, 58, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Hombach, S.; Degen, J.; Odermatt, B. The oligodendroglial precursor cell line Oli-neu represents a cell culture system to examine functional expression of the mouse gap junction gene connexin29 (Cx29). Front. Pharmacol. 2013, 4, 83. [Google Scholar] [CrossRef] [Green Version]
- Basu, R.; Sarma, J.D. Connexin 43/47 channels are important for astrocyte/ oligodendrocyte cross-talk in myelination and demyelination. J. Biosci. 2018, 43, 1055–1068. [Google Scholar] [CrossRef]
- Menichella, D.M.; Majdan, M.; Awatramani, R.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J. Neurosci. 2006, 26, 10984–10991. [Google Scholar] [CrossRef] [Green Version]
- Eiberger, J.; Kibschull, M.; Strenzke, N.; Schober, A.; Büssow, H.; Wessig, C.; Djahed, S.; Reucher, H.; Koch, D.A.; Lautermann, J.; et al. Expression pattern and functional characterization of connexin29 in transgenic mice. Glia 2006, 53, 601–611. [Google Scholar] [CrossRef]
- Sutor, B.; Schmolke, C.; Teubner, B.; Schirmer, C.; Willecke, K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin32-deficient mice. Cereb. Cortex. 2000, 10, 684–697. [Google Scholar] [CrossRef] [Green Version]
- Menichella, D.M.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Connexins are critical for normal myelination in the CNS. J. Neurosci. 2003, 23, 5963–5973. [Google Scholar] [CrossRef] [PubMed]
- Tress, O.; Maglione, M.; May, D.; Pivneva, T.; Richter, N.; Seyfarth, J.; Binder, S.; Zlomuzica, A.; Seifert, G.; Theis, M.; et al. Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J. Neurosci. 2012, 32, 7499–7518. [Google Scholar] [CrossRef] [PubMed]
- Bergoffen, J.; Scherer, S.S.; Wang, S.; Scott, M.O.; Bone, L.J.; Paul, D.L.; Chen, K.; Lensch, M.W.; Chance, P.F.; Fischbeck, K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 1993, 262, 2039–2042. [Google Scholar] [CrossRef]
- Sargiannidou, I.; Kim, G.H.; Kyriakoudi, S.; Eun, B.L.; Kleopa, K.A. A start codon CMT1X mutation associated with transient encephalomyelitis causes complete loss of Cx32. Neurogenetics 2015, 16, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Orthmann-Murphy, J.L.; Enriquez, A.D.; Abrams, C.K.; Scherer, S.S. Loss-of-function GJA12/Connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol. Cell. Neurosci. 2007, 34, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, L.; Inoue, K.; Helman, G.; Mora, S.; Maski, K.; Soul, J.S.; Bloom, M.; Evans, S.H.; Goto, Y.I.; Caldovic, L.; et al. GJC2 promoter mutations causing Pelizaeus-Merzbacher-like disease. Mol. Genet. Metab. 2014, 111, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Rash, J.E.; Yasumura, T.; Dudek, F.E. Ultrastructure, histological distribution, and freeze-fracture immunocytochemistry of gap junctions in rat brain and spinal cord. Cell Biol. Int. 1998, 22, 731–749. [Google Scholar] [CrossRef] [Green Version]
- De Bock, M.; Vandenbroucke, R.E.; Decrock, E.; Culot, M.; Cecchelli, R.; Leybaert, L. A new angle on blood-CNS interfaces: A role for connexins? FEBS Lett. 2014, 588, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, K.; Chiba, H.; Fujita, H.; Kojima, T.; Saito, T.; Endo, T.; Sawada, N. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell Physiol. 2006, 208, 123–132. [Google Scholar] [CrossRef]
- Li, H.; Yu, S.; Wang, R.; Sun, Z.; Zhou, X.; Zheng, L.; Yin, Z.; Sun, Y. Polymorphism of CONNEXIN37 gene is a risk factor for ischemic stroke in Han Chinese population. Lipids Health Dis. 2018, 17, 72. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, N.B.; Attwell, D.; Hall, C.N. Pericyte-mediated regulation of capillary diameter: A component of neurovascular coupling in health and disease. Front. Neuroenergetics 2010, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Dalkara, T.; Gursoy-Ozdemir, Y.; Yemisci, M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [Green Version]
- Dobrenis, K.; Chang, H.Y.; Pina-Benabou, M.H.; Woodroffe, A.; Lee, S.C.; Rozental, R.; Spray, D.C.; Scemes, E. Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. J. Neurosci. Res. 2005, 82, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, S.B.; Uy, B.; Perera, A.; Nicholson, L.F. AGEs-RAGE mediated up-regulation of connexin43 in activated human microglial CHME-5 cells. Neurochem. Int. 2012, 60, 640–651. [Google Scholar] [CrossRef]
- Eugenín, E.A.; Eckardt, D.; Theis, M.; Willecke, K.; Bennett, M.V.; Saez, J.C. Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-gamma and tumor necrosis factor-alpha. Proc. Natl. Acad. Sci. USA 2001, 98, 4190–4195. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Syed, M.; Kielian, T. Staphylococcus aureus-derived peptidoglycan induces Cx43 expression and functional gap junction intercellular communication in microglia. J. Neurochem. 2005, 95, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Orellana, J.A.; Shoji, K.F.; Abudara, V.; Ezan, P.; Amigou, E.; Sáez, P.J.; Jiang, J.X.; Naus, C.C.; Sáez, J.C.; Giaume, C. Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. 2011, 31, 4962–4977. [Google Scholar] [CrossRef]
- Goings, G.E.; Kozlowski, D.A.; Szele, F.G. Differential activation of microglia in neurogenic versus non-neurogenic regions of the forebrain. Glia 2006, 54, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, O.; Gutierrez-Fernandez, F.; Lopez-Virgen, V.; Collas-Aguilar, J.; Quinones-Hinojosa, A.; Garcia-Verdugo, J.M. Immunological regulation of neurogenic niches in the adult brain. Neuroscience 2012, 226, 270–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosher, K.I.; Andres, R.H.; Fukuhara, T.; Bieri, G.; Hasegawa-Moriyama, M.; He, Y.; Guzman, R.; Wyss-Coray, T. Neural progenitor cells regulate microglia functions and activity. Nat. Neurosci. 2012, 15, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, P.; Battista, D.; Depino, A.; Roca, V.; Graciarena, M.; Pitossi, F. The more you have, the less you get: The functional role of inflammation on neuronal differentiation of endogenous and transplanted neural stem cells in the adult brain. J. Neurochem. 2010, 112, 1368–1385. [Google Scholar] [CrossRef]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Goldman, J.E.; Sekino, Y.; Sato, K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J. Neurosci. 2014, 34, 2231–2243. [Google Scholar] [CrossRef] [Green Version]
- Pang, B.; Neijssen, J.; Qiao, X.; Janssen, L.; Janssen, H.; Lippuner, C.; Neefjes, J. Direct antigen presentation and gap junction mediated cross-presentation during apoptosis. J. Immunol. 2009, 183, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Temple, S. The development of neural stem cells. Nature 2001, 414, 112–117. [Google Scholar] [CrossRef]
- Heng, J.I.; Chariot, A.; Nguyen, L. Molecular layers underlying cytoskeletal remodelling during cortical development. Trends Neurosci. 2010, 33, 38–47. [Google Scholar] [CrossRef]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Kempermann, G.; Wiskott, L.; Gage, F.H. Functional significance of adult neurogenesis. Curr. Opin. Neurobiol. 2004, 14, 186–191. [Google Scholar] [CrossRef]
- Hsieh, J. Orchestrating transcriptional control of adult neurogenesis. Genes Dev. 2012, 26, 1010–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, M.; Magnanti, B.L.; Correia Carreira, S.; Yang, A.; Alamo-Hernández, U.; Riojas-Rodriguez, H.; Calamandrei, G.; Koppe, J.G.; Krayer von Krauss, M.; Keune, H.; et al. Chlorpyrifos and neurodevelopmental effects: A literature review and expert elicitation on research and policy. Environ. Health 2012, 11 (Suppl. 1), S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, M. Gap junctional communication in morphogenesis. Prog. Biophys. Mol. Biol. 2007, 94, 186–206. [Google Scholar] [CrossRef] [Green Version]
- Nadarajah, B.; Jones, AM.; Evans, W.H.; Parnavelas, J.G. Differential expression of connexins during neocortical development and neuronal circuit formation. J. Neurosci. 1997, 17, 3096–3111. [Google Scholar] [CrossRef] [Green Version]
- Cina, C.; Bechberger, J.F.; Ozog, M.A.; Naus, C.C. Expression of connexins in embryonic mouse neocortical development. J. Comp. Neurol. 2007, 504, 298–313. [Google Scholar] [CrossRef]
- Gulisano, M.; Parenti, R.; Spinella, F.; Cicirata, F. Cx36 is dynamically expressed during early development of mouse brain and nervous system. Neuroreport 2000, 11, 3823–3828. [Google Scholar] [CrossRef]
- Söhl, G.; Eiberger, J.; Jung, Y.T.; Kozak, C.A.; Willecke, K. The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. Biol. Chem. 2001, 382, 973–978. [Google Scholar] [CrossRef]
- Lo, C.W. The role of gap junction membrane channels in development. J. Bioenerg. Biomembr. 1996, 28, 379–385. [Google Scholar] [CrossRef]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef] [Green Version]
- Elias, L.A.; Wang, D.D.; Kriegstein, A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nature 2007, 448, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.F.; Alcami, P.; Striedinger, K.M.; Spray, D.; Scemes, E. The carboxyl-terminal domain of connexin43 is a negative modulator of neuronal differentiation. J. Biol. Chem. 2010, 285, 11836–11845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartfield, E.M.; Rinaldi, F.; Glover, CP.; Wong, L.F.; Caldwell, M.A.; Uney, J.B. Connexin 36 expression regulates neuronal differentiation from neural progenitor cells. PLoS ONE 2011, 6, e14746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söhl, G.; Degen, J.; Teubner, B.; Willecke, K. The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Lett. 1998, 428, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, D.F.; Trovato-Salinaro, A.; Mudò, G.; Mirone, M.B.; Belluardo, N. Cellular expression of connexins in the rat brain: Neuronal localization, effects of kainate-induced seizures and expression in apoptotic neuronal cells. Eur. J. Neurosci. 2003, 18, 1807–1827. [Google Scholar] [CrossRef]
- Deans, M.R.; Gibson, J.R.; Sellitto, C.; Connors, B.W.; Paul, D.L. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron 2001, 31, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Elias, L.A.; Kriegstein, A.R. Gap junctions: Multifaceted regulators of embryonic cortical development. Trends Neurosci. 2008, 31, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Cina, C.; Maass, K.; Theis, M.; Willecke, K.; Bechberger, J.F.; Naus, C.C. Involvement of the cytoplasmic C-terminal domain of connexin43 in neuronal migration. J. Neurosci. 2009, 29, 2009–2021. [Google Scholar] [CrossRef] [Green Version]
- Qi, G.J.; Chen, Q.; Chen, L.J.; Shu, Y.; Bu, L.L.; Shao, X.Y.; Zhang, P.; Jiao, F.J.; Shi, J.; Tian, B. Phosphorylation of Connexin 43 by Cdk5 Modulates Neuronal Migration During Embryonic Brain Development. Mol. Neurobiol. 2016, 53, 2969–2982. [Google Scholar] [CrossRef]
- Vejar, S.; Oyarzún, J.E.; Retamal, M.A.; Ortiz, F.C.; Orellana, J.A. Connexin and Pannexin-Based Channels in Oligodendrocytes: Implications in Brain Health and Disease. Front. Cell. Neurosci. 2019, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- Peinado, A. Immature neocortical neurons exist as extensive syncytial networks linked by dendrodendritic electrical connections. J. Neurophysiol. 2001, 85, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Peinado, A.; Yuste, R.; Katz, L.C. Extensive dye coupling between rat neocortical neurons during the period of circuit formation. Neuron 1993, 10, 103–114. [Google Scholar] [CrossRef]
- Song, H.J.; Stevens, C.F.; Gage, F.H. Neural stem cells from adult hippocampus develop essential properties of functional CNS neurons. Nat. Neurosci. 2002, 5, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Weissman, T.A.; Riquelme, P.A.; Ivic, L.; Flint, A.C.; Kriegstein, A.R. Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 2004, 43, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuste, R.; Peinado, A.; Katz, L.C. Neuronal domains in developing neocortex. Science 1992, 257, 665–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuste, R.; Nelson, D.A.; Rubin, W.W.; Katz, L.C. Neuronal domains in developing neocortex: Mechanisms of coactivation. Neuron 1995, 14, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Kandler, K.; Katz, L.C. Coordination of neuronal activity in developing visual cortex by gap junction-mediated biochemical communication. J. Neurosci. 1998, 18, 1419–1427. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, H. Purinergic signaling in neural development. Semin. Cell. Dev. Biol. 2011, 22, 194–204. [Google Scholar] [CrossRef]
- Cavaliere, F.; Donno, C.; D’Ambrosi, N. Purinergic signaling: A common pathway for neural and mesenchymal stem cell maintenance and differentiation. Front. Cell. Neurosci. 2015, 9, 211. [Google Scholar] [CrossRef] [Green Version]
- Messemer, N.; Kunert, C.; Grohmann, M.; Sobottka, H.; Nieber, K.; Zimmermann, H.; Franke, H.; Nörenberg, W.; Straub, I.; Schaefer, M.; et al. P2X7 receptors at adult neural progenitor cells of the mouse subventricular zone. Neuropharmacology 2013, 73, 122–137. [Google Scholar] [CrossRef]
- Delarasse, C.; Gonnord, P.; Galante, M.; Auger, R.; Daniel, H.; Motta, I.; Kanellopoulos, J.M. Neural progenitor cell death is induced by extracellular ATP via ligation of P2X7 receptor. J. Neurochem. 2009, 109, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, M.D.; Gu, B.J.; Eamegdool, S.S.; Weible, M.W., 2nd.; Wiley, J.S.; Allen, D.G.; Chan-Ling, T. P2X7 receptors mediate innate phagocytosis by human neural precursor cells and neuroblasts. Stem Cells 2015, 33, 526–541. [Google Scholar] [CrossRef] [PubMed]
- Wiencken-Barger, A.E.; Djukic, B.; Casper, K.B.; McCarthy, K.D. A role for Connexin43 during neurodevelopment. Glia 2007, 55, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Pilaz, L.J.; Silver, D.L. Post-transcriptional regulation in corticogenesis: How RNA-binding proteins help build the brain. Wiley Interdiscip. Rev. RNA 2015, 6, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, F.A.; Carvalho, L.R.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.; Leite, R.E.; Jacob Filho, W.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The Human Brain in Numbers: A Linearly Scaled-up Primate Brain. Front Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Mayorquin, L.C.; Rodriguez, A.V.; Sutachan, J.J.; Albarracín, S.L. Connexin-Mediated Functional and Metabolic Coupling Between Astrocytes and Neurons. Front. Mol. Neurosci. 2018, 11, 118. [Google Scholar] [CrossRef]
- Johanson, C.E.; Stopa, E.G.; McMillan, P.N. The blood-cerebrospinal fluid barrier: Structure and functional significance. Methods Mol. Biol. 2011, 686, 101–131. [Google Scholar]
- Bruni, J.E. Ependymal development, proliferation, and functions: A review. Microsc. Res. Tech. 1998, 41, 2–13. [Google Scholar] [CrossRef]
- Del Bigio, M.R. The ependyma: A protective barrier between brain and cerebrospinal fluid. Glia 1995, 14, 1–13. [Google Scholar] [CrossRef]
- Del Bigio, M.R. Ependymal cells: Biology and pathology. Acta Neuropathol. 2010, 119, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Battefeld, A.; Klooster, J.; Kole, M.H. Myelinating satellite oligodendrocytes are integrated in a glial syncytium constraining neuronal high-frequency activity. Nat. Commun. 2016, 7, 11298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chever, O.; Dossi, E.; Pannasch, U.; Derangeon, M.; Rouach, N. Astroglial networks promote neuronal coordination. Sci Signal. 2016, 9, ra6. [Google Scholar] [CrossRef] [PubMed]
- Magnotti, L.M.; Goodenough, D.A.; Paul, D.L. Functional heterotypic interactions between astrocyte and oligodendrocyte connexins. Glia 2011, 59, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasseff, S.K.; Scherer, S.S. Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol. Dis. 2011, 42, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereda, A.E. Electrical synapses and their functional interactions with chemical synapses. Nat. Rev. Neurosci. 2014, 15, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E.; Staines, W.A.; Yasumura, T.; Patel, D.; Furman, C.S.; Stelmack, G.L.; Nagy, J.I. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc. Natl. Acad. Sci. USA 2000, 97, 7573–7578. [Google Scholar] [CrossRef] [Green Version]
- Rozental, R.; Morales, M.; Mehler, M.F.; Urban, M.; Kremer, M.; Dermietzel, R.; Kessler, J.A.; Spray, D.C. Changes in the properties of gap junctions during neuronal differentiation of hippocampal progenitor cells. J. Neurosci. 1998, 18, 1753–1762. [Google Scholar] [CrossRef] [Green Version]
- Vis, J.C.; Nicholson, L.F.; Faull, R.L.; Evans, W.H.; Severs, N.J.; Green, C.R. Connexin expression in Huntington’s diseased human brain. Cell. Biol. Int. 1998, 22, 837–847. [Google Scholar] [CrossRef]
- Weickert, S.; Ray, A.; Zoidl, G.; Dermietzel, R. Expression of neural connexins and pannexin1 in the hippocampus and inferior olive: A quantitative approach. Mol. Brain Res. 2005, 133, 102–109. [Google Scholar] [CrossRef]
- Kosaka, T. Neuronal gap junctions in the polymorph layer of the rat dentate gyrus. Brain Res. 1983, 277, 347–351. [Google Scholar] [CrossRef]
- Fukuda, T.; Kosaka, T. Gap junctions linking the dendritic network of GABAergic interneurons in the hippocampus. J. Neurosci. 2000, 20, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kosaka, T. The dual network of GABAergic interneurons linked by both chemical and electrical synapses: A possible infrastructure of the cerebral cortex. Neurosci. Res. 2000, 38, 123–130. [Google Scholar] [CrossRef]
- Apostolides, P.F.; Trussell, L.O. Regulation of interneuron excitability by gap junction coupling with principal cells. Nat. Neurosci. 2013, 16, 1764–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galarreta, M.; Hestrin, S. A network of fast-spiking cells in the neocortex connected by electrical synapses. Nature 1999, 402, 72–75. [Google Scholar] [CrossRef]
- Sevetson, J.; Haas, J.S. Asymmetry and modulation of spike timing in electrically coupled neurons. J. Neurophysiol. 2015, 113, 1743–1751. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.H.; Chen, N.; Wang, J.H. The coupling features of electrical synapses modulate neuronal synchrony in hypothalamic superachiasmatic nucleus. Brain Res. 2014, 1550, 9–17. [Google Scholar] [CrossRef]
- Landisman, C.E.; Long, M.A.; Beierlein, M.; Deans, M.R.; Paul, D.L.; Connors, B.W. Electrical synapses in the thalamic reticular nucleus. J. Neurosci. 2002, 22, 1002–1009. [Google Scholar] [CrossRef] [Green Version]
- Vandecasteele, M.; Glowinski, J.; Venance, L. Connexin mRNA expression in single dopaminergic neurons of substantia nigra pars compacta. Neurosci. Res. 2006, 56, 419–426. [Google Scholar] [CrossRef]
- Leussis, M.P.; Bolivar, V.J. Habituation in rodents: A review of behavior, neurobiology, and genetics. Neurosci. Biobehav. Rev. 2006, 30, 1045–1064. [Google Scholar] [CrossRef]
- Pannasch, U.; Rouach, N. Emerging role for astroglial networks in information processing: From synapse to behavior. Trends Neurosci. 2013, 36, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Orthmann-Murphy, J.L.; Freidin, M.; Fischer, E.; Scherer, S.S.; Abrams, C.K. Two Distinct Heterotypic Channels Mediate Gap Junction Coupling between Astrocyte and Oligodendrocyte Connexins. J. Neurosci. 2007, 27, 13949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Ochalski, A.; Hertzberg, E.L.; Nagy, J.I. LM and EM immunolocalization of the gap junctional protein connexin 43 in rat brain. Brain Res. 1990, 508, 313–319. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ochalski, A.; Hertzberg, E.L.; Nagy, J.I. On the organization of astrocytic gap junctions in rat brain as suggested by LM and EM immunohistochemistry of connexin43 expression. J. Comp. Neurol. 1990, 302, 853–883. [Google Scholar] [CrossRef]
- Giaume, C.; McCarthy, K.D. Control of gap-junctional communication in astrocytic networks. Trends Neurosci. 1996, 19, 319–325. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Urban-Maldonado, M.; Iacobas, S.; Scemes, E.; Spray, D.C. Array analysis of gene expression in connexin-43 null astrocytes. Physiol. Genom. 2003, 15, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Griemsmann, S.; Höft, S.P.; Bedner, P.; Zhang, J.; von Staden, E.; Beinhauer, A.; Degen, J.; Dublin, P.; Cope, D.W.; Richter, N.; et al. Characterization of Panglial Gap Junction Networks in the Thalamus, Neocortex, and Hippocampus Reveals a Unique Population of Glial Cells. Cereb. Cortex 2015, 25, 3420–3433. [Google Scholar] [CrossRef] [Green Version]
- Wolburg, H.; Rohlmann, A. Structure—Function relationships in gap junctions. Int. Rev. Cytol. 1995, 157, 315–373. [Google Scholar]
- Rash, J.E.; Duffy, H.S.; Dudek, F.E.; Bilhartz, B.L.; Whalen, L.R.; Yasumura, T. Grid-mapped freeze-fracture analysis of gap junctions in gray and white matter of adult rat central nervous system, with evidence for a “panglial syncytium” that is not coupled to neurons. J. Comp. Neurol. 1997, 388, 265–292. [Google Scholar] [CrossRef]
- Rouach, N.; Koulakoff, A.; Abudara, V.; Willecke, K.; Giaume, C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008, 322, 1551–1555. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Buckalew, R.; Du, Y.; Kiyoshi, C.M.; Alford, C.C.; Wang, W.; McTigue, D.M.; Enyeart, J.J.; Terman, D.; Zhou, M. Gap junction coupling confers isopotentiality on astrocyte syncytium. Glia 2016, 64, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibino, H.; Fujita, A.; Iwai, K.; Yamada, M.; Kurachi, Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J. Biol. Chem. 2004, 279, 44065–44073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rash, J.E.; Davidson, K.G.; Yasumura, T.; Furman, C.S. Freeze-fracture and immunogold analysis of aquaporin-4 (AQP4) square arrays, with models of AQP4 lattice assembly. Neuroscience 2004, 129, 915–934. [Google Scholar] [CrossRef] [Green Version]
- Benfenati, V.; Caprini, M.; Nicchia, G.P.; Rossi, A.; Dovizio, M.; Cervetto, C.; Nobile, M.; Ferroni, S. Carbenoxolone inhibits volume-regulated anion conductance in cultured rat cortical astroglia. Channels 2009, 3, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Ventura, R.; Harris, K.M. Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Kreczmanski, P.; Heinsen, H.; Mantua, V.; Woltersdorf, F.; Masson, T.; Ulfig, N.; Schmidt-Kastner, R.; Korr, H.; Steinbusch, H.W.M.; Hof, P.R.; et al. Microvessel lengthdensity, total length, and length perneuron in five subcortical regionsin schizophrenia. Acta Neuropathol. 2009, 117, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Kreczmanski, P.; Schmidt-Kastner, R.; Heinsen, H.; Steinbusch, H.W.M.; Hof, P.R.; Schmitz, C. Stereological studies of capillarylength density in the frontal cortexof schizophrenics. Acta Neuropathol. 2005, 109, 510–518. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Shepro, D.; Morel, N.M. Pericyte physiology. FASEB J. 1993, 7, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, D.; Katyshev, V.; Balabanov, R.D.; Borisov, A.; Dore-Duffy, P. The CNS microvascular pericyte: Pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS 2011, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Tajes, M.; Ramos-Fernández, E.; Weng-Jiang, X.; Bosch-Morató, M.; Guivernau, B.; Eraso-Pichot, A.; Salvador, B.; Fernàndez-Busquets, X.; Roquer, J.; Muñoz, F.J. The blood-brain barrier: Structure, function and therapeutic approaches to cross it. Mol. Membr. Biol. 2014, 31, 152–167. [Google Scholar] [CrossRef] [Green Version]
- Boulay, A.C.; Cisternino, S.; Cohen-Salmon, M. Immunoregulation at the gliovascular unit in the healthy brain: A focus on Connexin 43. Brain Behav. Immun. 2016, 56, 1–9. [Google Scholar] [CrossRef]
- Johnson, A.M.; Roach, J.P.; Hu, A.; Stamatovic, S.M.; Zochowski, M.R.; Keep, R.F.; Andjelkovic, A.V. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 2018, 32, 2615–2629. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Wallerstein, A.; Abdul-Muneer, P.M. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 2019, 317, 260–270. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016, 64, 1097–1123. [Google Scholar] [CrossRef] [PubMed]
- Osipova, E.D.; Semyachkina-Glushkovskaya, O.V.; Morgun, A.V.; Pisareva, N.V.; Malinovskaya, N.A.; Boitsova, E.B.; Pozhilenkova, E.A.; Belova, O.A.; Salmin, V.V.; Taranushenko, T.E.; et al. Gliotransmitters and cytokines in the control of blood-brain barrier permeability. Rev. Neurosci. 2018, 29, 567–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaria, R.N. The blood-brain barrier and cerebrovascular pathology in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1999, 893, 113–125. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Deane, R.; Sallstrom, J.; Chow, N.; Miano, J.M. Neurovascular pathways and Alzheimer amyloid beta-peptide. Brain Pathol. 2005, 15, 78–83. [Google Scholar] [CrossRef]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: Implications for drug therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef]
- Meyer, E.P.; Ulmann-Schuler, A.; Staufenbiel, M.; Krucker, T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 3587–3592. [Google Scholar] [CrossRef] [Green Version]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Lee, H.; Pienaar, I.S. Disruption of the blood-brain barrier in Parkinson’s disease: Curse or route to a cure? Front. Biosci. 2014, 19, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Bartels, A.L.; Willemsen, A.T.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural. Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiffert, E.; Dreier, J.P.; Ivens, S.; Bechmann, I.; Tomkins, O.; Heinemann, U.; Friedman, A. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J. Neurosci. 2004, 24, 7829–7836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oby, E.; Janigro, D. The blood-brain barrier and epilepsy. Epilepsia 2006, 47, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006, 129, 18–35. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.C. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J. Anat. 2002, 200, 639–646. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Saadoun, S.; Binder, D.K.; Manley, G.T.; Krishna, S.; Verkman, A.S. Molecular mechanisms of brain tumor edema. Neuroscience 2004, 129, 1011–1020. [Google Scholar] [CrossRef]
- Bronger, H.; König, J.; Kopplow, K.; Steiner, H.H.; Ahmadi, R.; Herold-Mende, C.; Keppler, D.; Nies, T. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005, 65, 11419–11428. [Google Scholar] [CrossRef] [Green Version]
- Reddy, O.C.; van der Werf, Y.D. The Sleeping Brain: Harnessing the Power of the Glymphatic System through Lifestyle Choices. Brain Sci. 2020, 10, 868. [Google Scholar] [CrossRef]
- Watanabe, C.; Imaizumi, T.; Kawai, H.; Suda, K.; Honma, Y.; Ichihashi, M.; Ema, M.; Mizutani, K.I. Aging of the Vascular System and Neural Diseases. Front. Aging Neurosci. 2020, 12, 557384. [Google Scholar] [CrossRef]
- Andreotti, J.P.; Silva, W.N.; Costa, A.C.; Picoli, C.C.; Bitencourt, F.C.O.; Coimbra-Campos, L.M.C.; Resende, R.R.; Magno, L.A.V.; Romano-Silva, M.A.; Mintz, A.; et al. Neural stem cell niche heterogeneity. Semin. Cell Dev. Biol. 2019, 95, 42–53. [Google Scholar] [CrossRef]
- Yoo, S.; Blackshaw, S. Regulation and function of neurogenesis in the adult mammalian hypothalamus. Prog. Neurobiol. 2018, 170, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Bruni, J.E.; Del Bigio, M.R.; Clattenburg, R.E. Ependyma: Normal and pathological. A review of the literature. Brain Res. 1985, 356, 1–19. [Google Scholar] [CrossRef]
- Jacquet, B.V.; Patel, M.; Iyengar, M.; Liang, H.; Therit, B.; Salinas-Mondragon, R.; Lai, C.; Olsen, J.C.; Anton, E.S.; Ghashghaei, H.T. Analysis of neuronal proliferation, migration and differentiation in the postnatal brain using equine infectious anemia virus-based lentiviral vectors. Gene Ther. 2009, 16, 1021–1033. [Google Scholar] [CrossRef]
- Kyrousi, C.; Arbi, M.; Pilz, G.A.; Pefani, D.E.; Lalioti, M.E.; Ninkovic, J.; Götz, M.; Lygerou, Z.; Taraviras, S. Mcidas and GemC1 are key regulators for the generation of multiciliated ependymal cells in the adult neurogenic niche. Development 2015, 142, 3661–3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzadeh, Z.; Merkle, F.T.; Soriano-Navarro, M.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell 2008, 3, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Paez-Gonzalez, P.; Abdi, K.; Luciano, D.; Liu, Y.; Soriano-Navarro, M.; Rawlins, E.; Bennett, V.; Garcia-Verdugo, J.M.; Kuo, C.T. Ank3-dependent SVZ niche assembly is required for the continued production of new neurons. Neuron 2011, 71, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Spassky, N.; Merkle, F.T.; Flames, N.; Tramontin, A.D.; García-Verdugo, J.M.; Alvarez-Buylla, A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J. Neurosci. 2005, 25, 10–18. [Google Scholar] [CrossRef]
- Doetsch, F.; Caillé, I.; Lim, D.A.; García-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [Green Version]
- Doetsch, F.; García-Verdugo, J.M.; Alvarez-Buylla, A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J. Neurosci. 1997, 17, 5046–5061. [Google Scholar] [CrossRef]
- García-Verdugo, J.M.; Doetsch, F.; Wichterle, H.; Lim, D.A.; Alvarez-Buylla, A. Architecture and cell types of the adult subventricular zone: In search of the stem cells. J. Neurobiol. 1998, 36, 234–248. [Google Scholar] [CrossRef]
- Bozoyan, L.; Khlghatyan, J.; Saghatelyan, A. Astrocytes control the development of the migration-promoting vasculature scaffold in the postnatal brain via VEGF signaling. J. Neurosci. 2012, 32, 1687–1704. [Google Scholar] [CrossRef] [Green Version]
- Platel, J.C.; Dave, K.A.; Gordon, V.; Lacar, B.; Rubio, M.E.; Bordey, A. NMDA receptors activated by subventricular zone astrocytic glutamate are critical for neuroblast survival prior to entering a synaptic network. Neuron 2010, 65, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Mason, H.A.; Ito, S.; Corfas, G. Extracellular signals that regulate the tangential migration of olfactory bulb neuronal precursors: Inducers, inhibitors, and repellents. J. Neurosci. 2001, 21, 7654–7663. [Google Scholar] [CrossRef]
- Arshad, A.; Vose, L.R.; Vinukonda, G.; Hu, F.; Yoshikawa, K.; Csiszar, A.; Brumberg, J.C.; Ballabh, P. Extended Production of Cortical Interneurons into the Third Trimester of Human Gestation. Cereb. Cortex 2016, 26, 2242–2256. [Google Scholar] [CrossRef] [Green Version]
- Paredes, M.F.; James, D.; Gil-Perotin, S.; Kim, H.; Cotter, J.A.; Ng, C.; Sandoval, K.; Rowitch, D.H.; Xu, D.; McQuillen, P.S.; et al. Extensive migration of young neurons into the infant human frontal lobe. Science 2016, 354, aaf7073. [Google Scholar] [CrossRef] [Green Version]
- Sanai, N.; Nguyen, T.; Ihrie, R.A.; Mirzadeh, Z.; Tsai, H.H.; Wong, M.; Gupta, N.; Berger, M.S.; Huang, E.; Garcia-Verdugo, J.M.; et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nat. Cell Biol. 2011, 478, 382–386. [Google Scholar] [CrossRef]
- Bergmann, O.; Liebl, J.; Bernard, S.; Alkass, K.; Yeung, M.S.; Steier, P.; Kutschera, W.; Johnson, L.; Landén, M.; Druid, H.; et al. The age of olfactory bulb neurons in humans. Neuron 2012, 74, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Quiñones-Hinojosa, A.; Sanai, N.; Soriano-Navarro, M.; Gonzalez-Perez, O.; Mirzadeh, Z.; Gil-Perotin, S.; Romero-Rodriguez, R.; Berger, M.S.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Cellular composition and cytoarchitecture of the adult human subventricular zone: A niche of neural stem cells. J. Comp. Neurol. 2006, 494, 415–434. [Google Scholar] [CrossRef]
- Wang, C.; Liu, F.; Liu, Y.Y.; Zhao, C.H.; You, Y.; Wang, L.; Zhang, J.; Wei, B.; Ma, T.; Zhang, Q.; et al. Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res. 2011, 21, 1534–1550. [Google Scholar] [CrossRef] [Green Version]
- LaMonica, B.E.; Lui, J.H.; Wang, X.; Kriegstein, A.R. OSVZ progenitors in the human cortex: An updated perspective on neurodevelopmental disease. Curr. Opin. Neurobiol. 2012, 22, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 2006, 26, 7907–7918. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, O.; Romero-Rodriguez, R.; Soriano-Navarro, M.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Epidermal growth factor induces the progeny of subventricular zone type B cells to migrate and differentiate into oligodendrocytes. Stem Cells 2009, 27, 2032–2043. [Google Scholar] [CrossRef] [Green Version]
- Talaverón, R.; Fernández, P.; Escamilla, B.; Pastor, A.M.; Matarredona, E.R.; Sáez, J.C. Neural progenitor cells isolated from the subventricular zone present hemichannel activity and form functional gap junctions with glial cells. Front. Cell. Neurosci. 2015, 9, 411. [Google Scholar] [CrossRef] [Green Version]
- Marins, M.; Xavier, A.L.R.; Viana, N.B.; Fortes, F.S.A.; Fróes, M.M.; Menezes, J.R.L. Gap junctions are involved in cell migration in the early postnatal subventricular zone. Dev. Neurobiol. 2009, 69, 715–730. [Google Scholar] [CrossRef]
- Freitas, A.S.; Xavier, A.L.R.; Furtado, C.M.; Hedin-Pereira, C.; Fróes, M.M.; Menezes, J.R.L. Dye coupling and connexin expression by cortical radial glia in the early postnatal subventricular zone. Dev. Neurobiol. 2012, 72, 1482–1497. [Google Scholar] [CrossRef]
- Lacar, B.; Young, S.Z.; Platel, J.C.; Bordey, A. Gap junction-mediated calcium waves define communication networks among murine postnatal neural progenitor cells. Eur. J. Neurosci. 2011, 34, 1895–1905. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Bolteus, A.J.; Balkin, D.M.; Henschel, O.; Bordey, A. GFAP-expressing cells in the postnatal subventricular zone display a unique glial phenotype intermediate between radial glia and astrocytes. Glia 2006, 54, 394–410. [Google Scholar] [CrossRef]
- Miragall, F.; Albiez, P.; Bartels, H.; de Vries, U.; Dermietzel, R. Expression of the gap junction protein connexin43 in the subependymal layer and the rostral migratory stream of the mouse: Evidence for an inverse correlation between intensity of connexin43 expression and cell proliferation activity. Cell. Tissue Res. 1997, 287, 243–253. [Google Scholar]
- Khodosevich, K.; Zuccotti, A.; Kreuzberg, M.M.; Le Magueresse, C.; Frank, M.; Willecke, K.; Monyer, H. Connexin45 modulates the proliferation of transit-amplifying precursor cells in the mouse subventricular zone. Proc. Natl. Acad. Sci. USA 2012, 109, 20107–20112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duval, N.; Gomès, D.; Calaora, V.; Calabrese, A.; Meda, P.; Bruzzone, R. Cell coupling and Cx43 expression in embryonic mouse neural progenitor cells. J. Cell Sci. 2002, 115, 3241–3251. [Google Scholar] [PubMed]
- Talaverón, R.; Matarredona, E.R.; de la Cruz, R.R.; Macías, D.; Gálvez, V.; Pastor, A.M. Implanted neural progenitor cells regulate glial reaction to brain injury and establish gap junctions with host glial cells. Glia 2014, 62, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Tang, H.; Cai, J.; Zhu, M.; Zhang, X.; Rao, M.; Mattson, M.P. Gap junctional communication is required to maintain mouse cortical neural progenitor cells in a proliferative state. Dev. Biol. 2004, 272, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemcke, H.; Kuznetsov, S.A. Involvement of connexin43 in the EGF/EGFR signalling during self-renewal and differentiation of neural progenitor cells. Cell. Signal. 2013, 25, 2676–2684. [Google Scholar] [CrossRef]
- Imbeault, S.; Gauvin, L.G.; Toeg, H.D.; Pettit, A.; Sorbara, C.D.; Migahed, L.; DesRoches, R.; Menzies, A.S.; Nishii, K.; Paul, D.L.; et al. The extracellular matrix controls gap junction protein expression and function in postnatal hippocampal neural progenitor cells. BMC Neurosci. 2009, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Rozental, R.; Mehler, M.F.; Morales, M.; Andrade-Rozental, A.F.; Kessler, J.A.; Spray, D.C. Differentiation of hippocampal progenitor cells in vitro: Temporal expression of intercellular coupling and voltage- and ligand-gated responses. Dev. Biol. 1995, 167, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Rozental, R.; Srinivas, M.; Gökhan, S.; Urban, M.; Dermietzel, R.; Kessler, J.A.; Spray, D.C.; Mehler, M.F. Temporal expression of neuronal connexins during hippocampal ontogeny. Brain Res. Rev. 2000, 32, 57–71. [Google Scholar] [CrossRef]
- Kunze, A.; Congreso, M.R.; Hartmann, C.; Wallraff-Beck, A.; Hüttmann, K.; Bedner, P.; Requardt, R.; Seifert, G.; Redecker, C.; Willecke, K.; et al. Connexin expression by radial glia-like cells is required for neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. USA 2009, 106, 11336–11341. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Deng, Y.; Bao, X.; Reuss, L.; Altenberg, G.A. Mechanism of the defect in gap-junctional communication by expression of a connexin 26 mutant associated with dominant deafness. FASEB J. 2005, 19, 1516–1518. [Google Scholar] [CrossRef]
- Retamal, M.A.; Cortés, C.J.; Reuss, L.; Bennett, M.V.L.; Sáez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, J.; Das, G.D. Post-natal origin of microneurones in the rat brain. Nat. Cell Biol. 1965, 207, 953–956. [Google Scholar] [CrossRef]
- Bauer, S.; Patterson, P.H. The cell cycle-apoptosis connection revisited in the adult brain. J. Cell Biol. 2005, 171, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakic, P. Neurogenesis in adult primates. Prog. Brain Res. 2002, 138, 3–14. [Google Scholar] [PubMed]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Fuentealba, L.C.; Obernier, K.; Alvarez-Buylla, A. Adult neural stem cells bridge their niche. Cell Stem Cell 2012, 10, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wake, H.; Moorhouse, A.J.; Miyamoto, A.; Nabekura, J. Microglia: Actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 2013, 36, 209–217. [Google Scholar] [CrossRef]
- Wu, Y.; Dissing-Olesen, L.; MacVicar, B.A.; Stevens, B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015, 36, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Bessis, A.; Béchade, C.; Bernard, D.; Roumier, A. Microglial control of neuronal death and synaptic properties. Glia 2007, 55, 233–238. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [Green Version]
- Béchade, C.; Cantaut-Belarif, Y.; Bessis, A. Microglial control of neuronal activity. Front. Cell. Neurosci. 2013, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Parenti, R.; Campisi, A.; Vanella, A.; Cicirata, F. Immunocytochemical and RT-PCR analysis of connexin36 in cultures of mammalian glial cells. Arch. Ital. Biol. 2002, 140, 101–108. [Google Scholar] [PubMed]
- Cepeda, C.; Chang, J.W.; Owens, G.C.; Huynh, M.N.; Chen, J.Y.; Tran, C.; Vinters, H.; Levine, M.S.; Mathern, G.W. In Rasmussen encephalitis, hemichannels associated with microglial activation are linked to cortical pyramidal neuron coupling: A possible mechanism for cellular hyperexcitability. CNS Neurosci. Ther. 2015, 21, 152–163. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Talaverón, R.; Pastor, A.M. Interactions Between Neural Progenitor Cells and Microglia in the Subventricular Zone: Physiological Implications in the Neurogenic Niche and After Implantation in the Injured Brain. Front. Cell. Neurosci. 2018, 12, 268. [Google Scholar] [CrossRef]
- Wasseff, S.K.; Scherer, S.S. Activated microglia do not form functional gap junctions in vivo. J. Neuroimmunol. 2014, 269, 90–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, A.D.; Eugenín, E.A.; Brañes, M.C.; Bennett, M.V.L.; Sáez, J.C. Identification of second messengers that induce expression of functional gap junctions in microglia cultured from newborn rats. Brain Res. 2002, 943, 191–201. [Google Scholar] [CrossRef]
- Maezawa, I.; Jin, L.W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef] [Green Version]
- Sáez, P.J.; Shoji, K.F.; Retamal, M.A.; Harcha, P.A.; Ramírez, G.; Jiang, J.X.; von Bernhardi, R.; Sáez, J.C. ATP is required and advances cytokine-induced gap junction formation in microglia in vitro. Mediat. Inflamm. 2013, 2013, 216402. [Google Scholar] [CrossRef]
- Martino, G.; Pluchino, S. The therapeutic potential of neural stem cells. Nat. Rev. Neurosci. 2006, 7, 395–406. [Google Scholar] [CrossRef]
- Ribeiro Xavier, A.; Kress, B.T.; Goldman, S.A.; Lacerda de Menezes, J.R.; Nedergaard, M. A Distinct Population of Microglia Supports Adult Neurogenesis in the Subventricular Zone. J. Neurosci. 2015, 35, 11848–11861. [Google Scholar] [CrossRef]
- Xavier, A.L.; Lima, F.R.S.; Nedergaard, M.; Menezes, J.R.L. Ontogeny of CX3CR1-EGFP expressing cells unveil microglia as an integral component of the postnatal subventricular zone. Front. Cell. Neurosci. 2015, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solano Fonseca, R.; Mahesula, S.; Apple, D.M.; Raghunathan, R.; Dugan, A.; Cardona, A.; O’Connor, J.; Kokovay, E. Neurogenic Niche Microglia Undergo Positional Remodeling and Progressive Activation Contributing to Age-Associated Reductions in Neurogenesis. Stem Cells Dev. 2016, 25, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Aarum, J.; Sandberg, K.; Haeberlein, S.L.; Persson, M.A. Migration and differentiation of neural precursor cells can be directed by microglia. Proc. Natl. Acad. Sci. USA 2003, 100, 15983–15988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, N.M.; Sutter, B.M.; Laywell, E.D.; Levkoff, L.H.; Kearns, S.M.; Marshall, G.P.; Scheffler, B.; Steindler, D.A. Microglia instruct subventricular zone neurogenesis. Glia 2006, 54, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Chong, A.; Yusta-Boyo, M.J.; Vergaño-Vera, E.; Bulfone, A.; de Pablo, F.; Vicario-Abejón, C. IGF-I promotes neuronal migration and positioning in the olfactory bulb and the exit of neuroblasts from the subventricular zone. Eur. J. Neurosci. 2009, 30, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.B.; Braunstein, T.H.; Nielsen, M.S.; MacAulay, N. Distinct permeation profiles of the connexin 30 and 43 hemichannels. FEBS Lett. 2014, 588, 1446–1457. [Google Scholar] [CrossRef]
- Medina-Ceja, L.; Salazar-Sánchez, J.C.; Ortega-Ibarra, J.; Morales-Villagrán, A. Connexins-Based Hemichannels/Channels and Their Relationship with Inflammation, Seizures and Epilepsy. Int. J. Mol. Sci. 2019, 20, 5976. [Google Scholar] [CrossRef] [Green Version]
- Stehberg, J.; Moraga-Amaro, R.; Salazar, C.; Becerra, A.; Echeverría, C.; Orellana, J.A.; Bultynck, G.; Ponsaerts, R.; Leybaert, L.; Simon, F.; et al. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 2012, 26, 3649–3657. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Davidson, J.O.; Green, C.R.; Nicholson, L.F.B.; O’Carroll, S.J.; Zhang, J. Connexins and Pannexins in cerebral ischemia. Biochim. Biophys. Acta Biomembr. 2018, 1860, 224–236. [Google Scholar] [CrossRef]
- Froger, N.; Orellana, J.A.; Calvo, C.F.; Amigou, E.; Kozoriz, M.G.; Naus, C.C.; Sáez, J.C.; Giaume, C. Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol. Cell. Neurosci. 2010, 45, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Reactive astrocytes in neural repair and protection. Neuroscientist 2005, 11, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Basilico, B.; Marrone, M.C.; Ragozzino, D. Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Semin Cell Dev Biol. 2019, 94, 138–151. [Google Scholar] [CrossRef]
- Mandolesi, G.; Musella, A.; Gentile, A.; Grasselli, G.; Haji, N.; Sepman, H.; Fresegna, D.; Bullitta, S.; De Vito, F.; Musumeci, G.; et al. Interleukin-1β alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosci. 2013, 33, 12105–12121. [Google Scholar] [CrossRef]
- Takaki, J.; Fujimori, K.; Miura, M.; Suzuki, T.; Sekino, Y.; Sato, K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: The ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J. Neuroinflammation 2012, 9, 275. [Google Scholar] [CrossRef] [Green Version]
- Karpuk, N.; Burkovetskaya, M.; Fritz, T.; Angle, A.; Kielian, T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J. Neurosci. 2011, 31, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Turecek, J.; Yuen, G.S.; Han, V.Z.; Zeng, X.H.; Bayer, K.U.; Welsh, J.P. NMDA receptor activation strengthens weak electrical coupling in mammalian brain. Neuron 2014, 81, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef]
- Schiza, N.; Sargiannidou, I.; Kagiava, A.; Karaiskos, C.; Nearchou, M.; Kleopa, K.A. Transgenic replacement of Cx32 in gap junction-deficient oligodendrocytes rescues the phenotype of a hypomyelinating leukodystrophy model. Hum. Mol. Genet. 2015, 24, 2049–2064. [Google Scholar] [CrossRef] [Green Version]
- May, D.; Tress, O.; Seifert, G.; Willecke, K. Connexin47 protein phosphorylation and stability in oligodendrocytes depend on expression of Connexin43 protein in astrocytes. J. Neurosci. 2013, 33, 7985–7996. [Google Scholar] [CrossRef]
- Trosko, J.E.; Yotti, L.P.; Warren, S.T.; Tsushimoto, G.; Chang, C. Inhibition of cell-cell communication by tumour promoters. Carcinog. Compr. Surv. 1982, 7, 565–585. [Google Scholar]
- Budunova, I.V.; Williams, G.M. Cell culture assays for chemicals with tumour-promoting or tumour-inhibiting activity based on the modulation of intercellular communication. Cell. Biol. Toxicol. 1994, 10, 71–116. [Google Scholar] [CrossRef]
- Sun, Y.; Nakashima, M.N.; Takahashi, M.; Kuroda, N.; Nakashima, K. Determination of bisphenol A in rat brain by microdialysis and column switching high-performance liquid chromatography with fluorescence detection. Biomed. Chromatogr. 2002, 16, 319–326. [Google Scholar] [CrossRef]
- Miyazaki, W.; Fujiwara, Y.; Katoh, T. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the development and function of the blood-brain barrier. Neurotoxicology 2016, 52, 64–71. [Google Scholar] [CrossRef]
- Defamie, N.; Mesnil, M. The modulation of gap-junctional intercellular communication by lipid rafts. Biochim. Biophys. Acta 2012, 1818, 1866–1869. [Google Scholar] [CrossRef]
- Oh, S. Bisphenol A and 4-tert-Octylphenol Inhibit Cx46 Hemichannel. Currents Korean J. Physiol. Pharmacol. 2015, 19, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.Y.; Wong, E.W.; Lie, P.P.; Li, M.W.; Su, L.; Siu, E.R.; Yan, H.; Mannu, J.; Mathur, P.P.; Bonanomi, M.; et al. Environmental toxicants and male reproductive function. Spermatogenesis 2011, 1, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.Y.; Wong, E.W.; Lie, P.P.; Li, M.W.; Mruk, D.D.; Yan, H.H.; Mok, K.W.; Mannu, J.; Mathur, P.P.; Lui, W.Y.; et al. Regulation of blood-testis barrier dynamics by desmosome, gap junction, hemidesmosome and polarity proteins: An unexpected turn of events. Spermatogenesis 2011, 1, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Tsushimoto, G.; Chang, C.C.; Trosko, J.E.; Matsumura, E. Cytotoxic, Mutagenic, and Cell-Cell Communication Inhibitory Properties of DDT, Lindane, and Chlordane on Chinese Hamster Cells in vitro. Arch. Environ. Contain. Toxicol. 1983, 12, 721–730. [Google Scholar] [CrossRef]
- Kang, K.S.; Wilson, M.R.; Hayashi, T.; Chang, C.C.; Trosko, J.E. Inhibition of gap junctional intercellular communication in normal human breast epithelial cells after treatment with pesticides, PCBs, and PBBs, alone or in mixtures. Environ. Health Persp. 1996, 104, 192–200. [Google Scholar]
- Ruch, R.J.; Klaunig, J.E. Effects of tumor promoters, genotoxic carcinogens and hepatocytotoxins on mouse hepatocyte intercellular communication. Cell. Biol. Toxicol. 1986, 2, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Criswell, K.A.; Loch-Caruso, R.; Stuenkel, E.L. Lindane inhibition of gap junctional communication in myometrial myocytes is partially dependent on phosphoinositide-generated second messengers. Toxicol. Appl. Pharmacol. 1995, 130, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Bonney, W.J.; Ruch, R.J. Changes in gap junction permeability, gap junction number, and connexin43 expression in lindane-treated rat liver epithelial cells. Toxicol. Appl. Pharmacol. 1995, 130, 79–86. [Google Scholar] [CrossRef]
- Defamie, N.; Mograbi, B.; Roger, C.; Cronier, L.; Malassine, A.; Brucker-Davis, F.; Fenichel, P.; Segretain, D.; Pointis, G. Disruption of gap junctional intercellular communication by lindane is associated with aberrant localization of connexin43 and zonula occludens-1 in 42GPA9 Sertoli cells. Carcinogenesis 2001, 22, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- Leithe, E.; Kjenseth, A.; Bruun, J.; Sirnes, S.; Rivedal, E. Inhibition of connexin43 gap junction channels by the endocrine disruptor ioxynil. Toxicol. Appl. Pharmacol. 2010, 247, 10–17. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, S.; Liu, L.; Hou, X.; Jiang, J.; Wei, X.; Hao, W. Effects of bisphenol A on gap junctions in HaCaT cells as mediated by the estrogen receptor pathway. J. Appl. Toxicol. 2019, 39, 271–281. [Google Scholar] [CrossRef]
- Salian, S.; Doshi, T.; Vanage, G. Neonatal exposure of male rats to Bisphenol A impairs fertility and expression of sertoli cell junctional proteins in the testis. Toxicology 2009, 265, 56–67. [Google Scholar] [CrossRef]
- Simecková, P.; Vondrácek, J.; Andrysík, Z.; Zatloukalová, J.; Krcmár, P.; Kozubík, A.; Machala, M. The 2,2′,4,4′,5,5′-hexachlorobiphenyl-enhanced degradation of connexin 43 involves both proteasomal and lysosomal activities. Toxicol. Sci. 2009, 107, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.K.; Rhee, S.K. Inhibitory effect of bisphenol A on gap junctional intercellular communication in an epithelial cell line of rat mammary tissue. Arch Pharm Res. 2007, 30, 337–343. [Google Scholar] [CrossRef]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef] [PubMed]
- Sovadinova, I.; Babica, P.; Böke, H.; Kumar, E.; Wilke, A.; Park, J.S.; Trosko, J.E.; Upham, B.L. Phosphatidylcholine Specific PLC-Induced Dysregulation of Gap Junctions, a Robust Cellular Response to Environmental Toxicants, and Prevention by Resveratrol in a Rat Liver Cell Model. PLoS ONE 2015, 10, e0124454. [Google Scholar] [CrossRef] [Green Version]
- Babica, P.; Zurabian, R.; Kumar, E.R.; Chopra, R.; Mianecki, M.J.; Park, J.S.; Jaša, L.; Trosko, J.E.; Upham, B.L. Methoxychlor and Vinclozolin Induce Rapid Changes in Intercellular and Intracellular Signaling in Liver Progenitor Cells. Toxicol. Sci. 2016, 153, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Čtveráčková, L.; Jančula, D.; Raška, J.; Babica, P.; Sovadinová, I. Structure-Dependent Effects of Phthalates on Intercellular and Intracellular Communication in Liver Oval Cells. Int. J. Mol. Sci. 2020, 21, E6069. [Google Scholar] [CrossRef] [PubMed]
- Machala, M.; Bláha, L.; Vondrácek, J.; Trosko, J.E.; Scott, J.; Upham, B.L. Inhibition of gap junctional intercellular communication by noncoplanar polychlorinated biphenyls: Inhibitory potencies and screening for potential mode(s) of action. Toxicol. Sci. 2003, 76, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierucci, F.; Frati, A.; Squecco, R.; Lenci, E.; Vicenti, C.; Slavik, J.; Francini, F.; Machala, M.; Meacci, E. Non-dioxin-like organic toxicant PCB153 modulates sphingolipid metabolism in liver progenitor cells: Its role in Cx43-formed gap junction impairment. Arch. Toxicol. 2017, 91, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Upham, B.L.; Bláha, L.; Babica, P.; Park, J.S.; Sovadinova, I.; Pudrith, C.; Rummel, A.M.; Weis, L.M.; Sai, K.; Tithof, P.K.; et al. Tumor promoting properties of a cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidylcholine-specific phospholipase C. Cancer Sci. 2008, 99, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Gakhar, G.; Schrempp, D.; Nguyen, T.A. Regulation of gap junctional intercellular communication by TCDD in HMEC and MCF-7 breast cancer cells. Toxicol. Appl. Pharm. 2009, 235, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.M.; Telang, S.; Tong, C. Inhibition of intercellular communication between liver cells by the liver tumor promoter l,l,1-trichloro-2,2-bis(p-chlorophenyl) ethane. Cancer Lett. 1981, 11, 339–344. [Google Scholar] [CrossRef]
- Stern, A.H. Hazard identification of the potential for dieldrin carcinogenicity to humans. Environ. Res. 2014, 131, 188–214. [Google Scholar] [CrossRef]
- Telang, S.; Tong, C.; Williams, G.M. Epigenetic membrane effects of a possible tumor promoting type on cultured liver cells by the non-genotoxic organochlorine pesticides chlordane and heptachlor. Carcinogenesis 1982, 3, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Ruch, R.J.; Fransson, R.; Flodstrom, S.; Warngard, L.; Klaunig, J.E. Inhibition of hepatocyte gap junctional intercellular communication by endosulfan, chlordane and heptachlor. Carcinogenesis 1990, 11, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Tiemann, U.; Pohland, R. Inhibitory effects of organochlorine pesticides on intercellular transfer of lucifer yellow in cultured bovine oviductal cells. Reprod. Toxicol. 1999, 13, 123–130. [Google Scholar] [CrossRef]
- Mograbi, B.; Corcelle, E.; Defamie, N.; Samson, M.; Nebout, M.; Segretain, D.; Fénichel, P.; Pointis, G. Aberrant Connexin 43 endocytosis by the carcinogen lindane involves activation of the ERK/mitogen-activated protein kinase pathway. Carcinogenesis 2003, 24, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Tsushimoto, G.; Trosko, J.E.; Chang, C.C.; Matsumura, F. Inhibition of intercellular communication by chlordecone (kepone) and mirex in Chinese hamster V79 cells in vitro. Toxicol. Appl. Pharm. 1982, 64, 550–556. [Google Scholar] [CrossRef]
- Caldwell, V.; Loch-Caruso, R. Chlordecone rapidly and reversibly inhibits gap junctional communication in human embryonic palatal mesenchyme cells. In Vitro Toxicol. 1992, 5, 113–122. [Google Scholar]
- Campen, K.A.; McNatty, K.P.; Pitman, J.L. A protective role of cumulus cells after short-term exposure of rat cumulus cell-oocyte complexes to lifestyle or environmental contaminants. Reprod. Toxicol. 2017, 69, 19–33. [Google Scholar] [CrossRef]
- Acuña-Hernández, D.G.; Arreola-Mendoza, L.; Santacruz-Márquez, R.; García-Zepeda, S.P.; Parra-Forero, L.Y.; Olivares-Reyes, J.A.; Hernández-Ochoa, I. Bisphenol A alters oocyte maturation by prematurely closing gap junctions in the cumulus cell-oocyte complex. Toxicol. Appl. Pharmacol. 2018, 344, 13–22. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Ruch, R.J.; DeAngelo, A.B.; Kaylor, W.H. Inhibition of mouse hepatocyte intercellular communication by phthalate monoesters. Cancer Lett. 1988, 43, 65–71. [Google Scholar] [CrossRef]
- Smith, J.H.; Isenberg, J.S.; Pugh, G., Jr.; Kamendulis, L.M.; Ackley, D.; Lington, A.W.; Klaunig, J.E. Comparative in vivo hepatic effects of Di-isononyl phthalate (DINP) and related C7-C11 dialkyl phthalates on gap junctional intercellular communication (GJIC), peroxisomal beta-oxidation (PBOX), and DNA synthesis in rat and mouse liver. Toxicol. Sci. 2000, 54, 312–321. [Google Scholar] [CrossRef]
- Kamendulis, L.M.; Isenberg, J.S.; Smith, J.H.; Pugh, G.J.; Lington, A.W.; Klaunig, J.E. Comparative effects of phthalate monoesters on gap junctional intercellular communication and peroxisome proliferation in rodent and primate hepatocytes. J. Toxicol. Environ. Health A 2002, 65, 569–588. [Google Scholar] [CrossRef] [PubMed]
- Machala, M.; Bláha, L.; Lehmler, H.J.; Plísková, M.; Májková, Z.; Kapplová, P.; Sovadinová, I.; Vondrácek, J.; Malmberg, T.; Robertson, L.W. Toxicity of hydroxylated and quinoid PCB metabolites: Inhibition of gap junctional intercellular communication and activation of aryl hydrocarbon and estrogen receptors in hepatic and mammary cells. Chem. Res. Toxicol. 2004, 17, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Dawson, B.; Chang, C.C. PBB inhibits metabolic cooperation in Chinese hamster cells in vitro: Its potential as a tumor promoter. Environ. Health Perspect. 1981, 37, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Tsushimoto, G.; Trosko, J.E.; Chang, C.C.; Aust, S.D. Inhibition of metabolic cooperation in Chinese hamster V79 cells in culture by various polybrominated biphenyl (PBB) congeners. Carcinogenesis 1982, 3, 181–185. [Google Scholar] [CrossRef]
- Bláha, L.; Kapplová, P.; Vondrácek, J.; Upham, B.; Machala, M. Inhibition of gap-junctional intercellular communication by environmentally occurring polycyclic aromatic hydrocarbons. Toxicol. Sci. 2002, 65, 43–51. [Google Scholar] [CrossRef]
- Andrysík, Z.; Procházková, J.; Kabátková, M.; Umannová, L.; Simečková, P.; Kohoutek, J.; Kozubík, A.; Machala, M.; Vondráček, J. Aryl hydrocarbon receptor-mediated disruption of contact inhibition is associated with connexin43 downregulation and inhibition of gap junctional intercellular communication. Arch. Toxicol. 2013, 87, 491–503. [Google Scholar] [CrossRef]
- Wang, F.F.; Zheng, C.J.; Guo, X.B. Effect of PM2.5 collected during the dust and non-dust periods on the viability and gap junctional intercellular communication in human lung fibroblasts. Wei Sheng Yan Jiu 2006, 35, 26–30. [Google Scholar]
- Theiss, C.; Meller, K. Aluminum impairs gap junctional intercellular communication between astroglial cells in vitro. Cell Tissue Res. 2002, 310, 143–154. [Google Scholar] [CrossRef]
- Yoshida, M.; Kujiraoka, T.; Hara, M.; Nakazawa, H.; Sumi, Y. Methylmercury inhibits gap junctional intercellular communication in primary cultures of rat proximal tubular cells. Arch. Toxicol. 1998, 72, 192–196. [Google Scholar] [CrossRef]
- Mansouri, A.; Cregut, M.; Abbes, C.; Durand, M.J.; Landoulsi, A.; Thouand, G. The Environmental Issues of DDT Pollution and Bioremediation: A Multidisciplinary Review. Appl. Biochem. Biotechnol. 2017, 181, 309–339. [Google Scholar] [CrossRef]
- Matsushima, A. A Novel Action of Endocrine-Disrupting Chemicals on Wildlife; DDT and Its Derivatives Have Remained in the Environment. Int. J. Mol. Sci. 2018, 19, 1377. [Google Scholar] [CrossRef] [Green Version]
- Cummings, A.M. Methoxychlor as a model for environmental estrogens. Crit. Rev. Toxicol. 1997, 27, 367–379. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. DDT, Lindane, and 2,4-D. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Lyon, France, 2018; Volume 113. [Google Scholar]
- Brokamp, C.; Todd, J.; Montemagno, C.; Wendell, D. Electrophysiology of single and aggregate Cx43 hemichannels. PLoS ONE 2012, 7, e47775. [Google Scholar] [CrossRef]
- Epstein, S.S. Kepone—Hazard evaluation. Sci. Total Environ. 1978, 9, 1–62. [Google Scholar] [CrossRef]
- Dallaire, R.; Muckle, G.; Rouget, F.; Kadhel, P.; Bataille, H.; Guldner, L.; Seurin, S.; Chajès, V.; Monfort, C.; Boucher, O.; et al. Cognitive, visual, and motor development of 7-month-old Guadeloupean infants exposed to chlordecone. Environ. Res. 2012, 118, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Boucher, O.; Simard, M.N.; Muckle, G.; Rouget, F.; Kadhel, P.; Bataille, H.; Chajès, V.; Dallaire, R.; Monfort, C.; Thomé, J.P.; et al. Exposure to an organochlorine pesticide (chlordecone) and development of 18-month-old infants. Neurotoxicology 2013, 35, 162–168. [Google Scholar] [CrossRef]
- Nedellec, V.; Rabl, A.; Dab, W. Public health and chronic low chlordecone exposure in Guadeloupe, Part 1: Hazards, exposure-response functions, and exposures. Environ. Health 2016, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Kucklick, J.R.; Helm, P.A. Advances in the environmental analysis of polychlorinated naphthalenes and toxaphene. Anal. Bioanal. Chem. 2006, 386, 819–836. [Google Scholar] [CrossRef]
- McKinlay, R.; Plant, J.A.; Bell, J.N.; Voulvoulis, N. Endocrine disrupting pesticides: Implications for risk assessment. Environ. Int. 2008, 34, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Van Ravenzwaay, B.; Kolle, S.N.; Ramirez, T.; Kamp, H.G. Vinclozolin: A case study on the identification of endocrine active substances in the past and a future perspective. Toxicol. Lett. 2013, 223, 271–279. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef] [PubMed]
- Vinas, R.; Jeng, Y.J.; Watson, C.S. Non-genomic effects of xenoestrogen mixtures. Int. J. Environ. Res. Public Health 2012, 9, 2694–2714. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Tilloy-Ellul, A.; Chevalier, S.; Charuel, C.; Pointis, G. Sertoli cell junctional proteins as early targets for different classes of reproductive toxicants. Reprod. Toxicol. 2004, 18, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Li, M.W.; Mruk, D.D.; Lee, W.M.; Cheng, C.Y. Disruption of the blood-testis barrier integrity by bisphenol A in vitro: Is this a suitable model for studying blood-testis barrier dynamics? Int. J. Biochem Cell Biol. 2009, 41, 2302–2314. [Google Scholar] [CrossRef] [Green Version]
- Mohri, T.; Yoshida, S. Estrogen and bisphenol A disrupt spontaneous [Ca(2+)](i) oscillations in mouse oocytes. Biochem. Biophys. Res. Commun. 2005, 326, 166–173. [Google Scholar] [CrossRef]
- Zarean, M.; Keikha, M.; Poursafa, P.; Khalighinejad, P.; Amin, M.; Kelishadi, R. A systematic review on the adverse health effects of di-2-ethylhexyl phthalate. Environ. Sci. Pollut. Res. Int. 2016, 23, 24642–24693. [Google Scholar] [CrossRef]
- Kimber, I.; Dearman, R.J. An assessment of the ability of phthalates to influence immune and allergic responses. Toxicology 2010, 271, 73–82. [Google Scholar] [CrossRef]
- North, M.L.; Takaro, T.K.; Diamond, M.L.; Ellis, A.K. Effects of phthalates on the development and expression of allergic disease and asthma. Ann. Allergy Asthma Immunol. 2014, 112, 496–502. [Google Scholar] [CrossRef]
- Koch, H.M.; Preuss, R.; Angerer, J. Di(2-ethylhexyl)phthalate (DEHP): Human. Metabolism and internal exposure—An update and latest results. Int. J. Androl. 2006, 29, 155–165. [Google Scholar] [CrossRef]
- Safe, S. Polychlorinated biphenyls (PCBs) and polybrominated biphenyls (PBBs): Biochemistry, toxicology, and mechanism of action. Crit. Rev. Toxicol. 1984, 13, 319–395. [Google Scholar] [CrossRef]
- Baker, T.K.; Kwiatkowski, A.P.; Madhukar, B.V.; Klaunig, J.E. Inhibition of gap junctional intercellular communication by 2,3,78-tetrachlorodibenzo-p-dioxin (TCDD) in rat hepatocytes. Carcinogenesis 1995, 16, 2321–2326. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Gao, D.; Liao, F.; Zhou, F.; Wang, X. The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol. Environ. Saf. 2016, 128, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, T.; Zou, L.; Xiong, L.; Zhang, T.; Kong, L.; Xue, Y.; Tang, M. Genome-wide identification and functional analysis of long non-coding RNAs in human endothelial cell line after incubation with PM2.5. Chemosphere 2019, 216, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Klocke, C.; Allen, J.L.; Sobolewski, M.; Mayer-Pröschel, M.; Blum, J.L.; Lauterstein, D.; Zelikoff, J.T.; Cory-Slechta, D.A. Neuropathological Consequences of Gestational Exposure to Concentrated Ambient Fine and Ultrafine Particles in the Mouse. Toxicol. Sci. 2017, 156, 492–508. [Google Scholar] [CrossRef]
- Loch-Caruso, R.; Corcos, I.A.; Trosko, J.E. Inhibition of metabolic coupling by metals. J. Toxicol. Environ. Health 1991, 32, 33–48. [Google Scholar] [CrossRef]
- Yorifuji, T.; Takaoka, S.; Grandjean, P. Accelerated functional losses in ageing congenital Minamata disease patients. Neurotoxicol. Teratol. 2018, 69, 49–53. [Google Scholar] [CrossRef]
- Guxens, M.; Sunyer, J. A review of epidemiological studies on neuropsychological effects of air pollution. Swiss Med Wkly. 2012, 141, 13322. [Google Scholar]
- Dominici, F.; Peng, R.D.; Bell, M.L.; Pham, L.; McDermott, A.; Zeger, S.L.; Samet, J.M. Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA 2006, 295, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Rocha, I.I.; Narasimhalu, K.; De Silva, D.A. Impact of Air Pollution and Seasonal Haze on Neurological Conditions. Ann. Acad. Med. Singap. 2020, 49, 26–36. [Google Scholar] [CrossRef]
- Chen, J.C.; Schwartz, J. Neurobehavioral effects of ambient air pollution on cognitive performance in US adults. Neurotoxicology 2009, 30, 231–239. [Google Scholar] [CrossRef]
- Harris, M.H.; Gold, D.R.; Rifas-Shiman, S.L.; Melly, S.J.; Zanobetti, A.; Coull, B.A.; Schwartz, J.D.; Gryparis, A.; Kloog, I.; Koutrakis, P.; et al. Prenatal and Childhood Traffic-Related Pollution Exposure and Childhood Cognition in the Project Viva Cohort (Massachusetts, USA). Environ. Health Perspect. 2015, 123, 1072–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, F.; Li, T.Y.; Lin, C.; Tang, D. Effects of prenatal polycyclic aromatic hydrocarbon exposure and environmental tobacco smoke on child IQ in a Chinese cohort. Environ. Res. 2012, 114, 40–46. [Google Scholar] [CrossRef]
- Oudin, A.; Forsberg, B.; Adolfsson, A.N.; Lind, N.; Modig, L.; Nordin, M.; Nordin, S.; Adolfsson, R.; Nilsson, L.G. Traffic-Related Air Pollution and Dementia Incidence in Northern Sweden: A Longitudinal Study. Environ. Health Perspect. 2016, 124, 306–312. [Google Scholar] [CrossRef]
- Oudin, A.; Bråbäck, L.; Åström, D.O.; Strömgren, M.; Forsberg, B. Association between neighbourhood air pollution concentrations and dispensed medication for psychiatric disorders in a large longitudinal cohort of Swedish children and adolescents. BMJ Open 2016, 6, e010004. [Google Scholar] [CrossRef] [PubMed]
- Porta, D.; Narduzzi, S.; Badaloni, C.; Bucci, S.; Cesaroni, G.; Colelli, V.; Davoli, M.; Sunyer, J.; Zirro, E.; Schwartz, J.; et al. Air Pollution and Cognitive Development at Age 7 in a Prospective Italian Birth Cohort. Epidemiology 2016, 27, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Suglia, S.F.; Gryparis, A.; Wright, R.O.; Schwartz, J.; Wright, R.J. Association of black carbon with cognition among children in a prospective birth cohort study. Am. J. Epidemiol. 2008, 167, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Volk, H.E.; Lurmann, F.; Penfold, B.; Hertz-Picciotto, I.; McConnell, R. Traffic-related air pollution, particulate matter, and autism. JAMA Psychiatry 2013, 70, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, J.; Zeng, X.; Zeng, Y.; Wang, S.; Chen, S. Association of traffic-related air pollution with children’s neurobehavioral functions in Quanzhou, China. Environ. Health Perspect. 2009, 117, 1612–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zablotsky, B.; Black, L.I.; Maenner, M.J.; Schieve, L.A.; Blumberg, S.J. Estimated Prevalence of Autism and Other Developmental Disabilities Following Questionnaire Changes in the 2014 National Health Interview Survey. Natl. Health Stat Rep. 2015, 87, 1–20. [Google Scholar]
- Woodley Of Menie, M.A.; Dunkel, C.S. In France, are secular IQ losses biologically caused? A comment on Dutton and Lynn. Intelligence 2015, 53, 81–85. [Google Scholar] [CrossRef]
- Van den Bosch, M.; Meyer-Lindenberg, A. Environmental Exposures and Depression: Biological Mechanisms and Epidemiological Evidence. Annu. Rev. Public Health 2019, 40, 239–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Lim, Y.H.; Kim, K.N.; Choi, Y.H.; Hong, Y.C.; Lee, N. Urinary phthalate metabolites concentrations and symptoms of depression in an elderly population. Sci. Total Environ. 2018, 625, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.; Arseneault, L.; Barratt, B.; Beevers, S.; Danese, A.; Odgers, C.L.; Moffitt, T.E.; Reubeng, A.; Kelly, F.J.; Fisher, H.L. Exploration of NO2 and PM2.5 air pollution and mental health problems using high-resolution data in London-based children from a UK longitudinal cohort study. Psychiatry Res. 2019, 272, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Newbury, J.B.; Arseneault, L.; Beevers, S.; Kitwiroon, N.; Roberts, S.; Pariante, C.M.; Kelly, F.J.; Fisher, H.L. Association of Air Pollution Exposure with Psychotic Experiences During Adolescence. JAMA Psychiatry 2019, 76, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Landrigan, P.J. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Rana, H.K.; Akhtar, M.R.; Ahmed, M.B.; Liò, P.; Quinn, J.M.W.; Huq, F.; Moni, M.A. Genetic effects of welding fumes on the progression of neurodegenerative diseases. Neurotoxicology 2019, 71, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.; de Montigny, J.G.; Ofner, M.; Do, M.T. Environmental factors associated with autism spectrum disorder: A scoping review for the years 2003–2013. Health Promot. Chronic Dis. Prev. Can. 2017, 37, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Saeedi Saravi, S.S.; Dehpour, A.R. Potential role of organochlorine pesticides in the pathogenesis of neurodevelopmental, neurodegenerative, and neurobehavioral disorders: A review. Life Sci. 2016, 145, 255–264. [Google Scholar] [CrossRef]
- Ye, B.S.; Leung, A.O.W.; Wong, M.H. The association of environmental toxicants and autism spectrum disorders in children. Environ. Pollut. 2017, 227, 234–242. [Google Scholar] [CrossRef]
- Vrijheid, M.; Casas, M.; Gascon, M.; Valvi, D.; Nieuwenhuijsen, M. Environmental pollutants and child health-A review of recent concerns. Int. J. Hyg. Environ. Health 2016, 219, 331–342. [Google Scholar] [CrossRef]
- Carter, C.J.; Blizard, R.A. Autism genes are selectively targeted by environmental pollutants including pesticides, heavy metals, bisphenol A, phthalates and many others in food, cosmetics or household products. Neurochem. Int. 2016, 186, 30197–30198. [Google Scholar] [CrossRef] [PubMed]
- Fluegge, K.; Fluegge, K. Exposure to ambient PM10 and nitrogen dioxide and ADHD risk: A reply to Min & Min (2017). Environ. Int. 2017, 103, 109–110. [Google Scholar] [PubMed]
- Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Lee, S. Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse 2008, 62, 501–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, T.D.; Middleton, F.; Faraone, S.V. Environmental risk factors for attention-deficit hyperactivity disorder. Acta Paediatr. 2007, 96, 1269–1274. [Google Scholar] [CrossRef]
- Froehlich, T.E.; Anixt, J.S.; Loe, I.M.; Chirdkiatgumchai, V.; Kuan, L.; Gilman, R.C. Update on environmental risk factors for attention-deficit/hyperactivity disorder. Curr. Psychiatry Rep. 2011, 13, 333–344. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Bellinger, D.C.; Coull, B.A.; Anderson, S.; Barber, R.; Wright, R.O.; Wright, R.J. Associations between traffic-related black carbon exposure and attention in a prospective birth cohort of urban children. Environ. Health Perspect. 2013, 121, 859–864. [Google Scholar] [CrossRef]
- Newman, N.C.; Ryan, P.; Lemasters, G.; Levin, L.; Bernstein, D.; Hershey, G.K.; Lockey, J.E.; Villareal, M.; Reponen, T.; Grinshpun, S.; et al. Traffic-related air pollution exposure in the first year of life and behavioral scores at 7 years of age. Environ. Health Perspect. 2013, 121, 731–736. [Google Scholar] [CrossRef]
- Sunyer, J.; Esnaola, M.; Alvarez-Pedrerol, M.; Forns, J.; Rivas, I.; López-Vicente, M.; Suades-González, E.; Foraster, M.; Garcia-Esteban, R.; Basagaña, X.; et al. Association between traffic-related air pollution in schools and cognitive development in primary school children: A prospective cohort study. PLoS Med. 2015, 12, e1001792. [Google Scholar] [CrossRef]
- Min, J.Y.; Min, K.B. Exposure to ambient PM 10 and NO 2 and the incidence of attention-deficit hyperactivity disorder in childhood. Environ. Int. 2017, 99, 221–227. [Google Scholar] [CrossRef]
- Fluegge, K.R.; Fluegge, K.R. Glyphosate Use Predicts ADHD Hospital Discharges in the Healthcare Cost and Utilization Project Net (HCUPnet): A Two-Way Fixed-Effects Analysis. PLoS ONE 2015, 10, e0133525. [Google Scholar] [CrossRef] [Green Version]
- Banaschewski, T.; Becker, K.; Scherag, S.; Franke, B.; Coghill, D. Molecular genetics of attention-deficit/hyperactivity disorder: An overview. Eur. Child Adolesc. Psychiatry 2010, 19, 237–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakmak, S.; Dales, R.E.; Vidal, C.B. Air pollution and hospitalization for epilepsy in Chile. Environ. Int. 2010, 36, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Conway, G.A.; Jin, L.; Zhou, J.; Zhang, J.; Li, X.; Zhou, L.; Li, T.; Zhang, J. Increase in Medical Emergency Calls and Calls for Central Nervous System Symptoms During a Severe Air Pollution Event, January 2013, Jinan City, China. Epidemiology 2017, 28, S67–S73. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Tian, X.; Yang, C.; Li, Y.; Hu, Y. Association between ambient air pollution and hospital admission for epilepsy in Eastern China. Epilepsy Res. 2019, 152, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Fan, Y.N.; Kan, H.D.; Chen, R.J.; Liu, J.H.; Li, Y.F.; Zhang, Y.; Ji, A.L.; Cai, T.J. The Novel Relationship between Urban Air Pollution and Epilepsy: A Time Series Study. PLoS ONE 2016, 11, e0161992. [Google Scholar] [CrossRef]
- Poon, E.; Bandara, S.B.; Allen, J.B.; Sadowski, R.N.; Schantz, S.L. Developmental PCB exposure increases susceptibility to audiogenic seizures in adulthood. Neurotoxicology 2015, 46, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Bandara, S.B.; Eubig, P.A.; Sadowski, R.N.; Schantz, S.L. Developmental PCB Exposure Increases Audiogenic Seizures and Decreases Glutamic Acid Decarboxylase in the Inferior Colliculus. Toxicol. Sci. 2016, 149, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Aschengrau, A.; Winter, M.R.; Vieira, V.M.; Webster, T.F.; Janulewicz, P.A.; Gallagher, L.G.; Weinberg, J.; Ozonoff, D.M. Long-term health effects of early life exposure to tetrachloroethylene (PCE)-contaminated drinking water: A retrospective cohort study. Environ. Health 2015, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Bedner, P.; Dupper, A.; Hüttmann, K.; Müller, J.; Herde, M.K.; Dublin, P.; Deshpande, T.; Schramm, J.; Häussler, U.; Haas, C.A.; et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 2015, 138, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Khan, D.; Dupper, A.; Deshpande, T.; Graan, P.N.; Steinhäuser, C.; Bedner, P. Experimental febrile seizures impair interastrocytic gap junction coupling in juvenile mice. J. Neurosci. Res. 2016, 94, 804–813. [Google Scholar] [CrossRef]
- Collignon, F.; Wetjen, N.M.; Cohen-Gadol, A.A.; Cascino, G.D.; Parisi, J.; Meyer, F.B.; Marsh, W.R.; Roche, P.; Weigand, S.D. Altered expression of connexin subtypes in mesial temporal lobe epilepsy in humans. J. Neurosurg. 2006, 105, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Myung, W.; Cheong, H.K.; Yi, S.M.; Hong, Y.C.; Cho, S.I.; Kim, H. Ambient air pollution exposure and risk of migraine: Synergistic effect with high temperature. Environ. Int. 2018, 121, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Martins-Ferreira, H.; Nedergaard, M.; Nicholson, C. Perspectives on spreading depression. Brain Res. Rev. 2000, 32, 215–234. [Google Scholar] [CrossRef]
- Mustieles, V.; Pérez-Lobato, R.; Olea, N.; Fernández, M.F. Bisphenol A: Human exposure and neurobehavior. Neurotoxicology 2015, 49, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Vishnevetsky, J.; Herbstman, J.B.; Calafat, A.M.; Xiong, W.; Rauh, V.; Wang, S. Prenatal bisphenol a exposure and child behavior in an inner-city cohort. Environ. Health Perspect. 2012, 120, 1190–1194. [Google Scholar] [CrossRef] [Green Version]
- Perera, F.; Nolte, E.L.R.; Wang, Y.; Margolis, A.E.; Calafat, A.M.; Wang, S.; Garcia, W.; Hoepner, L.A.; Peterson, B.S.; Rauh, V.; et al. Bisphenol A exposure and symptoms of anxiety and depression among inner city children at 10–12 years of age. Environ. Res. 2016, 151, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Ejaredar, M.; Lee, Y.; Roberts, D.J.; Sauve, R.; Dewey, D. Bisphenol A exposure and children’s behavior: A systematic review. J. Expo. Sci. Environ. Epidemiol. 2017, 27, 175–183. [Google Scholar] [CrossRef]
- Shiue, I. Urinary heavy metals, phthalates and polyaromatic hydrocarbons independent of health events are associated with adult depression: USA NHANES, 2011–2012. Environ. Sci. Pollut. Res 2015, 22, 17095–17103. [Google Scholar] [CrossRef]
- Quesseveur, G.; Portal, B.; Basile, J.A.; Ezan, P.; Mathou, A.; Halley, H.; Leloup, C.; Fioramonti, X.; Déglon, N.; Giaume, C.; et al. Attenuated Levels of Hippocampal Connexin 43 and its Phosphorylation Correlate with Antidepressant- and Anxiolytic-Like Activities in Mice. Front. Cell. Neurosci. 2015, 9, 490. [Google Scholar] [CrossRef] [Green Version]
- Kasdagli, M.I.; Katsouyanni, K.; Dimakopoulou, K.; Samoli, E. Air pollution and Parkinson’s disease: A systematic review and meta-analysis up to 2018. Int. J. Hyg. Environ. Health 2019, 222, 402–409. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharm. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Landolfi, A.; Troisi, J.; Savanelli, M.C.; Vitale, C.; Barone, P.; Amboni, M. Bisphenol A glucuronidation in patients with Parkinson’s disease. Neurotoxicology 2017, 63, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.Y.; Cui, Y.; Deng, F.; Feng, J.C. Connexin: A potential novel target for protecting the central nervous system? Neural Regen. Res. 2015, 10, 659–666. [Google Scholar] [PubMed]
- Lu, C.; Meng, Z.; He, Y.; Xiao, D.; Cai, H.; Xu, Y.; Liu, X.; Wang, X.; Mo, L.; Liang, Z.; et al. Involvement of gap junctions in astrocyte impairment induced by manganese exposure. Brain Res. Bull. 2018, 140, 107–113. [Google Scholar] [CrossRef]
- Wang, J.; Ma, T.; Ma, D.; Li, H.; Hua, L.; He, Q.; Deng, X. The Impact of Air Pollution on Neurodegenerative Diseases. Ther. Drug Monit. 2020. [Google Scholar] [CrossRef]
- Kilian, J.; Kitazawa, M. The emerging risk of exposure to air pollution on cognitive decline and Alzheimer’s disease—Evidence from epidemiological and animal studies. Biomed. J. 2018, 41, 141–162. [Google Scholar] [CrossRef]
- Yi, C.; Mei, X.; Ezan, P.; Mato, S.; Matias, I.; Giaume, C.; Koulakoff, A. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 2016, 23, 1691–1701. [Google Scholar] [CrossRef]
- Harcha, P.A.; Vargas, A.; Yi, C.; Koulakoff, A.A.; Giaume, C.; Sáez, J.C. Hemichannels Are Required for Amyloid β-Peptide-Induced Degranulation and Are Activated in Brain Mast Cells of APPswe/PS1dE9 Mice. J. Neurosci. 2015, 35, 9526–9538. [Google Scholar] [CrossRef] [Green Version]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vienne-Jumeau, A.; Tafani, C.; Ricard, D. Environmental risk factors of primary brain tumors: A review. Rev. Neurol. 2019, 175, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Khuder, S.A.; Mutgi, A.B.; Schaub, E.A. Meta-analyses of brain cancer and farming. Am. J. Ind. Med. 1998, 34, 252–260. [Google Scholar] [CrossRef]
- Van Maele-Fabry, G.; Hoet, P.; Lison, D. Parental occupational exposure to pesticides as risk factor for brain tumors in children and young adults: A systematic review and meta-analysis. Environ. Int. 2013, 56, 19–31. [Google Scholar] [CrossRef]
- Van Maele-Fabry, G.; Gamet-Payrastre, L.; Lison, D. Residential exposure to pesticides as risk factor for childhood and young adult brain tumors: A systematic review and meta-analysis. Environ. Int. 2017, 106, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.S.; Park, J.E.; Ryu, D.Y.; Lee, Y.S. Effects and neuro-toxic mechanisms of 2, 2′, 4, 4′, 5, 5′-hexachlorobiphenyl and endosulfan in neuronal stem cells. J. Vet. Med Sci. 2001, 63, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Seifert, G.; Schilling, K.; Steinhäuser, C. Astrocyte dysfunction in neurological disorders: A molecular perspective. Nat. Rev. Neurosci. 2006, 7, 194–206. [Google Scholar] [CrossRef]
- John Lin, C.C.; Deneen, B. Astrocytes: The missing link in neurologic disease? Semin Pediatr. Neurol. 2013, 20, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Herbert, M.R. Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr. Opin. Neurol. 2010, 23, 103–110. [Google Scholar] [CrossRef]
- Stamou, M.; Streifel, K.M.; Goines, P.E.; Lein, P.J. Neuronal connectivity as a convergent target of gene × environment interactions that confer risk for Autism Spectrum Disorders. Neurotoxicol. Teratol. 2013, 36, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Sarrouilhe, D.; Dejean, C.; Mesnil, M. Connexin43- and Pannexin-Based Channels in Neuroinflammation and Cerebral Neuropathies. Front. Mol. Neurosci. 2017, 10, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Pierce, K. Brain overgrowth in autism during a critical time in development: Implications for frontal pyramidal neuron and interneuron development and connectivity. Int. J. Dev. Neurosci. 2005, 23, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H.; Townsend, J.; Courchesne, E.; Lincoln, A.J.; Schreibman, L.; Yeung-Courchesne, R. Neurologic abnormalities in infantile autism. J. Child Neurol. 1996, 11, 84–92. [Google Scholar] [CrossRef]
- Townsend, J.; Harris, N.S.; Courchesne, E. Visual attention abnormalities in autism: Delayed orienting to location. J. Int. Neuropsychol. Soc. 1996, 2, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Trivedi, M.H.; Garver, C.R.; Grannemann, B.D.; Andrews, A.A.; Savla, J.S.; Johnson, D.G.; Mehta, J.A.; Schroeder, J.L. The pattern of sensory processing abnormalities in autism. Autism 2006, 10, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Trivedi, M.H.; Grannemann, B.D.; Garver, C.R.; Johnson, D.G.; Andrews, A.A.; Savla, J.S.; Mehta, J.A.; Schroeder, J.L. Sensory correlations in autism. Autism 2007, 11, 123–134. [Google Scholar] [CrossRef]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism—Comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Grubišc, V.; Parpura, V. The second brain in autism spectrum disorder: Could connexin43 expressed in enteric glial cells play a role? Front. Cell. Neurosci. 2015, 9, 242. [Google Scholar]
- Hong, Y.Z.; Zhang, X.J.; Hong, L.; Huang, Q.R.; Wu, Q. Influence of acupuncture of “Changqiang” (GV 1) on learning-memory ability and gap junction-related protein expression in the prefrontal cortex in autism rats. Zhen Ci Yan Jiu 2014, 39, 173–179. [Google Scholar]
- Dickerson, F.; Adamos, M.B.; Katsafanas, E.; Khushalani, S.; Origoni, A.; Savage, C.L.G.; Schroeder, J.; Schweinfurth, L.A.B.; Stallings, C.; Sweeney, K.; et al. The association among smoking, HSV-1 exposure, and cognitive functioning in schizophrenia, bipolar disorder, and non-psychiatric controls. Schizophr. Res. 2016, 176, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti-Pierri, N.; Berg, J.S.; Scaglia, F.; Belmont, J.; Bacino, C.A.; Sahoo, T.; Lalani, S.R.; Graham, B.; Lee, B.; Shinawi, M.; et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Steinman, K.J.; Reilly, B.; Wallace, A.S.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, E.; et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 2016, 18, 341–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollob, M.H.; Jones, D.L.; Krahn, A.D.; Danis, L.; Gong, X.Q.; Shao, Q.; Liu, X.; Veinot, J.P.; Tang, A.S.; Stewart, A.F.; et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 2006, 354, 2677–2688. [Google Scholar] [CrossRef]
- Christiansen, J.; Dyck, J.D.; Elyas, B.G.; Lilley, M.; Bamforth, J.S.; Hicks, M.; Sprysak, K.A.; Tomaszewski, R.; Haase, S.M.; Vicen-Wyhony, L.M.; et al. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ. Res. 2004, 94, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Soemedi, R.; Topf, A.; Wilson, I.J.; Darlay, R.; Rahman, T.; Glen, E.; Hall, D.; Huang, N.; Bentham, J.; Bhattacharya, S.; et al. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum. Mol. Genet. 2012, 21, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Guida, V.; Ferese, R.; Rocchetti, M.; Bonetti, M.; Sarkozy, A.; Cecchetti, S.; Gelmetti, V.; Lepri, F.; Copetti, M.; Lamorte, G.; et al. A variant in the carboxyl-terminus of connexin 40 alters GAP junctions and increases risk for tetralogy of Fallot. Eur. J. Hum. Genet. 2013, 21, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Mackay, D.S.; Bennett, T.M.; Culican, S.M.; Shiels, A. Exome sequencing identifies novel and recurrent mutations in GJA8 and CRYGD associated with inherited cataract. Hum. Genom. 2014, 8, 19. [Google Scholar] [CrossRef]
- Huang, X.; Xiao, X.; Jia, X.; Li, S.; Li, M.; Guo, X.; Liu, X.; Zhang, Q. Mutation analysis of the genes associated with anterior segment dysgenesis, microcornea and microphthalmia in 257 patients with glaucoma. Int. J. Mol. Med. 2015, 36, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Prokudin, I.; Simons, C.; Grigg, J.R.; Storen, R.; Kumar, V.; Phua, Z.Y.; Smith, J.; Flaherty, M.; Davila, S.; Jamieson, R.V. Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1. Eur. J. Hum. Genet. 2014, 22, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.S. The New Classification of Seizures by the International League Against Epilepsy 2017. Curr. Neurol. Neurosci. Rep. 2017, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Han, Y.; Li, Q.; Zhang, J. Endogenous sulfur dioxide regulates hippocampal neuron apoptosis in developing epileptic rats and is associated with the PERK signaling pathway. Neurosci. Lett. 2018, 665, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Bell, I.R.; Hardin, E.E.; Baldwin, C.M.; Schwartz, G.E. Increased limbic system symptomatology and sensitizability of young adults with chemical and noise sensitivities. Environ. Res. 1995, 70, 84–97. [Google Scholar] [CrossRef]
- Fernandes, M.J.S.; Carletti, C.O.; Sierra de Araújo, L.F.; Santos, R.C.; Reis, J. Respiratory gases, air pollution and epilepsy. Rev. Neurol. 2019, 175, 604–613. [Google Scholar] [CrossRef]
- Ramsdell, J.S. Neurological disease rises from ocean to bring model for human epilepsy to life. Toxins 2010, 2, 1646–1675. [Google Scholar] [CrossRef]
- Yamada, J.; Inoue, K.; Furukawa, T.; Fukuda, A. Low-concentration tributyltin perturbs inhibitory synaptogenesis and induces neuronal death in immature but not mature neurons. Toxicol. Lett. 2010, 198, 282–288. [Google Scholar] [CrossRef]
- Sato, I.; Kawamoto, K.; Nishikawa, Y.; Tsuda, S.; Yoshida, M.; Yaegashi, K.; Saito, N.; Liu, W.; Jin, Y. Neurotoxicity of perfluorooctane sulfonate (PFOS) in rats and mice after single oral exposure. J. Toxicol. Sci. 2009, 34, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, K.; Sato, I.; Tsuda, S.; Yoshida, M.; Yaegashi, K.; Saito, N.; Liu, W.; Jin, Y. Ultrasonic-induced tonic convulsion in rats after subchronic exposure to perfluorooctane sulfonate (PFOS). J. Toxicol. Sci. 2011, 36, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, E.; Eftekhari, A.; Samiei, M.; Maleki Dizaj, S.; Vinken, M. The role and therapeutic potential of connexins, pannexins and their channels in Parkinson’s disease. Cell. Signal. 2019, 58, 111–118. [Google Scholar] [CrossRef]
- Kawasaki, A.; Hayashi, T.; Nakachi, K.; Trosko, J.; Sugihara, K.; Kotake, Y.; Ohta, S. Modulation of connexin 43 in rotenone-induced model of Parkinson’s disease. Neuroscience 2009, 160, 61–68. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.; Liu, X.; Fu, Q. Gastrodin ameliorates Parkinson’s disease by downregulating connexin 43. Mol. Med. Rep. 2013, 8, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzún, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 hemichannels and pannexin-1 channels contribute to the α-synuclein-induced dysfunction and death of astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Mhatre, I.; Richardson, J.R. Gene-environment interactions in Alzheimer’s disease: A potential path to precision medicine. Pharmacol. Ther. 2019, 199, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease beyond 1994: The path to therapeutics. Neurobiol. Aging 1994, 15, S131–S133. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Fajardo, V.A.; LeBlanc, P.J. MacPherson REK. Examining the Relationship between Trace Lithium in Drinking Water and the Rising Rates of Age-Adjusted Alzheimer’s Disease Mortality in Texas. J. Alzheimers Dis. 2018, 61, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Amtul, Z.; Nikolova, S.; Gao, L.; Keeley, R.J.; Bechberger, J.F.; Fisher, A.L.; Bartha, R.; Munoz, D.G.; McDonald, R.J.; Naus, C.C.; et al. Comorbid Aβ toxicity and stroke: Hippocampal atrophy, pathology, and cognitive deficit. Neurobiol. Aging 2014, 35, 1605–1614. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.I.; Li, W.; Hertzberg, E.L.; Marotta, C.A. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996, 717, 173–178. [Google Scholar] [CrossRef]
- Begcevic, I.; Kosanam, H.; Martínez-Morillo, E.; Dimitromanolakis, A.; Diamandis, P.; Kuzmanov, U.; Hazrati, L.N.; Diamandis, E.P. Semiquantitative proteomic analysis of human hippocampal tissues from Alzheimer’s disease and age-matched control brains. Clin. Proteom. 2013, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Hokama, M.; Oka, S.; Leon, J.; Ninomiya, T.; Honda, H.; Sasaki, K.; Iwaki, T.; Ohara, T.; Sasaki, T.; LaFerla, F.M.; et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: The Hisayama study. Cereb. Cortex 2014, 24, 2476–2488. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Ezan, P.; Giaume, C.; Koulakoff, A. Astroglial connexin immunoreactivity is specifically altered at β-amyloid plaques in β-amyloid precursor protein/presenilin1 mice. Neuroscience 2010, 171, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Koulakoff, A.; Mei, X.; Orellana, J.A.; Sáez, J.C.; Giaume, C. Glial connexin expression and function in the context of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1818, 2048–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.; Ezan, P.; Fernández, P.; Schmitt, J.; Sáez, J.C.; Giaume, C.; Koulakoff, A. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017, 65, 1607–1625. [Google Scholar] [CrossRef] [PubMed]
- Biswas, J.; Gupta, S.; Verma, D.K.; Gupta, P.; Singh, A.; Tiwari, S.; Goswami, P.; Sharma, S.; Singh, S. Involvement of glucose related energy crisis and endoplasmic reticulum stress: Insinuation of streptozotocin induced Alzheimer’s like pathology. Cell. Signal. 2018, 42, 211–226. [Google Scholar] [CrossRef]
- Khalil, M.; Ronda, J.; Weintraub, M.; Jain, K.; Silver, R.; Silverman, A.J. Brain mast cell relationship to neurovasculature during development. Brain Res. 2007, 1171, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Maslinska, D.; Laure-Kamionowska, M.; Maslinski, K.T.; Gujski, M.; Maslinski, S. Distribution of tryptase-containing mast cells and metallothionein reactive astrocytes in human brains with amyloid deposits. Inflamm. Res. 2007, 56, S17–S18. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef]
- Niederhoffer, N.; Levy, R.; Sick, E.; Andre, P.; Coupin, G.; Lombard, Y.; Gies, J.P. Amyloid beta peptides trigger CD47-dependent mast cell secretory and phagocytic responses. Int. J. Immunopathol Pharm. 2009, 22, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Orellana, J.A.; Figueroa, X.F.; Sánchez, H.A.; Contreras-Duarte, S.; Velarde, V.; Sáez, J.C. Hemichannels in the neurovascular unit and white matter under normal and inflamed conditions. CNS Neurol. Disord. Drug Targets 2011, 10, 404–414. [Google Scholar] [CrossRef]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sánchez-Gómez, M.V.; Cavaliere, F.; Rodríguez, J.J.; Verkhratsky, A.; Matute, C. Ca(2+) -dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell. 2013, 12, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-β and Alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stridh, M.H.; Tranberg, M.; Weber, S.G.; Blomstrand, F.; Sandberg, M. Stimulated efflux of amino acids and glutathione from cultured hippocampal slices by omission of extracellular calcium: Likely involvement of connexin hemichannels. J. Biol. Chem. 2008, 283, 10347–10356. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Guan, J.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 17042–17051. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Shen, H.; Zhang, J.; Zhu, Y.G.; Ransom, B.R.; Chen, X.C.; Ye, Z.C. Dual pathways mediate β-amyloid stimulated glutathione release from astrocytes. Glia 2015, 63, 2208–2219. [Google Scholar] [CrossRef]
- Kozoriz, M.G.; Bechberger, J.F.; Bechberger, G.R.; Suen, M.W.; Moreno, A.P.; Maass, K.; Willecke, K.; Naus, C.C. The connexin43 C-terminal region mediates neuroprotection during stroke. J. Neuropathol. Exp. Neurol. 2010, 69, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Le, H.T.; Sin, W.C.; Lozinsky, S.; Bechberger, J.; Vega, J.L.; Guo, X.Q.; Sáez, J.C.; Naus, C.C. Gap junction intercellular communication mediated by connexin43 in astrocytes is essential for their resistance to oxidative stress. J. Biol. Chem. 2014, 289, 1345–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakase, T.; Fushiki, S.; Söhl, G.; Theis, M.; Willecke, K.; Naus, C.C. Neuroprotective role of astrocytic gap junctions in ischemic stroke. Cell. Commun. Adhes. 2003, 10, 413–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakase, T.; Söhl, G.; Theis, M.; Willecke, K.; Naus, C.C. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am. J. Pathol. 2004, 164, 2067–2075. [Google Scholar] [CrossRef] [Green Version]
- Bookheimer, S.Y.; Strojwas, M.H.; Cohen, M.S.; Saunders, A.M.; Pericak-Vance, M.A.; Mazziotta, J.C.; Small, G.W. Patterns of brain activation in people at risk for Alzheimer’s disease. N. Engl. J. Med. 2000, 343, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiroz, Y.T.; Budson, A.E.; Celone, K.; Ruiz, A.; Newmark, R.; Castrillón, G.; Lopera, F.; Stern, C.E. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann. Neurol. 2010, 68, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Kajiwara, Y.; Wang, E.; Wang, M.; Sin, W.C.; Brennand, K.J.; Schadt, E.; Naus, C.C.; Buxbaum, J.; Zhang, B. GJA1 (connexin43) is a key regulator of Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2018, 6, 144. [Google Scholar] [CrossRef]
- Theodoric, N.; Bechberger, J.F.; Naus, C.C.; Sin, W.C. Role of gap junction protein connexin43 in astrogliosis induced by brain injury. PLoS ONE 2012, 7, e47311. [Google Scholar] [CrossRef] [Green Version]
- Bakker, A.; Krauss, G.L.; Albert, M.S.; Speck, C.L.; Jones, L.R.; Stark, C.E.; Yassa, M.A.; Bassett, S.S.; Shelton, A.L.; Gallagher, M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 2012, 74, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Esposito, Z.; Belli, L.; Toniolo, S.; Sancesario, G.; Bianconi, C.; Martorana, A. Amyloid β, glutamate, excitotoxicity in Alzheimer’s disease: Are we on the right track? CNS Neurosci. Ther. 2013, 19, 549–555. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; Tanaka, K.; Dawe, G.S.; Ittner, L.M.; Farooqui, A.A. Slow excitotoxicity in Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 643–668. [Google Scholar] [CrossRef] [Green Version]
- Majoul, I.V.; Ernesti, J.S.; Butkevich, E.V.; Duden, R. Drebrins and Connexins: A Biomedical Perspective. Adv. Exp. Med. Biol. 2017, 1006, 225–247. [Google Scholar]
- Ren, R.; Zhang, L.; Wang, M. Specific deletion connexin43 in astrocyte ameliorates cognitive dysfunction in APP/PS1 mice. Life Sci. 2018, 208, 175–191. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, N.; Saini, A.; Nehru, B. Carbenoxolone Reverses the Amyloid Beta 1–42 Oligomer-Induced Oxidative Damage and Anxiety-Related Behavior in Rats. Neurotox. Res. 2019, 35, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Gajardo-Gómez, R.; Labra, V.C.; Maturana, C.J.; Shoji, K.F.; Santibañez, C.A.; Sáez, J.C.; Giaume, C.; Orellana, J.A. Cannabinoids prevent the amyloid β-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia 2017, 65, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.C.; Baca, S.M. Cortical spreading depression and migraine. Nat. Rev. Neurol. 2013, 9, 637–644. [Google Scholar] [CrossRef]
- Costa, C.; Tozzi, A.; Rainero, I.; Cupini, L.M.; Calabresi, P.; Ayata, C.; Sarchielli, P. Cortical spreading depression as a target for anti-migraine agents. J. Headache Pain 2013, 14, 62. [Google Scholar] [CrossRef] [Green Version]
- Sarrouilhe, D.; Dejean, C.; Mesnil, M. Involvement of gap junction channels in the pathophysiology of migraine with aura. Front. Physiol. 2014, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Sarrouilhe, D.; Mesnil, M.; Dejean, C. Targeting Gap Junctions: New Insights into the Treatment of Major Depressive Disorder. Curr. Med. Chem. 2019, 26, 3775–3791. [Google Scholar] [CrossRef]
- Palanza, P.; Nagel, S.C.; Parmigiani, S.; Vom Saal, F.S. Perinatal exposure to endocrine disruptors: Sex, timing and behavioral endpoints. Curr. Opin. Behav. Sci. 2016, 7, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Xin, F.; Fischer, E.; Krapp, C.; Krizman, E.N.; Lan, Y.; Mesaros, C.; Snyder, N.W.; Bansal, A.; Robinson, M.B.; Simmons, R.A.; et al. Mice exposed to bisphenol A exhibit depressive-like behavior with neurotransmitter and neuroactive steroid dysfunction. Horm. Behav. 2018, 102, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Matsuyoshi, C.; Miyazaki, W.; Benner, S.; Hosokawa, M.; Yokoyama, K.; Kakeyama, M.; Tohyama, C. Prenatal exposure to bisphenol A impacts neuronal morphology in the hippocampal CA1 region in developing and aged mice. Arch. Toxicol. 2016, 90, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespin, S.; Fromont, G.; Wager, M.; Levillain, P.; Cronier, L.; Monvoisin, A.; Defamie, N.; Mesnil, M. Expression of a gap junction protein, connexin43, in a large panel of human gliomas: New insights. Cancer Med. 2016, 5, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.P.; Hossain, M.Z.; Sehgal, A.; Boynton, A.L. Reduced connexin43 expression in high-grade human brain glioma cells. J. Surg. Oncol. 1999, 70, 21–24. [Google Scholar] [CrossRef]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Reduced expression of connexin-43 and functional gap junction coupling in human gliomas. Glia 2001, 33, 107–117. [Google Scholar] [CrossRef]
- Herrero-González, S.; Gangoso, E.; Giaume, C.; Naus, C.C.; Medina, J.M.; Tabernero, A. Connexin43 inhibits the oncogenic activity of c-Src in C6 glioma cells. Oncogene 2010, 29, 5712–5723. [Google Scholar] [CrossRef] [Green Version]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef]
- Strale, P.O.; Clarhaut, J.; Lamiche, C.; Cronier, L.; Mesnil, M.; Defamie, N. Down-regulation of Connexin43 expression reveals the involvement of caveolin-1 containing lipid rafts in human U251 glioblastoma cell invasion. Mol. Carcinog. 2012, 51, 845–860. [Google Scholar] [CrossRef]
- Aftab, Q.; Sin, W.C.; Naus, C.C. Reduction in gap junction intercellular communication promotes glioma migration. Oncotarget 2015, 6, 11447–11464. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.C.; Sin, W.C.; Aftab, Q.; Naus, C.C. Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia 2007, 55, 1554–1564. [Google Scholar] [CrossRef] [PubMed]
- Crespin, S.; Bechberger, J.; Mesnil, M.; Naus, C.C.; Sin, W.C. The carboxy-terminal tail of connexin43 gap junction protein is sufficient to mediate cytoskeleton changes in human glioma cells. J. Cell. Biochem. 2010, 110, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Chepied, A.; Daoud-Omar, Z.; Meunier-Balandre, A.C.; Laird, D.W.; Mesnil, M.; Defamie, N. Involvement of the Gap Junction Protein, Connexin43, in the Formation and Function of Invadopodia in the Human U251 Glioblastoma Cell Line. Cells 2020, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitomi, M.; Deleyrolle, L.P.; Mulkearns-Hubert, E.E.; Jarrar, A.; Li, M.; Sinyuk, M.; Otvos, B.; Brunet, S.; Flavahan, W.A.; Hubert, C.G.; et al. Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 2015, 11, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Sin, W.C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta 2012, 1818, 2058–2067. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Li, H.X.; Meng, H.Y.; Peng, X.X.; Zong, Q.; Zhang, K.; Han, G.L. A Meta-Analysis of Association Between Pesticides Exposure and Glioma Risk in Adults. J. Craniofac. Surg. 2015, 26, e672–e673. [Google Scholar]
- Woodley, M.A.; Te Nijenhuis, J.; Murphy, R. Were the Victorians cleverer than us? The decline in general intelligence estimated from a meta-analysis of the slowing of simple reaction time. Intelligence 2013, 41, 843–850. [Google Scholar] [CrossRef]
- Schönfelder, G.; Wittfoht, W.; Hopp, H.; Talsness, C.E.; Paul, M.; Chahoud, I. Parent bisphenol a accumulation in the human maternal-fetal-placental unit. Environ. Health Perspect. 2002, 110, A703–A707. [Google Scholar] [CrossRef]
- Fernandez, M.F.; Arrebola, J.P.; Taoufiki, J.; Navalón, A.; Ballesteros, O.; Pulgar, R.; Vilchez, J.L.; Olea, N. Bisphenol-A and chlorinated derivatives in adipose tissue of women. Reprod. Toxicol. 2007, 24, 259–264. [Google Scholar] [CrossRef]
- Koch, H.M.; Kolossa-Gehring, M.; Schröter-Kermani, C.; Angerer, J.; Brüning, T. Bisphenol A in 24 h urine and plasma samples of the German Environmental Specimen Bank from 1995 to 2009: A retrospective exposure evaluation. J. Expo. Sci. Environ. Epidemiol. 2012, 22, 610–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genuis, S.J.; Beesoon, S.; Lobo, R.A.; Birkholz, D. Human elimination of phthalate compounds: Blood, urine, and sweat (BUS) study. Sci. World J. 2012, 2012, 615068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikezuki, Y.; Tsutsumi, O.; Takai, Y.; Kamei, Y.; Taketani, Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum. Reprod. 2002, 17, 2839–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Pierik, F.H.; Angerer, J.; Meltzer, H.M.; Jaddoe, V.W.; Tiemeier, H.; Hoppin, J.A.; Longnecker, M.P. Levels of metabolites of organophosphate pesticides, phthalates, and bisphenol A in pooled urine specimens from pregnant women participating in the Norwegian Mother and Child Cohort Study (MoBa). Int. J. Hyg. Environ. Health 2009, 212, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Pirard, C.; Sagot, C.; Deville, M.; Dubois, N.; Charlier, C. Urinary levels of bisphenol A, triclosan and 4-nonylphenol in a general Belgian population. Environ. Int. 2012, 48, 78–83. [Google Scholar] [CrossRef]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Kuklenyik, Z.; Needham, L.L.; Calafat, A.M. Measuring environmental phenols and chlorinated organic chemicals in breast milk using automated on-line column-switching-high performance liquid chromatography-isotope dilution tandem mass spectrometry. Analyt. Technol. Biomed. Life Sci. 2006, 831, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Geens, T.; Neels, H.; Covaci, A. Distribution of bisphenol-A, triclosan and n-nonylphenol in human adipose tissue, liver and brain. Chemosphere 2012, 87, 796–802. [Google Scholar] [CrossRef]
- Calafat, A.M.; Weuve, J.; Ye, X.; Jia, L.T.; Hu, H.; Ringer, S.; Huttner, K.; Hauser, R. Exposure to bisphenol A and other phenols in neonatal intensive care unit premature infants. Environ. Health Perspect. 2009, 117, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Wu, C.; Zhang, J.; Qi, X.; Lv, S.; Jiang, S.; Zhou, T.; Lu, D.; Feng, C.; Chang, X.; et al. Prenatal exposure to mixture of heavy metals, pesticides and phenols and IQ in children at 7 years of age: The SMBCS study. Environ. Int. 2020, 139, 105692. [Google Scholar] [CrossRef]
- Tatsuta, N.; Nakai, K.; Kasanuma, Y.; Iwai-Shimada, M.; Sakamoto, M.; Murata, K.; Satoh, H. Prenatal and postnatal lead exposures and intellectual development among 12-year-old Japanese children. Environ. Res. 2020, 189, 109844. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.X.; Wang, Z.H.; Cheng, X.T.; Li, J.; Sang, Z.P.; Zhang, X.D.; Han, L.L.; Qiao, X.Y.; Wu, Z.M.; Wang, Z.Q. Arsenic and fluoride exposure in drinking water: children’s IQ and growth in Shanyin county, Shanxi province, China. Environ. Health Perspect. 2007, 115, 643–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Yanguas, S.C.; Willebrords, J.; Vinken, M. Models and methods for in vitro testing of hepatic gap junctional communication. Toxicol In Vitro 2015, 30, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandy, M.; Nyirenda, M. Developmental Origins of Health and Disease: The relevance to developing nations. Int. Health 2018, 10, 66–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martorell, R. Improved nutrition in the first 1000 days and adult human capital and health. Am. J. Hum. Biol. 2017, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, B.J.; Galvan, A.; Hare, T.A. Changes in cerebral functional organization during cognitive development. Curr. Opin. Neurobiol. 2005, 15, 239–244. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T.; Latz, E.; Manfredi, A. Mechanisms of sterile inflammation. Front. Immunol. 2013, 4, 398. [Google Scholar] [CrossRef] [Green Version]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Krämer-Albers, E.M. Extracellular vesicles as mediators of neuron-glia communication. Front. Cell. Neurosci. 2013, 7, 182. [Google Scholar] [CrossRef] [Green Version]
- Fróes, M.M.; Correia, A.H.; Garcia-Abreu, J.; Spray, D.C.; Campos de Carvalho, A.C.; Neto, M.V. Gap-junctional coupling between neurons and astrocytes in primary central nervous system cultures. Proc. Natl. Acad. Sci. USA 1999, 96, 7541–7546. [Google Scholar] [CrossRef] [Green Version]
- Rozental, R.; Andrade-Rozental, A.F.; Zheng, X.; Urban, M.; Spray, D.C.; Chiu, F.C. Gap junction-mediated bidirectional signaling between human fetal hippocampal neurons and astrocytes. Dev. Neurosci. 2001, 23, 420–431. [Google Scholar] [CrossRef]
- Wang, X.; Bukoreshtliev, N.V.; Gerdes, H.H. Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS ONE 2012, 7, e47429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schraufnagel, D.E.; Balmes, J.R.; De Matteis, S.; Hoffman, B.; Kim, W.J.; Perez-Padilla, R.; Rice, M.; Sood, A.; Vanker, A.; Wuebbles, D.J. Health Benefits of Air Pollution Reduction. Ann. Am. Thorac. Soc. 2019, 16, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Nielsen, O.; Koch, H.M.; Skakkebaek, N.E.; Juul, A.; Jørgensen, N.; Andersson, A.M. Changes in urinary excretion of phthalates, phthalate substitutes, bisphenols and other polychlorinated and phenolic substances in young Danish men; 2009–2017. Int. J. Hyg. Environ. Health 2020, 223, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The Blood-Brain Barrier Interface in Diabetes Mellitus: Dysfunctions, Mechanisms and Approaches to Treatment. Curr. Pharm. Des. 2020, 26, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
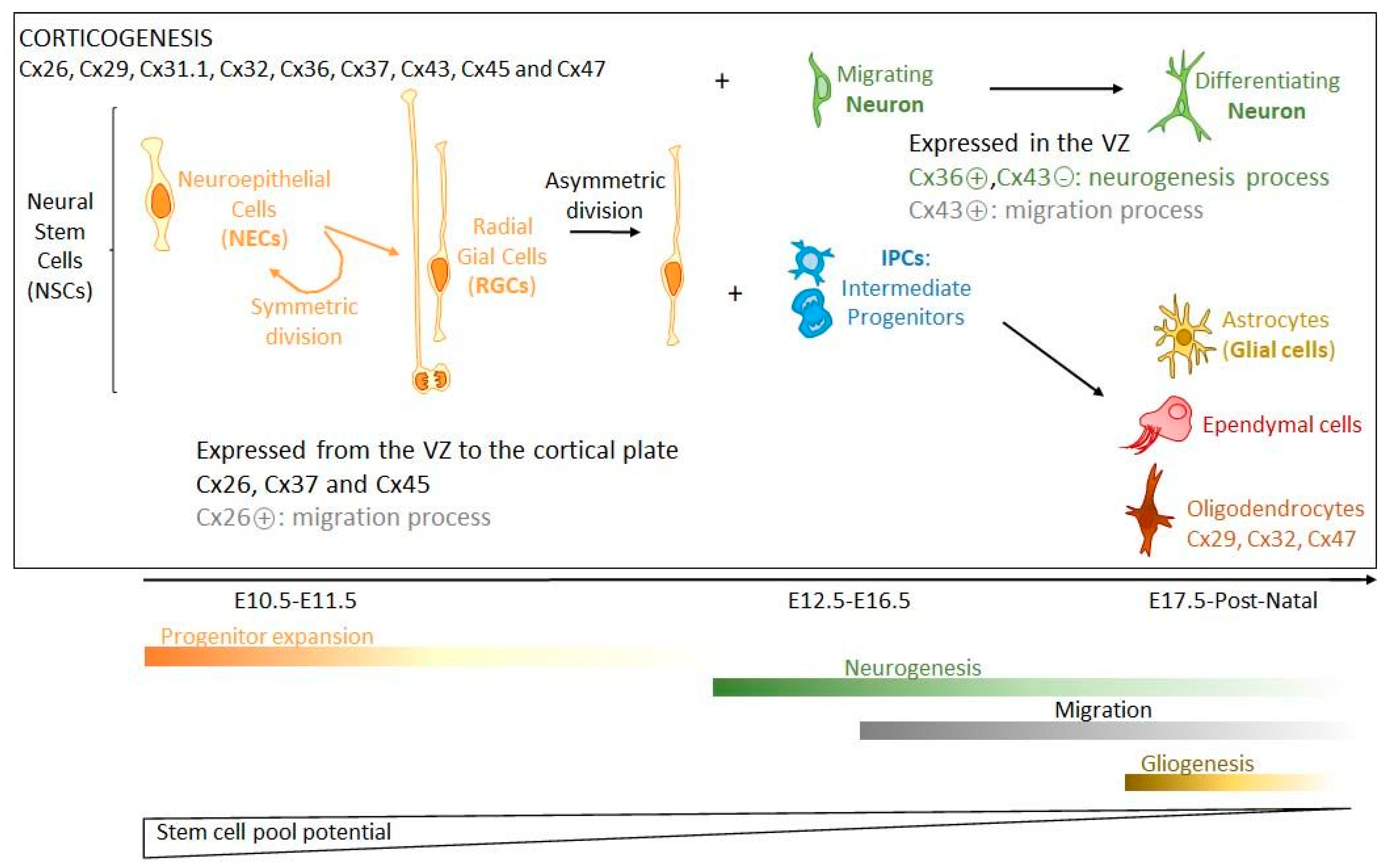
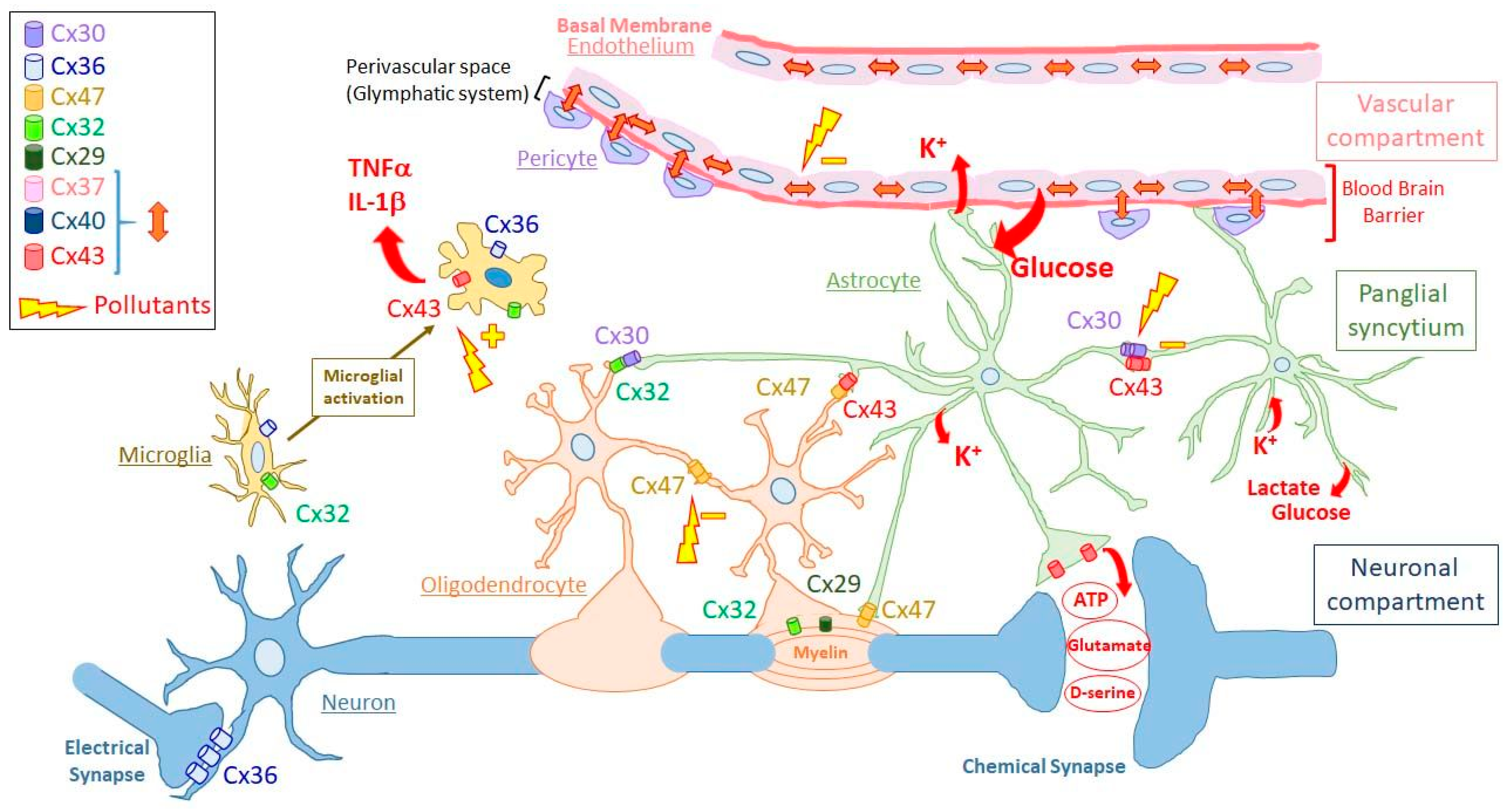
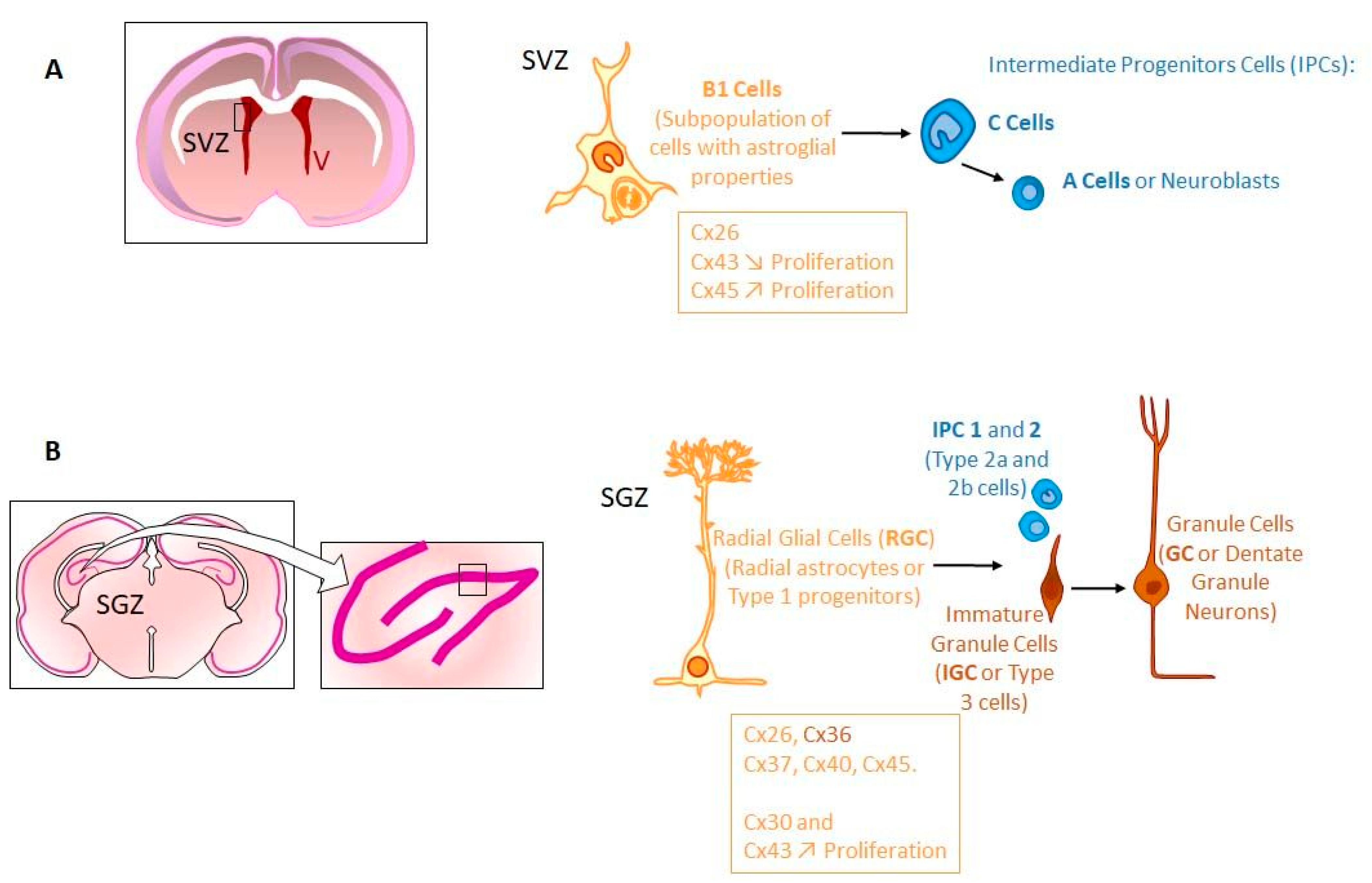

{kind=link}
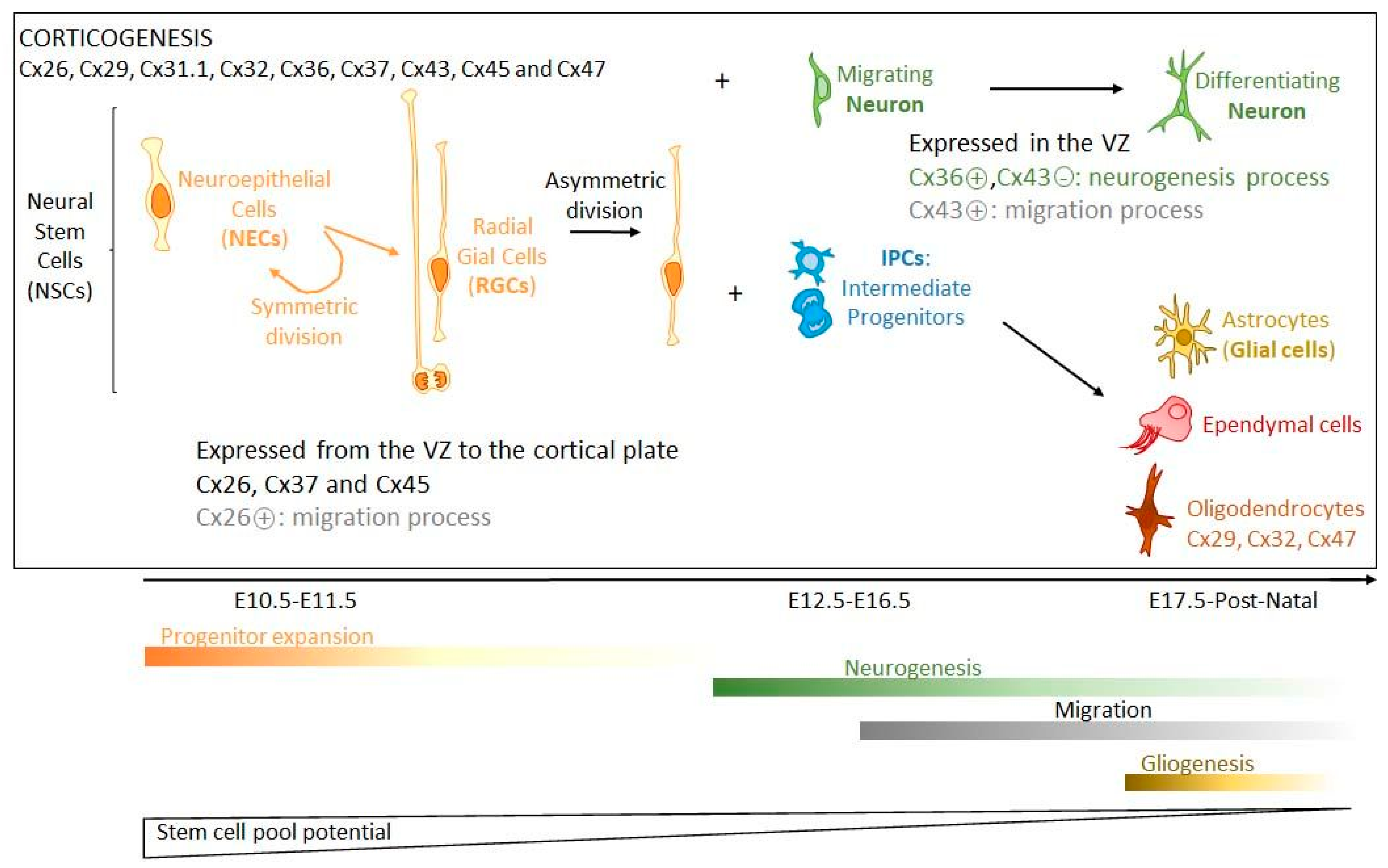{kind=link}
{kind=link}
{kind=link}
| Brain Pathologies [References] | Age at Diagnosis (Average) | Prevalence (% of Indicated Population) | Symptoms | Probable Causes |
|---|---|---|---|---|
| Neurodevelopmental | ||||
| ADHD 1 [6,7] | 9–10 years old (may appear as early as 3 years old) | 8–12% (of children worldwide) | -Attention deficit -Hyperactivity -Impulsivity | -Genetics -Exposures at pregnancy and young age (cigarette smoking, alcohol and drug uses, lead, …) -Brain injuries |
| ASD 2 [8,9,10] | From 2 years old or earlier | 1% (of total population) Prevalence increasing globally over past 50 years | -Lack of social interaction -Restricted interest -Repetitive behavior | -Genetics -Prenatal and postnatal exposures to environmental factors |
| Epilepsy [11] | Any age | 0.4–1% (of total population) | -Recurrent and brief episodes of involuntary movements that may be partial or generalized | -Genetics -Brain damage from prenatal or perinatal causes -Head injury -Brain infection (meningitis, encephalitis or neurocysticercosis, …) -Brain tumour |
| Neurodegenerative | ||||
| Alzheimer’s disease [2,12] | 65 years old | 10% (at age 65 and older in US) 146.2% increase in deaths from 2000 to 2018 | -Decline in memory, language, problem- solving -Decline in performing everyday activities through judgment, reasoning, and other cognitive abilities | -Age -Genetics -Environmental factors (cigarette smoking, alcohol use, …) |
| Parkinson’s disease [13,14] | 58 years old (1 patient/2) | 1% (above 60 years) | -Movement disorder (tremor, rigidity and bradykinesia) -Depression, anxiety | -Age -Genetics (5–10% of cases) -Environmental factors (pesticide exposure, synthetic heroine use, etc.) |
| Neurobehavioral | ||||
| MDD 3 [15,16] | 41.4 years | 28.2% (life time prevalence) | -Persistent sad mood -Anxiety -Irritability -Loss of interest -Loss of energy -Fatigue | -Genetics -Major life change, trauma, stress -Physical illness, medications |
| Migraines [17] | Any age | 12% (of total population) | -Recurrent attacks of moderate or severe headache lasting from 4 to 72 h | -Changes in hormonal levels in women -Circadian disruption -Genetics -Environmental factors (alcohol use, nitrates, carbon monoxide, etc…) |
| Other | ||||
| Cancer 4 [4] | Children and adults (45–70 years old) | 0.004–0.008% (of total population) | -Headache -Vertigo -Vision trouble (depends on tumor location) | -Genetics -Therapeutic irradiation -Electromagnetic fields (?) -N-nitroso compounds (food) -Unknown |
| GJ Compart. /GJ Networks | Cellular Components | Connexin Expression | Brain-Related Physiological Effects of Cx Deficiency in: | |||||
|---|---|---|---|---|---|---|---|---|
| Cell Types | Major Cell Functions | Cx Types (Major Cxs) | Cx Functions | Principal Physiological Roles of Major Cxs | Mice (Cx Knock out) | Human (Cx Gene Defect) | ||
| GJIC | HC | |||||||
| Neuronal | Neurons | Action potential transmission | Cx36 [25,26,27] Cx31.1 [23,29] Cx45 [30,31,32] Cx57 [33,34,35] | + [27] | + [28] | Electrical synapses (no synaptic delay): -Synchronization of interconnected neurons [27] -Excitability limitation in inhibitory networks [27] -Role in learning and memory [27] | Single KO Cx36: -Impaired short- and long-term memories [36,37] -Reduced motor learning [36,37] | Cx36: -Juvenile myoclonic epilepsy [38,39] |
| Panglial syncytium | Astrocytes | -Component of neuronal synapses (tripartite synapses) -BBB component (astrocytic endfeet) | Cx43 [40] Cx30 [40] Cx26 [41] | + [42] + [42] ? [43] | + [44] ? | -Glucose/lactate diffusion from capillaries to neurons and panglial syncytium (oligo., ependym.) [45] -K+ diffusion from neuron periphery and oligo. to capillaries [46,47] -Synaptic clearing of neurotransmitters, diffusion and recycling towards neurons [45] -Modulation of neuronal activity [48] -BBB maturation [49] -Gliosis and modulation of neuron activity through Cx43 HC activated by IL-1β/TNF-α (neuroinflam.) [50,51] or Ca2+/K+ [52,53,54]. Such activation leads to ATP release leading to paracrine/GJIC-mediated IP3/Ca2+ wave propagation (gliosis) [55] and gliotransmitters (glutamate) release (synaptic modulation) [56] | Single KO Cx43 (astro.): -Accelerated hippocampal spreading depression and enhanced locomotory activity [57] -Increased exploratory activity [58] -Anxiolytic-like effect in open field [58] Cx30: -Reduced exploratory activity -Anxiogenic behavior [59] Double KO Cx43 (astro.)/x30 [60]: -Vacuolated oligo. -Edematous astrocytes -Myelination abnormalities -Spatial memory impair -Sensimotor impair | Cx43: ODDD patients (according to mutations) [61]: -White matter change -Psychomotor delay -Epilepsy -Language disorder |
| Oligo. | -Axon myelination to facilitate propagation of action potentials | Cx2 9[62] Cx32 [63] Cx47 [63] | ? [64] + [62,65,66,67] + [62,65,66,67] | ? [68] | -Myelin integrity [69] -K+ buffering from neuron periphery to astrocytes [63,70] | Single KO Cx29: -No myelin defect [71] Cx32 [72]: -Enhanced intrinsic excitability of neurons -Myelination defects in neocortex -Dysfunction of inhibitory synaptic transmission Cx47 [66]: -No behavior abnormality -Vacuolation of nerve fibers -Myelination abnormality Double KO Cx32/Cx47 [73]: -Myelination abnormality -Astrogliosis -Microglia activation Cx30/Cx47 [74]: -Myelination defects -Severe motor impairment -Decreased number of oligodendrocytes -Microglia activation | Cx32: CMT1X patients: -Mostly peripheral neuropathy [75] -Encephalopathy linked to metabolic stress (white matter lesions) [76] Cx47: Pelizaeus-Merzbacher-like disease [77,78]: -Myelination abnormality -Impaired psychomotor development | |
| Ependym. | -Lining cerebral cavities -Restrictive barrier between cerebrospinal and interstitial fluids -Facilitate circulation of cerebrospinal fluid by ciliated cells | Cx43 [79] | + [79] | |||||
| Vascular | Endo. cells | -Lining brain capillaries | Cx37 [80] Cx40 [80] Cx43 [80] | + + + | -BBB integrity [81] -Maintenance and control of endothelial barrier function [81] | Cx37 polymorphism: -Ischemic stroke risk? [82] | ||
| Pericytes | -Regulation of blood flow through contraction capacity | Cx37 [80] Cx40 [80] Cx43 [80] | + + + | -Pericyte contraction? [83,84,85,86] -Maintenance of vascular homeostasis [81] | ||||
| Other | Microglia | -Inflammation control -Phagocyte micro-organisms and damaged cells | Cx32 [87] Cx36 [88] Cx43 [89] (When activated) | ? ? [88] (With neurons when activated) + [90,91] | + [87] + [92] | -Neuron death [87] -Control of NPC proliferation, differentiation and migration [93,94,95,96,97] -Exchange of antigenic peptides? [98] | ||
| Pollutants | GJIC | Cell Lines and Animal Models | References |
|---|---|---|---|
| Insecticides | |||
| Organochlorine insecticides | |||
| DDT 1 | I | Mammalian cells | [305,312,331] |
| Human breast epithelial cells | [313] | ||
| Rat liver epithelial cells (WB-F344) | [324] | ||
| Dieldrin | I | Mammalian cells | [305,332] |
| Chlordane, endosulfan, heptachlor, aldrin | I | Mammalian liver cells | [305,333,334] |
| Methoxychlor | I | Oviductal cells | [335] |
| Rat liver epithelial cells (WB-F344) | [325] | ||
| Lindane | I | Chinese hamster lung cells (V79) | [312] |
| Mouse hepatocytes (B6C3F1) | [314] | ||
| Rat myometrial smooth muscle cells | [315] | ||
| Rat liver epithelial cells (WB-F344) | [316] | ||
| Murine Sertoli cells (42GPA9) | [317,336] | ||
| Chlordecone | I | Chinese hamster lung cells (V79) | [337] |
| Human embryonic palatal mesenchymal cells | [338] | ||
| Mirex | I | Chinese hamster lung cells (V79) | [337] |
| Toxaphene | |||
| I | Human breast epithelial cells | [313] | |
| Herbicides | |||
| Ioxynil | I | Rat liver epithelial cells (IAR20) | [318] |
| Alachlor | I | Rat liver epithelial cells (WB-F344) | [324] |
| Fungicides | |||
| Vinclozolin | I | Rat liver epithelial cells (WB-F344) | [325] |
| Pentachlorophenol | I | Rat liver epithelial cells (WB-F344) | [324] |
| Bisphenol A | |||
| I | Rat epithelial mammary cells (BICRM1Rk) | [322] | |
| I | Cx46 HCs expressed in Xenopus oocytes | [309] | |
| A | Rat cumulus cell-oocyte complex | [339] | |
| I | Mouse cumulus cell-oocyte complex | [340] | |
| I | Human keratinocytes (HaCaT) | [319] | |
| Phthalates | |||
| Branched chain phthalate monoesters | I | Mouse hepatocytes (B6C3F1) | [341] |
| Straight chain phthalates | None | ||
| DINP 2 | I | Rat liver epithelial cells (WB-F344) Mouse hepatocytes (B6C3F1) | [342] |
| C7-C11 dialkyl phthalates | |||
| 8 phthalate monoesters | I | Rat and mouse hepatocytes | [343] |
| None | Hamster, cynomolgus and human hepatocytes | ||
| Medium-length side chain (3–6C) | I | Rat liver epithelial cells (WB-F344) | [326] |
| Others | |||
| PCB153 | I | Rat liver epithelial cells (WB-F344) | [324,327,328] |
| Derivatives of hydroxylated PCB 3 | I | Rat liver epithelial cells (WB-F344) | [344] |
| PBB 4 | I | Chinese hamster lung cells (V79) | [345,346] |
| DCP 5, PFDA 6, PFOA 7, PFOS 8, 1-monolaurin | I | Rat liver epithelial cells (WB-F344) | [324] |
| PAH 9 | I | Rat liver epithelial cells (WB-F344) | [329,347] |
| TCDD 10 | I | Human breat cells (MCF-7 and HMEC) | [330] |
| I | Rat liver epithelial cells (WB-F344) | [348] | |
| PM2.5 11 | I | Human lung fibroblasts | [349] |
| Aluminium | I | Rat astroglial cells | [350] |
| Mercury | I | Rat proximal tubular cells | [351] |
| Brain Disorders | Pathologies | Putative Environmental Causes | Connexin Characteristics and/or Possible Involvements |
|---|---|---|---|
| Neuro developmental | Autism spectrum disorders | Prenatal exposures to: -Traffic-related air pollutants [400] -Organochlorine pesticides [401] -Heavy metals (Hg and Pb) -Organic pollutants (DDT, PBDEs, PCBs) -Phthalates, BPA [402,403,404] -Nitrous oxide (N2O) [405] | Increased Cx43 expression (superior frontal cortex and less significantly in parietal region) [406] |
| Attention deficit hyperactivity disorders | Prenatal exposures to: -Cigarette smoke -Environmental toxins -Heavy metals (Pb) [407,408] -Air pollution [409,410,411,412] -Nitrous oxide [405] -Glyphosate [413] | In 10–25% of cases: 1q21.1 microdeletion (Cx40 and Cx50 gene loci?) [414] | |
| Epilepsy | -Air pollution (gaseous, fine particles) [415] -Air pollution (PM10) [416,417] -NO2 and SO2 pollution [418] -PCB (audiogenic seizures) [419,420] -PCE (in gestation, early childhood) [421] | Lack of GJIC between astrocytes [422,423] Increased Cx43 expression in hippocampus [424] | |
| Neurobehavioral | Migraines | -Traffic air pollution -PM2.5 [425] | GJ-mediated Ca2+ waves and Cx43 HC activation in astrocytes [426] |
| Major depressive disorders | Prenatal and childhood exposure: -BPA [427,428,429,430] Postnatal exposures: -Phthalates [415,431] -Heavy metals (Mn, Sn, Cd, Pb, Hg) [394,431] -PAHs [431] Prenatal and postnatal exposures: -Pesticides [394,395] -Air pollution (PM2.5) [396] | GJIC and Cx43 HC function in astrocytes [432] | |
| Neurodegenerative | Parkinson disease | -Traffic air pollution [433,434] -BPA [435] -Pesticides [401,436] -Metals (Pb, Hg, Al, Cd, Mn, As) [434,436] -PCBs [436] | Cx43 upregulation in rodent striatum [437] GJIC inhibition and decreased expression of Cx43 among astrocytes (Mn exposure) [438] |
| Alzheimer disease | -Air pollution [439,440] | Cx43 HC activation [441,442,443,444] | |
| Cancer | Glioma | -Pesticides (?) [445,446,447,448] | GJIC inhibition and decreased Cx43 expression in neuronal stem cells [449] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesnil, M.; Defamie, N.; Naus, C.; Sarrouilhe, D. Brain Disorders and Chemical Pollutants: A Gap Junction Link? Biomolecules 2021, 11, 51. https://doi.org/10.3390/biom11010051
Mesnil M, Defamie N, Naus C, Sarrouilhe D. Brain Disorders and Chemical Pollutants: A Gap Junction Link? Biomolecules. 2021; 11(1):51. https://doi.org/10.3390/biom11010051
Chicago/Turabian StyleMesnil, Marc, Norah Defamie, Christian Naus, and Denis Sarrouilhe. 2021. "Brain Disorders and Chemical Pollutants: A Gap Junction Link?" Biomolecules 11, no. 1: 51. https://doi.org/10.3390/biom11010051
APA StyleMesnil, M., Defamie, N., Naus, C., & Sarrouilhe, D. (2021). Brain Disorders and Chemical Pollutants: A Gap Junction Link? Biomolecules, 11(1), 51. https://doi.org/10.3390/biom11010051