Functional Evaluation of a Rare Variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) Gene Associated with Nonsyndromic Hearing Loss


{kind=link}
{kind=link}
{kind=link}
{kind=link}
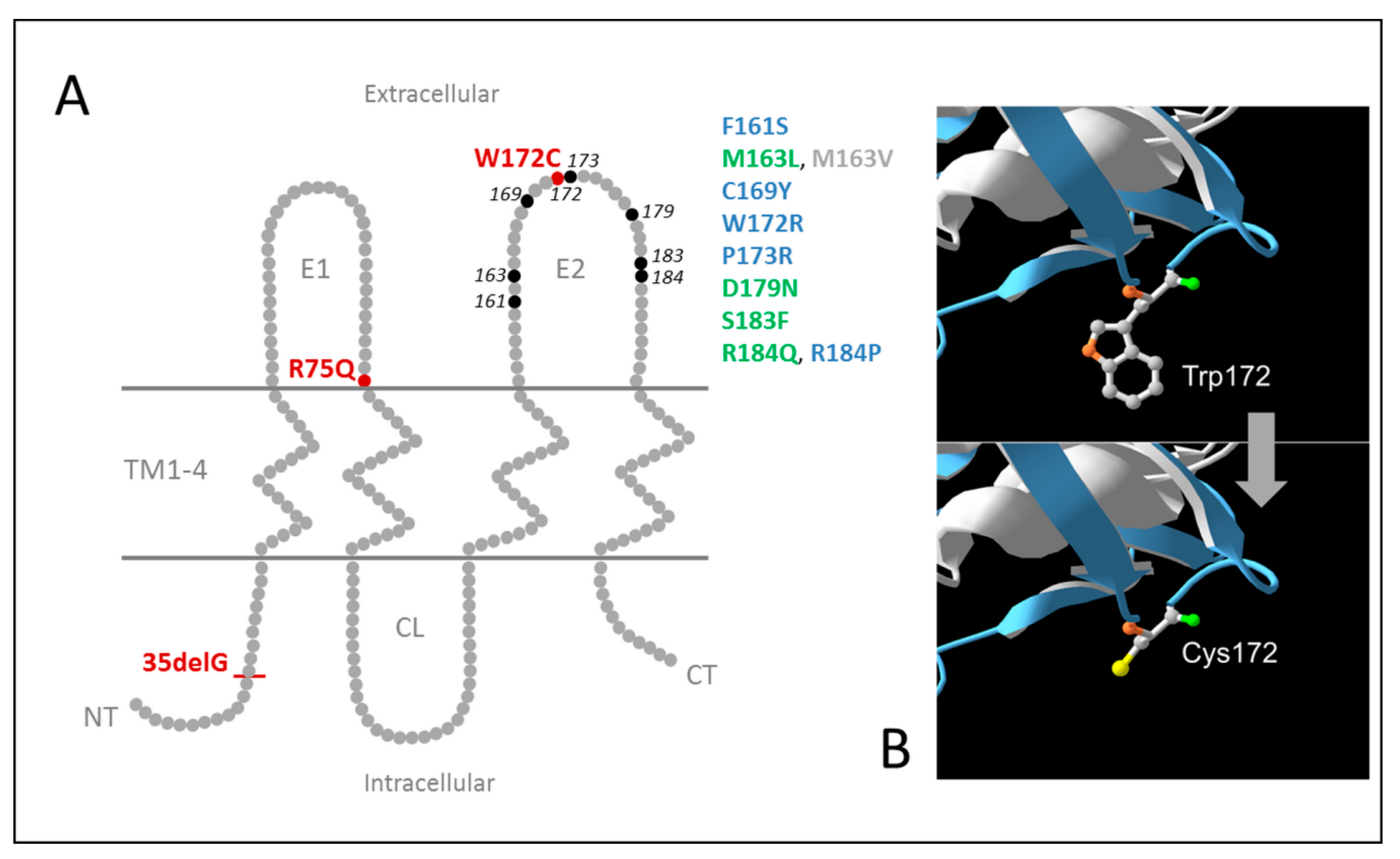{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. GJB2 Cloning Strategy
2.2. Cell Culture and Generating Cell Lines Expressing WT Cx26 or Cx26 Mutants
2.3. Immunostaining and Image Acquisition
2.4. Western Blot
2.5. Cx26-Hemichannels Permeability Assay
3. Results
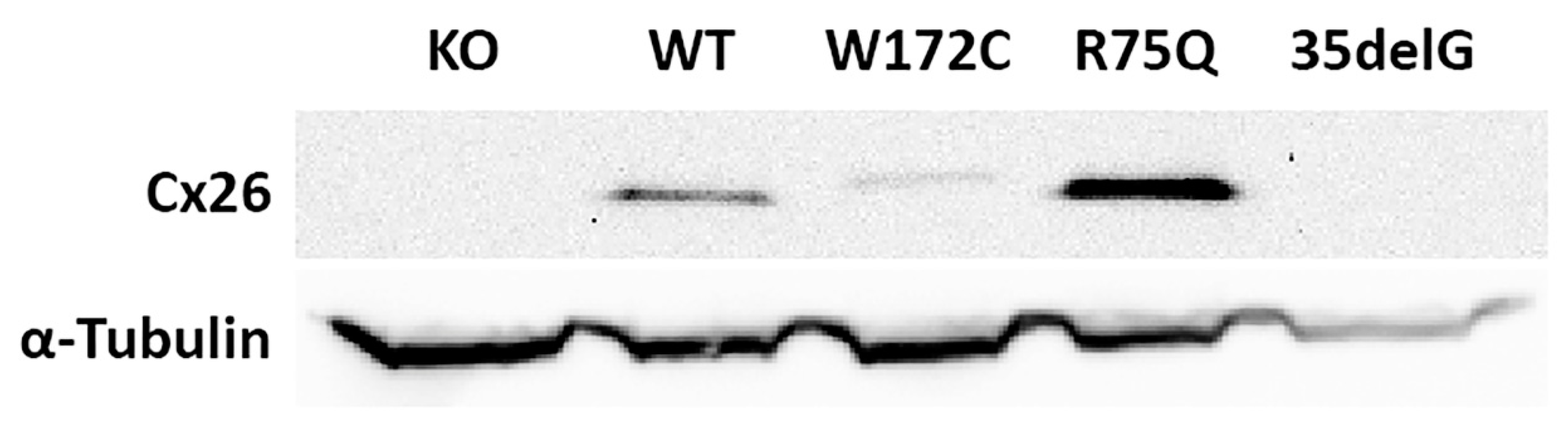
3.1. Generation of GJB2-Transgenic HeLa Cell Lines
3.2. Functional Studies
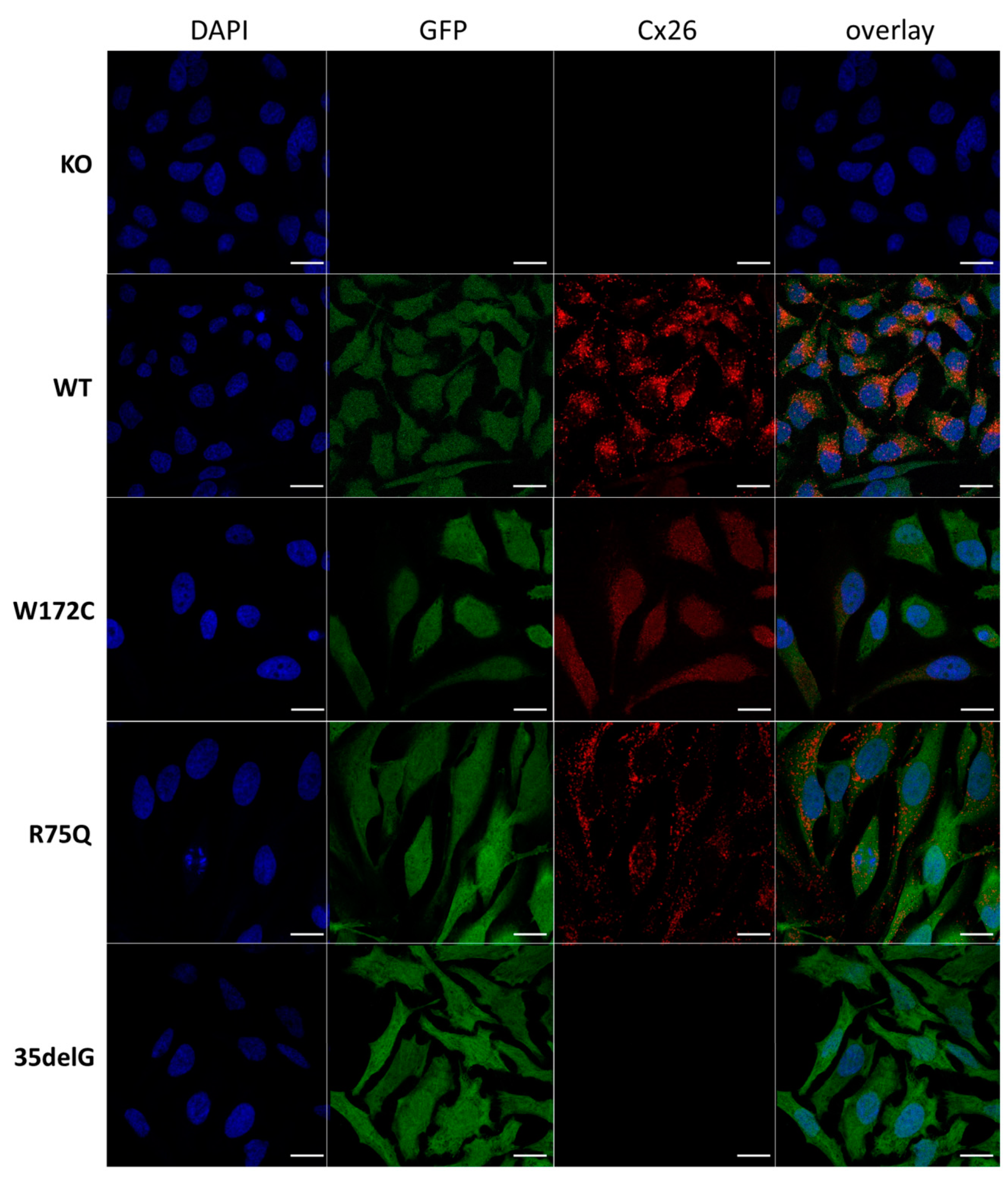
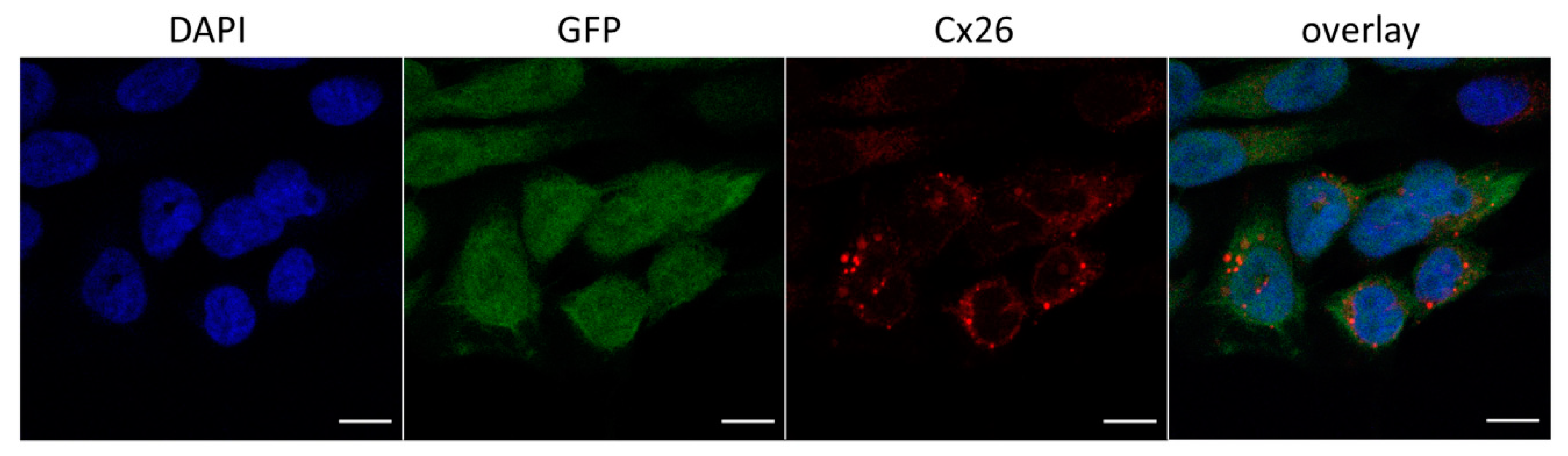
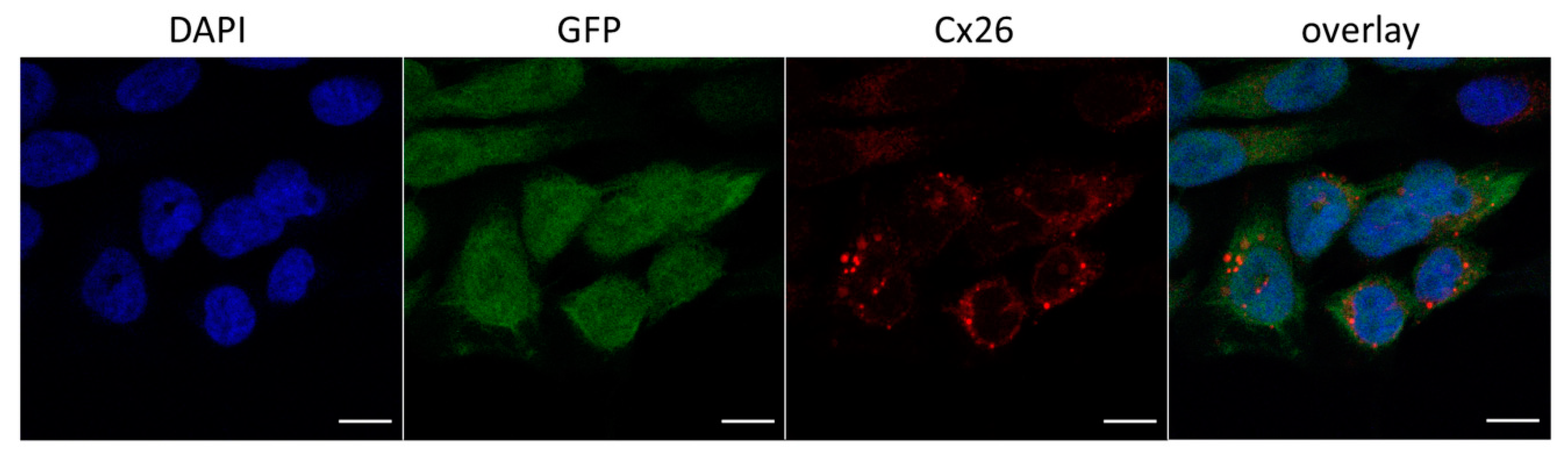
3.2.1. Cellular Localization of Cx26 Variants
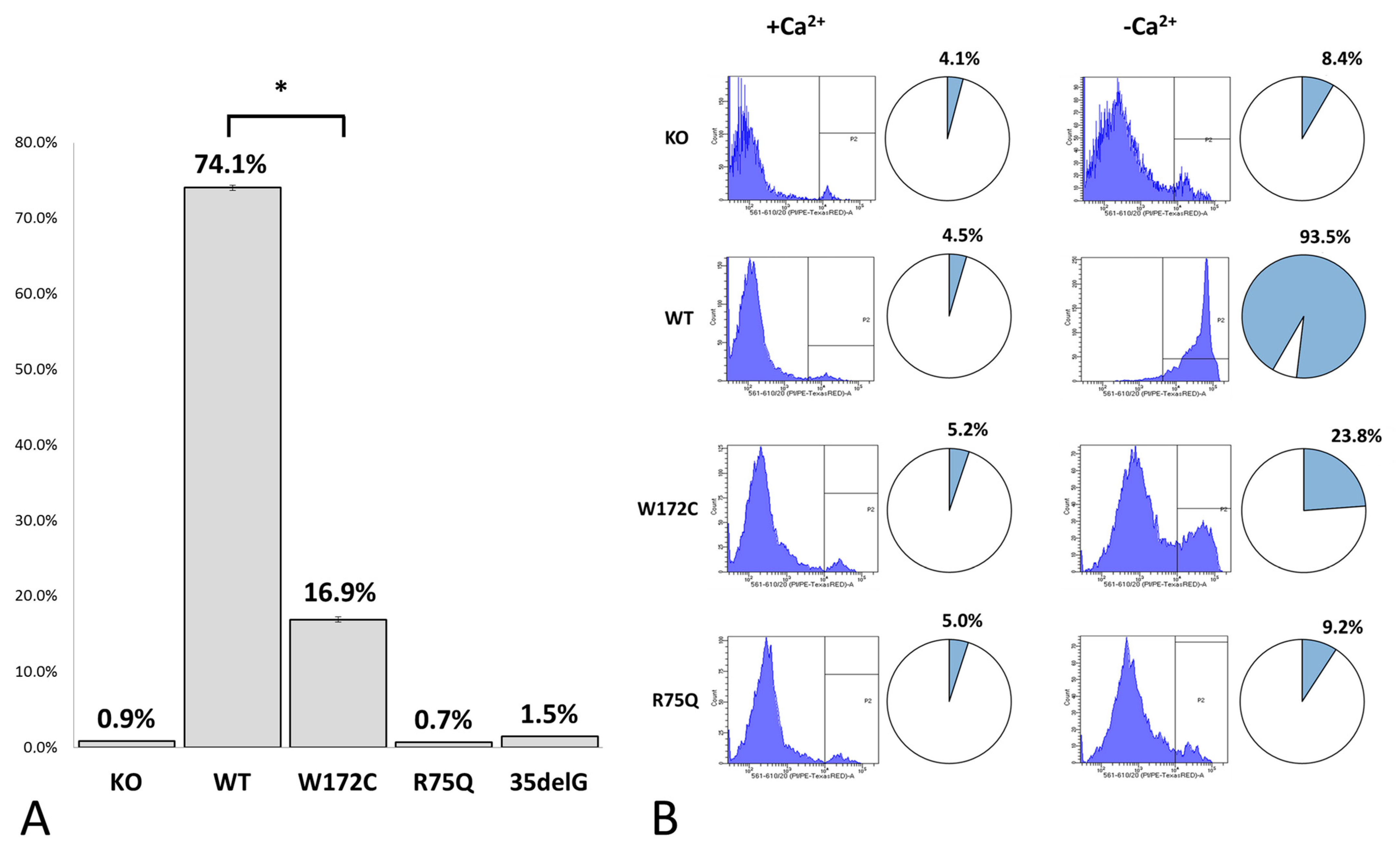
3.2.2. Evaluation of Cx26-Hemichannels Permeability by PI Dye Loading Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 4 April 2019).
- Toriello, H.V.; Smith, S.D. Hereditary Hearing Loss and Its Syndromes, 3rd ed.; Oxford University Press: Oxford, UK, 2013; p. 756. [Google Scholar]
- Del Castillo, F.J.; del Castillo, I. DFNB1 non-syndromic hearing impairment: Diversity of mutations and associated phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 2009, 458, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Stout, C.; Goodenough, D.A.; Paul, D.L. Connexins: Functions without junctions. Curr. Opin. Cell Biol. 2004, 16, 507–512. [Google Scholar] [CrossRef]
- Vinken, M. Connexin hemichannels: Novel mediators of toxicity. Arch. Toxicol. 2015, 89, 143–145. [Google Scholar] [CrossRef] [Green Version]
- Bruzzone, R.; White, T.W.; Paul, D.L. Connections with connexins: The molecular basis of direct intercellular signaling. Eur. J. Biochem. 1996, 238, 1–27. [Google Scholar] [CrossRef]
- Kikuchi, T.; Adams, J.C.; Miyabe, Y.; So, E.; Kobayashi, T. Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness. Med. Electron. Microsc. 2000, 33, 51–56. [Google Scholar] [CrossRef]
- Beltramello, M.; Piazza, V.; Bukauskas, F.F.; Pozzan, T.; Mammano, F. Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat. Cell Biol. 2005, 7, 63–69. [Google Scholar] [CrossRef]
- Jagger, D.J.; Forge, A. Connexins and gap junctions in the inner ear—It’s not just about K+ recycling. Cell Tissue Res. 2015, 360, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.B. Hypothesis of K+-Recycling Defect Is Not a Primary Deafness Mechanism for Cx26 (GJB2) Deficiency. Front. Mol. Neurosci. 2017, 10, 162. [Google Scholar] [CrossRef] [Green Version]
- Wiszniewski, L.; Limat, A.; Saurat, J.H.; Meda, P.; Salomon, D. Differential expression of connexins during stratification of human keratinocytes. J. Investig. Dermatol. 2000, 115, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, W.L.; Common, J.E.; Kelsell, D.P. Connexin 26 expression and mutation analysis in epidermal disease. Cell Commun. Adhes. 2001, 8, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.A.; Kelsell, D.P. Key functions for gap junctions in skin and hearing. Biochem. J. 2011, 438, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Meşe, G.; Richard, G.; White, T.W. Gap junctions: Basic structure and function. J. Investig. Dermatol. 2007, 127, 2516–2524. [Google Scholar] [CrossRef] [Green Version]
- Wingard, J.C.; Zhao, H.B. Cellular and Deafness Mechanisms Underlying Connexin Mutation-Induced Hearing Loss—A Common Hereditary Deafness. Front. Cell Neurosci. 2015, 9, 202. [Google Scholar] [CrossRef] [Green Version]
- Beach, R.; Abitbol, J.M.; Allman, B.L.; Esseltine, J.L.; Shao, Q.; Laird, D.W. GJB2 Mutations Linked to Hearing Loss Exhibit Differential Trafficking and Functional Defects as Revealed in Cochlear-Relevant Cells. Front. Cell Dev. Biol. 2020, 8, 215. [Google Scholar] [CrossRef]
- Denoyelle, F.; Lina-Granade, G.; Plauchu, H.; Bruzzone, R.; Chaïb, H.; Lévi-Acobas, F.; Weil, D.; Petit, C. Connexin 26 gene linked to a dominant deafness. Nature 1998, 393, 319–320. [Google Scholar] [CrossRef]
- Maestrini, E.; Korge, B.P.; Ocaña-Sierra, J.; Calzolari, E.; Cambiaghi, S.; Scudder, P.M.; Hovnanian, A.; Monaco, A.P.; Munro, C.S. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum. Mol. Genet. 1999, 8, 1237–1243. [Google Scholar] [CrossRef] [Green Version]
- Kelsell, D.P.; Wilgoss, A.L.; Richard, G.; Stevens, H.P.; Munro, C.S.; Leigh, I.M. Connexin mutations associated with palmoplantar keratoderma and profound deafness in a single family. Eur. J. Hum. Genet. 2000, 8, 469–472. [Google Scholar]
- Richard, G.; Rouan, F.; Willoughby, C.E.; Brown, N.; Chung, P.; Ryynänen, M.; Jabs, E.W.; Bale, S.J.; DiGiovanna, J.J.; Uitto, J.; et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am. J. Hum. Genet. 2002, 70, 1341–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marziano, N.K.; Casalotti, S.O.; Portelli, A.E.; Becker, D.L.; Forge, A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum. Mol. Genet. 2003, 12, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Scherer, S.S.; Yum, S.W. Dominant Cx26 mutants associated with hearing loss have dominant-negative effects on wild type Cx26. Mol. Cell Neurosci. 2011, 47, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Qual. Life Res. 2017, 136, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Thönnissen, E.; Rabionet, R.; Arbonès, M.L.; Estivill, X.; Willecke, K.; Ott, T. Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum. Genet. 2002, 111, 190–197. [Google Scholar] [CrossRef]
- Bruzzone, R.; Veronesi, V.; Gomès, D.; Bicego, M.; Duval, N.; Marlin, S.; Petit, C.; D’Andrea, P.; White, T.W. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003, 533, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Oshima, A.; Doi, T.; Mitsuoka, K.; Maeda, S.; Fujiyoshi, Y. Roles of Met-34, Cys-64, and Arg-75 in the assembly of human connexin 26. Implication for key amino acid residues for channel formation and function. J. Biol. Chem. 2003, 278, 1807–1816. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.L.; Chang, W.T.; Li, A.H.; Yeh, T.H.; Wu, C.Y.; Chen, M.S.; Huang, P.C. Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. J. Neurochem. 2003, 84, 735–742. [Google Scholar] [CrossRef]
- Thomas, T.; Telford, D.; Laird, D.W. Functional domain mapping and selective trans-dominant effects exhibited by Cx26 disease-causing mutations. J. Biol. Chem. 2004, 279, 19157–19168. [Google Scholar] [CrossRef] [Green Version]
- Skerrett, I.M.; Di, W.L.; Kasperek, E.M.; Kelsell, D.P.; Nicholson, B.J. Aberrant gating, but a normal expression pattern, underlies the recessive phenotype of the deafness mutant Connexin26M34T. FASEB J. 2004, 18, 860–862. [Google Scholar] [CrossRef] [Green Version]
- Melchionda, S.; Bicego, M.; Marciano, E.; Franzè, A.; Morgutti, M.; Bortone, G.; Zelante, L.; Carella, M.; D’Andrea, P. Functional characterization of a novel Cx26 (T55N) mutation associated to non-syndromic hearing loss. Biochem. Biophys. Res. Commun. 2005, 337, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Piazza, V.; Beltramello, M.; Menniti, M.; Colao, E.; Malatesta, P.; Argento, R.; Chiarella, G.; Gallo, L.V.; Catalano, M.; Perrotti, N.; et al. Functional analysis of R75Q mutation in the gene coding for Connexin 26 identified in a family with nonsyndromic hearing loss. Clin. Genet. 2005, 68, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Palmada, M.; Schmalisch, K.; Böhmer, C.; Schug, N.; Pfister, M.; Lang, F.; Blin, N. Loss of function mutations of the GJB2 gene detected in patients with DFNB1-associated hearing impairment. Neurobiol. Dis. 2006, 22, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Bicego, M.; Beltramello, M.; Melchionda, S.; Carella, M.; Piazza, V.; Zelante, L.; Bukauskas, F.F.; Arslan, E.; Cama, E.; Pantano, S.; et al. Pathogenetic role of the deafness-related M34T mutation of Cx26. Hum. Mol. Genet. 2006, 15, 2569–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerido, D.A.; DeRosa, A.M.; Richard, G.; White, T.W. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am. J. Physiol. Cell Physiol. 2007, 293, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, T.D.; Caria, H.; Simões-Teixeira, H.; Aasen, T.; Dias, O.; Andrea, M.; Kelsell, D.P.; Fialho, G. A novel M163L mutation in connexin 26 causing cell death and associated with autosomal dominant hearing loss. Hear. Res. 2008, 240, 87–92. [Google Scholar] [CrossRef]
- Choi, S.Y.; Park, H.J.; Lee, K.Y.; Dinh, E.H.; Chang, Q.; Ahmad, S.; Lee, S.H.; Bok, J.; Lin, X.; Kim, U.K. Different functional consequences of two missense mutations in the GJB2 gene associated with non-syndromic hearing loss. Hum. Mutat. 2009, 30, E716–E727. [Google Scholar] [CrossRef]
- Mani, R.S.; Ganapathy, A.; Jalvi, R.; Srikumari Srisailapathy, C.R.; Malhotra, V.; Chadha, S.; Agarwal, A.; Ramesh, A.; Rangasayee, R.R.; Anand, A. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur. J. Hum. Genet. 2009, 17, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Yum, S.W.; Zhang, J.; Scherer, S.S. Dominant connexin26 mutants associated with human hearing loss have trans-dominant effects on connexin30. Neurobiol. Dis. 2010, 38, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.-Y.; Lee, K.Y.; Kim, H.-J.; Kim, H.-K.; Chang, Q.; Park, H.-J.; Jeon, C.-J.; Lin, X.; Bok, J.; Kim, U.-K. Functional evaluation of GJB2 variants in nonsyndromic hearing loss. Mol. Med. 2011, 17, 550–556. [Google Scholar] [CrossRef]
- Su, C.C.; Li, S.Y.; Su, M.C.; Chen, W.C.; Yang, J.J. Mutation R184Q of connexin 26 in hearing loss patients has a dominant-negative effect on connexin 26 and connexin 30. Eur. J. Hum. Genet. 2010, 18, 1061–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosi, C.; Walker, A.E.; Depriest, A.D.; Cone, A.C.; Lu, C.; Badger, J.; Skerrett, I.M.; Sosinsky, G.E. Analysis of trafficking, stability and function of human connexin 26 gap junction channels with deafness-causing mutations in the fourth transmembrane helix. PLoS ONE 2013, 8, e70916. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, J.; Lee, M.G.; Choi, J.Y.; Lee, K.A. Non-syndromic hearing loss caused by the dominant cis mutation R75Q with the recessive mutation V37I of the GJB2 (Connexin 26) gene. Exp. Mol. Med. 2015, 47, e169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zonta, F.; Girotto, G.; Buratto, D.; Crispino, G.; Morgan, A.; Abdulhadi, K.; Alkowari, M.; Badii, R.; Gasparini, P.; Mammano, F. The p.Cys169Tyr variant of connexin 26 is not a polymorphism. Hum. Mol. Genet. 2015, 24, 2641–2648. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Paris, J.; Waldhaus, J.; Gordhandas, J.A.; Pique, L.; Schrijver, I. Comparative functional characterization of novel non-syndromic GJB2 gene variant p.Gly45Arg and lethal syndromic variant p.Gly45Glu. Peer J. 2016, 4, e2494. [Google Scholar] [CrossRef]
- Press, E.R.; Shao, Q.; Kelly, J.J.; Chin, K.; Alaga, A.; Laird, D.W. Induction of cell death and gain-of-function properties of connexin26 mutants predict severity of skin disorders and hearing loss. J. Biol. Chem. 2017, 292, 9721–9732. [Google Scholar] [CrossRef] [Green Version]
- Posukh, O.L.; Pallares-Ruiz, N.; Tadinova, V.; Osipova, L.P.; Claustres, M.; Roux, A.-F. First molecular screening of deafness in the Altai Republic population. BMC Med. Genet. 2005, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Posukh, O.L.; Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Bondar, A.A.; Morozov, I.V.; Maximov, V.N.; Voevoda, M.I. Unique mutational spectrum of the GJB2 Gene and its pathogenic contribution to deafness in Tuvinians (Southern Siberia, Russia): A high prevalence of rare variant c.516G>C (p.Trp172Cys). Genes 2019, 10, 429. [Google Scholar] [CrossRef] [Green Version]
- Posukh, O.L.; Institute of Cytology and Genetics, Novosibirsk, Russia. Personal communication, 2019.
- Tekin, M.; Xia, X.-J.; Erdenetungalag, R.; Cengiz, F.B.; White, T.W.; Radnaabazar, J.; Dangaasuren, B.; Tastan, H.; Nance, W.E.; Pandya, A. GJB2 Mutations in Mongolia: Complex Alleles, Low Frequency, and Reduced Fitness of the Deaf. Ann. Hum. Genet. 2010, 74, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. High Rates of Three Common GJB2 Mutations c.516G>C, c.-23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect. Genes 2020, 11, 833. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Distefano, M.T.; Hemphill, S.E.; Oza, A.M.; Siegert, R.K.; Grant, A.R.; Hughes, M.Y.; Cushman, B.J.; Azaiez, H.; Booth, K.T.; Chapin, A.; et al. ClinGen expert clinical validity curation of 164 hearing loss gene–disease pairs. Genet. Med. 2019, 21, 2239–2247. [Google Scholar] [CrossRef]
- Kowarz, E.; Loescher, D.; Marschalek, R. Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol. J. 2015, 10, 647–653. [Google Scholar] [CrossRef]
- Okunev, A.G.; Nartova, A.V.; Matveev, A.V. Recognition of Nanoparticles on Scanning Probe Microscopy Images using Computer Vision and Deep Machine Learning. In Proceedings of the IEEE International Multi-Conference on Engineering, Computer and Information Sciences (SIBIRCON), Novosibirsk, Russia, 25–27 October 2019. [Google Scholar]
- Elfgang, C.; Eckert, R.; Lichtenberg-Fraté, H.; Butterweck, A.; Traub, O.; Klein, R.A.; Hülser, D.F.; Willecke, K. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J. Cell Biol. 1995, 129, 805–817. [Google Scholar] [CrossRef]
- Cao, F.; Eckertm, R.; Elfgang, C.; Nitsche, J.M.; Snyder, S.A.; Hulser, D.F.; Willecke, K.; Nicholson, B.J. A quantitative analysis of connexin-specific permeability differences of gap junctions expressed in HeLa transfectants and Xenopus oocytes. J. Cell Sci. 1998, 111, 31–43. [Google Scholar] [PubMed]
- Sahu, G.; Sukumaran, S.; Bera, A.K. Pannexins form gap junctions with electrophysiological and pharmacological properties distinct from connexins. Sci. Rep. 2014, 4, 4955. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Palacios-Prado, N.; Sáez, J.C.; Lee, J. Confirmation of Connexin45 Underlying Weak Gap Junctional Intercellular Coupling in HeLa Cells. Biomolecules 2020, 10, E1389. [Google Scholar] [CrossRef]
- Uyguner, O.; Tukel, T.; Baykal, C.; Eris, H.; Emiroglu, M.; Hafiz, G.; Ghanbari, A.; Baserer, N.; Yuksel-Apak, M.; Wollnik, B. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clin. Genet. 2002, 62, 306–309. [Google Scholar] [CrossRef]
- Feldmann, D.; Denoyelle, F.; Blons, H.; Lyonnet, S.; Loundon, N.; Rouillon, I.; Hadj-Rabia, S.; Petit, C.; Couderc, R.; Garabédian, E.N.; et al. The GJB2 mutation R75Q can cause nonsyndromic hearing loss DFNA3 or hereditary palmoplantar keratoderma with deafness. Am. J. Med. Genet. A 2005, 137, 225–227. [Google Scholar] [CrossRef]
- Pavithra, A.; Selvakumari, M.; Nityaa, V.; Sharanya, N.; Ramakrishnan, R.; Narasimhan, M.; Srisailapathy, C.R. Autosomal dominant hearing loss resulting from p.R75Q mutation in the GJB2 gene: Nonsyndromic presentation in a South Indian family. Ann. Hum. Genet. 2015, 79, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparini, P.; Rabionet, R.; Barbujani, G.; Melchionda, S.; Petersen, M.; Brøndum-Nielsen, K.; Metspalu, A.; Oitmaa, E.; Pisano, M.; Fortina, P.; et al. High carrier frequency of the 35delG deafness mutation in European populations. Eur. J. Hum. Genet. 2000, 8, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.J.; Hand, G.M.; Engel, A.; Sosinsky, G.E. Conformational changes in surface structures of isolated connexin 26 gap junctions. EMBO J. 2002, 21, 3598–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimm, J.; Mechler, A.; Lin, H.; Rhee, S.; Lal, R. Calcium-dependent open/closed conformations and interfacial energy maps of reconstituted hemichannels. J. Biol. Chem. 2005, 280, 10646–10654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Zwart-Storm, E.A.; van Geel, M.; van Neer, P.A.; Steijlen, P.M.; Martin, P.E.; van Steensel, M.A. A novel missense mutation in the second extracellular domain of GJB2, p.Ser183Phe, causes a syndrome of focal palmoplantar keratoderma with deafness. Am. J. Pathol. 2008, 173, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuja, Z.; Li, L.; Gupta, S.; Meşe, G.; White, T.W. Connexin26 Mutations Causing Palmoplantar Keratoderma and Deafness Interact with Connexin43, Modifying Gap Junction and Hemichannel Properties. J. Investig. Dermatol. 2016, 136, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Martin, P.E.; Evans, W.H. Assembly of gap junction channels: Mechanism, effects of calmodulin antagonists and identification of connexin oligomerization determinants. Eur. J. Biochem. 2001, 268, 4544–4552. [Google Scholar] [CrossRef]
- Maza, J.; Sarma, J.D.; Koval, M. Defining a minimal motif required to prevent connexin oligomerization in the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 21115–21121. [Google Scholar] [CrossRef] [Green Version]
- Martínez, A.D.; Acuña, R.; Figueroa, V.; Maripillan, J.; Nicholson, B. Gap-junction channels dysfunction in deafness and hearing loss. Antioxid. Redox Signal. 2009, 11, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Nicholson, B.J. The role of connexins in ear and skin physiology—Functional insights from disease-associated mutations. Biochim. Biophys. Acta Biomembr. 2013, 1828, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Bai, D. Structural analysis of key gap junction domains—Lessons from genome data and disease-linked mutants. Semin. Cell Dev. Biol. 2016, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.C.; Purdy, M.D.; Baker, K.A.; Acharya, C.; McIntire, W.E.; Stevens, R.C.; Zhang, Q.; Harris, A.L.; Abagyan, R.; Yeager, M. An electrostatic mechanism for Ca2 þ–mediated regulation of gap junction channels. Nat. Commun. 2016, 7, 8770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, D.; Yue, B.; Aoyama, H. Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Biophys. Acta Biomembr. 2018, 1860, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maslova, E.A.; Orishchenko, K.E.; Posukh, O.L. Functional Evaluation of a Rare Variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) Gene Associated with Nonsyndromic Hearing Loss. Biomolecules 2021, 11, 61. https://doi.org/10.3390/biom11010061
Maslova EA, Orishchenko KE, Posukh OL. Functional Evaluation of a Rare Variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) Gene Associated with Nonsyndromic Hearing Loss. Biomolecules. 2021; 11(1):61. https://doi.org/10.3390/biom11010061
Chicago/Turabian StyleMaslova, Ekaterina A., Konstantin E. Orishchenko, and Olga L. Posukh. 2021. "Functional Evaluation of a Rare Variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) Gene Associated with Nonsyndromic Hearing Loss" Biomolecules 11, no. 1: 61. https://doi.org/10.3390/biom11010061
APA StyleMaslova, E. A., Orishchenko, K. E., & Posukh, O. L. (2021). Functional Evaluation of a Rare Variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) Gene Associated with Nonsyndromic Hearing Loss. Biomolecules, 11(1), 61. https://doi.org/10.3390/biom11010061