
Nicotinamide N-Methyltransferase: An Emerging Protagonist in Cancer Macro(r)evolution


Abstract
:1. Introduction
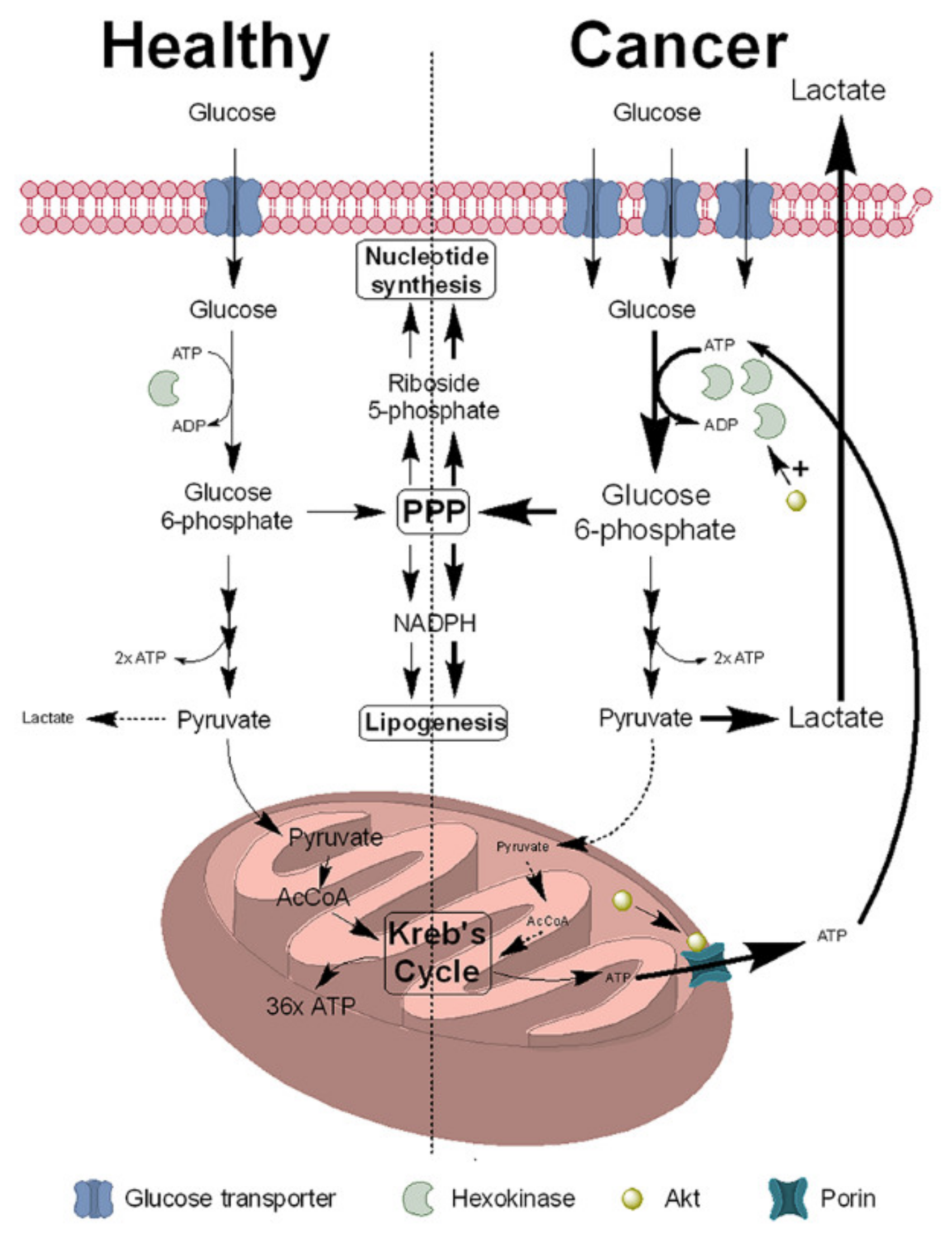
2. NNMT Expression Is Increased in Cancer Cells to Support the Warburg Effect
2.1. NAD+ Synthesis
2.2. NNMT Function and Regulation of NAD+ Synthesis
2.3. NNMT Promotes the Cancer Phenotype
2.4. NNMT Induces Sirtuin Expression and Activity
2.5. NNMT Increases Mitochondrial Function
2.6. NNMT Activates the Akt Signalling Pathway
3. NNMT Promotes the Epithelial-to-Mesenchymal Transition
4. Linking NNMT with Its Cellular Functions
4.1. Epigenetic Regulation
4.2. Regulation of NAD+-Dependent Pathways
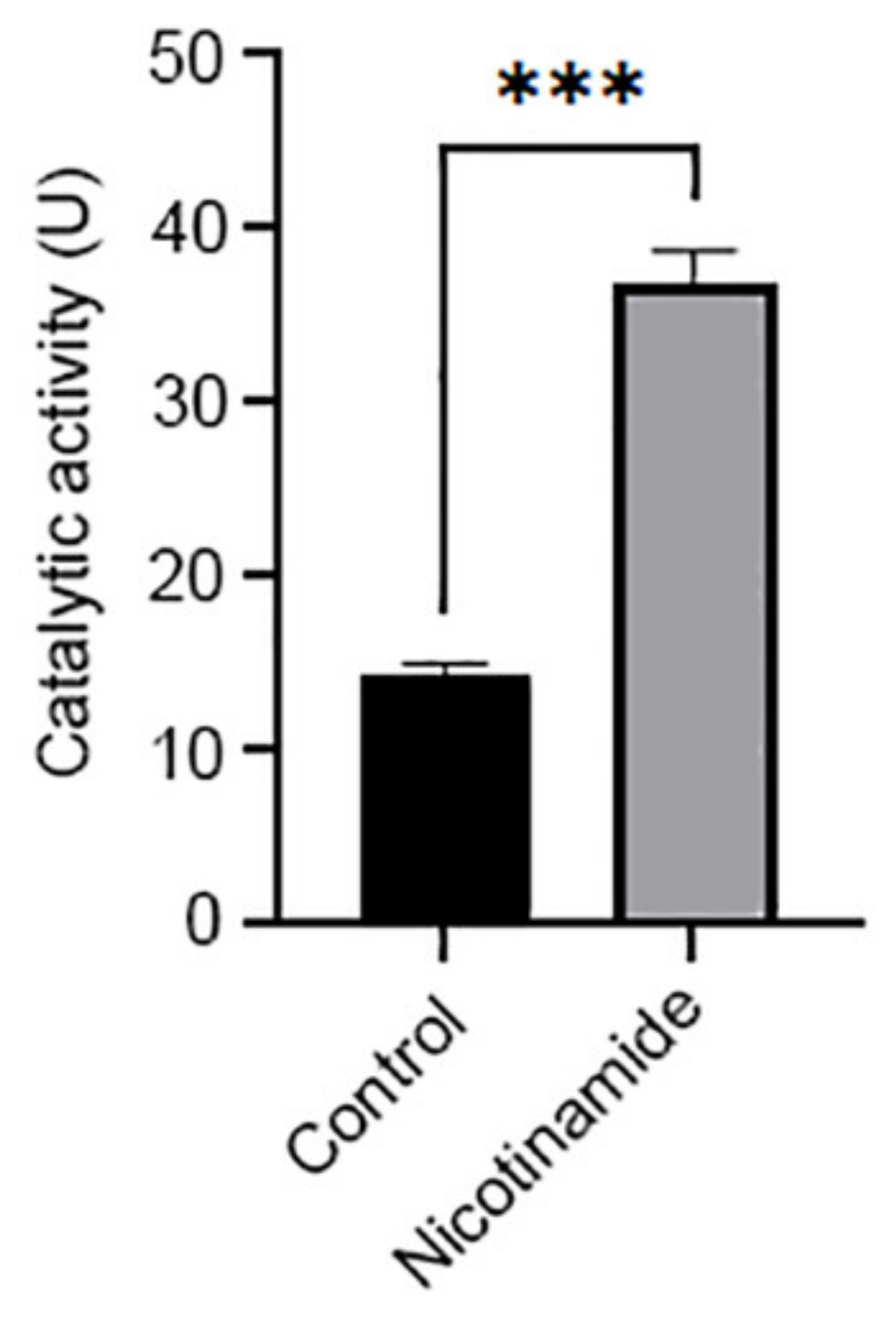
4.3. Synthesis of 1-Methylnicotinamide
5. Mechanisms Underlying Increased NNMT Activity in Cancer Cells
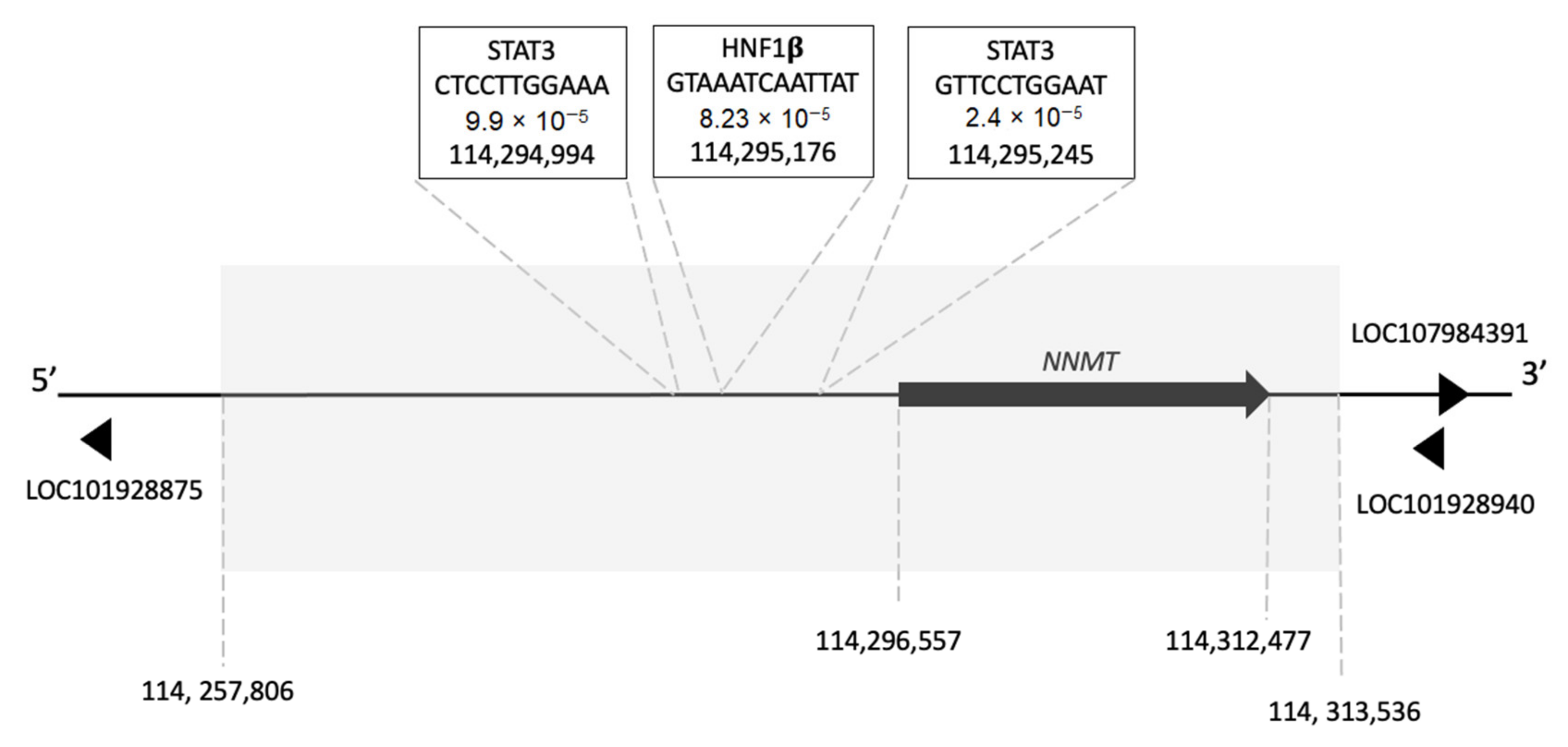
5.1. Transcription Factor Binding Sites in the NNMT Gene
5.2. NNMT Single-Nucleotide Polymorphisms and Cancer
5.3. Post-Translational Modifications
6. NNMT and Cancer—Cause or Consequence?
7. Therapeutic Targeting of NNMT
7.1. Small-Molecule Inhibitors of NNMT Activity
7.2. Nicotinamide Analogues
7.3. Covalent Inhibitors
7.4. Bisubstrate Inhibitors
7.5. Small-Molecule Inhibitors of NNMT Expression

8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.A.; Brand, M.D. Quantifying intracellular rated of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer metabolism. Nat. Rev. 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Cell Biology. Warburg effect and redox balance. Science 2011, 334, 1219–1220. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjelemand, A.B.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppicelli, S.; Bianchini, F.; Toti, A.; Laurenzana, A.; Fibbi, G.; Calorini, L. Extracellular acidity strengthens mesenchymal stem cells to promote melanoma progression. Cell Cycle 2015, 14, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Draoui, N.; Feron, O. Lactate shuttle at a glance: From physiological paradigms to anti-cancer treatments. Dis. Model. Mech. 2011, 4, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Alquraishi, M.; Puckett, D.L.; Alani, D.S.; Humidat, A.S.; Frankel, V.D.; Donohoe, D.R.; Whelan, J.; Bettaieb, A. Pyruvate kinase M2: A simple molecule with complex functions. Free Radic. Biol. Med. 2019, 143, 176–192. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The pentose phosphate pathway dynamics in cancer and its dependency on intracellular pH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumours. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [PubMed] [Green Version]
- Vyas, S.; Zaganjor, W.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Zampieri, L.X.; Capeloa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Glycolysis in cancer: A potential target for therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1358–1366. [Google Scholar] [CrossRef]
- Kim, J.-W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [Green Version]
- Ancey, P.-B.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 286, 2926–2943. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.D.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and mitochondrial ROS in cancer: Novel targets for anticancer therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer metabolism: Phenotype, signaling and therapeutic targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.R.; Marechal, A. The mitochondrial respiratory chain. Essays Biochem. 2010, 47, 1–23. [Google Scholar] [PubMed] [Green Version]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Rep. 2018, 71, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Liu Haigis, M.C.; Guarente, L. Mammalian sirtuins—Emerging roles in physiology, aging and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Skukiewicz, D. Sirtuins, epigenetics and longevity. Aging Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of SIRT in vitro, but can be a stimulator in cells. Cell Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Park, J.S.; Chung, G.J.; Lee, I.H.; Hwang, E.S. Diverse therapeutic efficacies and more diverse mechanisms of nicotinamide. Metabolomics 2019, 15, 137. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.D.; Denu, J.M. Structural identification of 2′- and 3′-O-acetyl-ADP-ribose as novel metabolites derived from the Sir2 family of β-NAD+-dependent histone/protein deacetylases. J. Biol. Chem. 2002, 277, 18535–18544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meszaros, L.G.; Bak, J.; Chu, A. Cyclic ADP-ribose as an endogenous regulator of the non-skeletal type ryanodine receptor Ca2+ channel. Nature 1993, 364, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.; Orsomando, G.; Raffelli, N.; Ruggieri, S. Enzymology of mammalian NAD metabolism in health and disease. Front. Biosci. 2008, 13, 6135–6154. [Google Scholar] [CrossRef] [Green Version]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Canto, C.; Menzies, K.; Auwerx, J. NAD+ metabolism and the control of energy homeostasis—A balancing act between mitochondria and the nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide N-methyltransferase: cDNA cloning, expression and biochemical characterisation. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef]
- Smith, M.-L.; Burnett, D.; Bennett, P.; Waring, R.H.; Brown, H.M.; Williams, A.C.; Ramsden, D.B. A direct correlation between nicotinamide N-methyltransferase activity and protein levels in human liver cytosol. Biochem. Biophys. Acta 1998, 1422, 238–244. [Google Scholar] [CrossRef]
- Seifert, R.; Hoshino, J.; Kroger, H. Nicotinamide methylation. Tissue distribution, developmental and neoplastic changes. Biochem. Biophys. Acta 1984, 801, 259–264. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, L.; Wojnar-Lason, K.; Kaczara, P.; Tworzydlo, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef] [PubMed]
- Riederer, M.; Erwa, W.; Zimmerman, R.; Frank, S.; Zechner, R. Adipose tissue as a source of nicotinaminde N-methyltransferase and homocysteine. Atherosclerosis 2009, 204, 412–417. [Google Scholar] [CrossRef]
- Xu, J.; Capezzone, M.; Xu, X.; Hershman, J.M. Activation of nicotinamide N-methyltransferase gene promoter by hepatocyte nuclear factor-1β in human papillary thyroid cancer cells. Mol. Endocrinol. 2005, 19, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, R.B.; Aravindan, S.; Kadampeswaran, A.; Evans, E.A.; Sandhu, K.K.; Levy, E.R.; Thomas, M.G.; Austen, B.M.; Ramsden, D.B. The expression of nicotinamide N-methyltransferase increases ATP synthesis and protects SH-SY5Y neuroblastoma cells against the toxicity of Complex I inhibitors. Biochem. J. 2011, 436, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Kocinaj, A.; Chaudhury, T.; Uddin, M.S.; Junaid, R.R.; Ramsden, D.B.; Hondhamuni, G.; Klamt, F.; Parsons, L.; Parsons, R.B. High expression of nicotinamide N-methyltransferase in patients with sporadic Alzheimer’s disease. Mol. Neurobiol. 2021, 58, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.B.; Smith, M.-L.; Williams, A.C.; Waring, R.H.; Ramsden, D.B. Expression of nicotinamide N-methyltransferase (E.C. 2.1.1.1) in the Parkinsonian brain. J. Neuropathol. Exp. Neurol. 2002, 61, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Parsons, R.B.; Smith, S.W.; Waring, R.H.; Williams, A.C.; Ramsden, D.B. High expression of nicotinamide N-methyltransferase in patients with idiopathic Parkinson’s disease. Neurosci. Lett. 2003, 342, 13–16. [Google Scholar] [CrossRef]
- Pumpo, M.D.; Sarnelli, G.; Spinella, S.; Budillon, G.; Cuomo, R. The metabolism of nicotinamide in human liver cirrhosis: A study on N-methylnicotinamide and 2-pyridone-5-carboxamide production. Am. J. Gastroenterol. 2001, 96, 1183–1187. [Google Scholar] [CrossRef]
- Mateuszuk, L.; Komich, T.I.; Slominska, E.; Gajda, M.; Wojcik, L.; Lomnicka, M.; Gwozdz, P.; Chlopicki, S. Activation of nicotinamide N-methyltransferase and increased formation of 1-methylnicotinamide (MNA) in atherosclerosis. Pharmacol. Rep. 2009, 61, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Fedorowicz, A.; Mateuszuk, L.; Kopec, G.; Skorka, T.; Kutryb-Zajac, B.; Zakrzewska, A.; Walczak, M.; Jakubowski, A.; Lomnicka, M.; Slominska, E.; et al. Activation of nicotinamide N-methyltransferase (NNMT)-1-methylnicotinamide (MNA) pathway in pulmonary hypertension. Respir. Res. 2016, 17, 108. [Google Scholar] [CrossRef] [Green Version]
- Sternak, M.; Khomich, T.I.; Jakubowski, A.; Szafarz, M.; Sczcepanski, W.; Bialas, M.; Stojak, M.; Szymura-Oleksiak, J.; Chlopicki, S. Nicotinamide N-methyltransferase (NNMT) and 1-methylnicotinamide (MNA) in experimental hepatitis induced by concanavalin A in the mouse. Pharmacol. Rep. 2010, 62, 483–493. [Google Scholar] [CrossRef]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD+ metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef] [Green Version]
- Giuliante, R.; Sartini, D.; Bacchetti, T.; Rocchetti, R.; Kloting, I.; Polidori, C.; Ferretti, G.; Emanuelli, M. Potential involvement of nicotinamide N-methyltransferase in the pathogenesis of metabolic syndrome. Metab. Syndr. Relat. Disord. 2015, 13, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.-C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubenek, S.; Natase, A.; Niculescu, A.M.; Tomescu, S.; Popescu, I.; Dima, S. Asssessment of gene expression profiles in peripheral occlusive arterial disease. Can. J. Cardiol. 2012, 28, 712–720. [Google Scholar] [CrossRef]
- Kim, H.C.; Mofarrahi, M.; Vassilakopoulos, T.; Maltais, F.; Sigala, I.; Debigare, R.; Bellenis, I.; Hussain, S.N.A. Expression and functional significance of nicotinamide N-methyltransferase in skeletal muscles of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 181, 797–805. [Google Scholar] [CrossRef]
- Ramsden, D.B.; Waring, R.H.; Parsons, R.B.; Barlow, D.J.; Williams, A.C. Nicotinamide N-methyltransferase: Genomic connection to disease. Int. J. Tryptophan Res. 2020, 13, 1178646920919770. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.B.; Waring, R.H.; Barlow, D.J.; Parsons, R.B. Nicotinamide N-methyltransferase in health and cancer. Int. J. Tryptophan Res. 2017, 10, 1178646917691739. [Google Scholar] [CrossRef] [Green Version]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marion, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Munoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, V.; Salvolini, E.; Lucarini, G.; Salvucci, A.; Campagna, R.; Rubini, C.; Sartini, D.; Emanuelli, M. Cancer stem cell enrichment is associated with enhancement of nicotinamide N-methyltransferase expression. IUBMB Life 2020, 72, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naïve and primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, L.J.Y.; Wang, X.; Wu, Q.; Sanvoranart, T.; Hubert, C.G.; Prager, B.C.; Wallace, L.C.; Jin, X.; Mack, S.C.; et al. Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2017, 2, e90019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, V.; Sartini, D.; Rochetti, R.; Santarelli, A.; Rubini, C.; Morganti, S.; Giuliante, R.; Calabrese, S.; Di Ruscio, G.; Orlando, F.; et al. Identification and characterization of cancer stem cells from head and neck squamous cell carcinoma cell lines. Cell Physiol. Biochem. 2015, 36, 784–798. [Google Scholar] [CrossRef] [PubMed]
- Kujundzic, R.N.; Prpic, M.; Dakovic, N.; Dabelic, N.; Tomljanovic, M.; Mojzes, A.; Frobe, A.; Troelj, K.G. Nicotinamide N-methyltransferase in acquisition of stem cell properties and therapy resistance to cancer. Int. J. Mol. Sci. 2021, 22, 5681. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blazenovic, I.; et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef]
- Matei, D.; Nephew, K.P. Epigenetic attire in ovarian cancer: The Emperor’s new clothes. Cancer Res. 2020, 80, 3775–3785. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Bockwoldt, M.; Houry, D.; Niere, M.; Gossmann, T.I.; Reinartz, I.; Schug, A.; Ziegler, M.; Heiland, I. Identification of evolutionary and kinetic drivers of NAD-dependent signalling. Proc. Natl. Acad. Sci. USA 2019, 116, 15957–15966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Haren, M.J.; Torano, J.S.; Sartini, D.; Emanuelli, M.; Parsons, R.B.; Martin, N.I. A rapid and efficient assay for the characterisation of substrates and inhibitors for nicotinamide N-methyltransferase. Biochemistry 2016, 55, 5307–5315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gossmann, T.I.; Ziegler, M.; Puntervoll, P.; de Figueiredo, L.P.; Schuster, S.; Heiland, I. NAD+ biosynthesis and salvage—A phylogenetic perspective. FEBS J. 2012, 279, 3355–3363. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, D.; Rose, G.; Cavalcante, P.; Covello, G.; Dato, S.; De Rango, F.; Greco, V.; Maggiolini, M.; Feraco, E.; Mari, V.; et al. A novel VNTR enhancer within the SIRT gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 2005, 85, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Lee, J.-H.; Lee, H.-Y.; Min, K.-J. Sirtuin signalling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.-H.; Kim, H.-S.; Song, S.; Lee, I.H.; Vassilopoulos, A.; Deng, C.-X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Estep, P.W., II; Warner, J.B.; Bulyk, M.L. Short-term calorie restriction in male mice feminizes gene expression and alters key regulators of conserved aging regulatory pathways. PLoS ONE 2009, 4, e5242. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.Y.; Mistry, R.J.; Aguirre, C.A.; Fasouli, E.S.; Thomas, M.G.; Klamt, F.; Ramsden, D.B.; Parsons, R.B. Nicotinamide N-methyltransferase increases complex I activity in SH-SY5Y cells via sirtuin-3. Biochem. Biophys. Res. Commun. 2015, 467, 491–496. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, J.; Wu, W.; Xie, S.; Yu, H.; Li, G.; Zhu, T.; Li, F.; Lu, J.; Wang, G.Y.; et al. Nicotinamide N-methyltransferase enhances chemoresistance in breast cancer through SIRT1 protein stabilization. Breast Cancer Res. 2019, 21, 64. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Moreno-Navarette, J.M.; Wei, X.; Kikukawa, Y.; Tzameli, I.; Prasad, D.; Lee, Y.; Asara, J.M.; Fernandez-Real, J.M.; Maratos-Flier, E.; et al. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat. Med. 2015, 21, 887–894. [Google Scholar] [CrossRef] [Green Version]
- You, Z.; Liu, Y.; Liu, X. Nicotinamide N-methyltransferase enhances the progression of prostate cancer by stabilizing sirtuin 1. Oncol. Lett. 2018, 15, 9195–9201. [Google Scholar] [CrossRef]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Ann. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Wang, J.-Y.; Yu, B.; Cong, X.; Zhang, W.-G.; Li, L.; Liu, L.-M.; Zhou, Y.; Zhang, C.-L.; Gu, P.-L.; et al. Peroxisome proliferator-activated receptor-γ coactivator 1-α inhibits vascular calcification through sirtuin 3-mediated reduction of mitochondrial oxidative stress. Antiox. Redox. Signal. 2019, 31, 75–91. [Google Scholar] [CrossRef]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, A.; Oliver, J.; Roca, P. Role of uncoupling proteins in cancer. Cancers 2010, 2, 567–591. [Google Scholar] [CrossRef] [PubMed]
- Rekar, A.K.; Goswami, M.T.; Krishnapuram, R.; Standiford, T.J.; Keshamouni, V.G. Molecular cross-regulation between PPAR-γ and other signaling pathways: Implications for lung cancer therapy. Lung Cancer 2011, 72, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Vella, V.; Nicoloso, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-γ agonists as antineoplastic agents in cancers with dysregulated IGF axis. Front. Endocrinol. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-α. Int. J. Obes. 2005, 29, S5–S9. [Google Scholar] [CrossRef] [Green Version]
- Thirupathi, A.; de Souza, C.T. Multi-regulatory network of ROS: The interconnection of ROS, PGC-alpha, and AMPK-SIRT1 during exercise. J. Physiol. Biochem. 2017, 73, 487–494. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-family regulatory network. Biochem. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signalling for suicide and survival. J. Cell Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Chaterjee, R.; Chatterjee, J. ROS and oncogenesis with special reference to EMT and stemness. Eur. J. Cell Biol. 2020, 99, 151073. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Abdulfattah, A.Y.; Warren, B.O.; Shu, J.; Said, N. Redox homeostasis and metabolism in cancer: A complex mechanism and potential targeted therapeutics. Int. J. Mol. Sci. 2020, 21, 3100. [Google Scholar] [CrossRef] [PubMed]
- Salganiak, R.I. The benefits and hazards of antioxidants: Controlling apoptosis and other protective mechanisms in cancer patients and the human population. J. Am. Coll. Nutr. 2001, 20, 464S–472S. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumaker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.; Winterford, C.M.; Harmon, B.V. Apoptosis. Its significance in cancer and cancer therapy. Cancer 1994, 73, 2013–2026. [Google Scholar] [CrossRef]
- Rodic, S.; Vincent, M.D. Reactive oxygen species (ROS) are a key determinant of cancer’s metabolic phenotype. Int. J. Cancer 2017, 142, 440–448. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, D. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Del Pilae Sosa Idelchik, M.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echtay, K.S. Mitochondrial uncoupling proteins—What is their physiological role? Free Radic. Biol. Med. 2007, 43, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachex, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Lee, D.S.M.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.F.; Keith, B.; Nissim, I.; Simon, M.C. Fructose-1, 6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, R.J.; Klamt, F.; Ramsden, D.B.; Parsons, R.B. Nicotinamide N-methyltransferase expression in SH-SY5Y human neuroblastoma cells decreases oxidative stress. J. Biochem. Mol. Toxicol. 2020, 34, e22439. [Google Scholar] [CrossRef]
- Yu, J.; Shi, L.; Lin, W.; Lu, B.; Zhao, Y. UCP2 promotes proliferation and chemoresistance through regulating the NF-κB/β-catenin axis and mitochondrial ROS in gallbladder cancer. Biochem. Pharmacol. 2020, 172, 113745. [Google Scholar] [CrossRef]
- Robinson, A.J.; Hopkins, G.L.; Rastogi, N.; Hodges, M.; Doyle, M.; Davies, S.; Hole, P.S.; Omidvar, N.; Darley, R.L.; Tonks, A. Reactive oxygen species drives proliferation in acute myeloid leukemia via the glycolytic regulator PFKFB3. Cancer Res. 2020, 80, 937–949. [Google Scholar] [CrossRef] [Green Version]
- Sanchis, D.; Busquets, S.; Alvarez, B.; Ricquier, D.; Lopez-Soriano, F.J.; Argiles, J.M. Skeletal muscle UCP2 and UCP3 gene expression in a rat cancer cachexia model. FEBS Lett. 1998, 436, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Bing, C.; Brown, M.; King, P.; Collins, P.; Tisdale, M.J.; Williams, G. Increased gene expression of brown fat uncoupling protein (UCP)1 and skeletal muscle UCP2 and UCP3 in MAC16-induced cancer cachexia. Cancer Res. 2000, 60, 2405–2410. [Google Scholar]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Narayanankutty, A. PI3K/Akt/mTOR pathway as a therapeutic target for colorectal cancer: A review of preclinical and clinical evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Tang, S.-W.; Yang, T.-C.; Lin, W.-C.; Chang, W.-H.; Wang, C.-C.; Lai, M.-K.; Lin, J.-Y. Nicotinamide N-methyltransferase induces cellular invasion through activating matrix metalloproteinase-expression in clear cell renal cell carcinoma cells. Carcinogenesis 2011, 32, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.G.; Saldanha, M.; Mistry, R.J.; Dexter, D.T.; Ramsden, D.B.; Parsons, R.B. Nicotinamide N-methyltransferase expression in SH-SY5Y neuroblastoma and N27 mesencephalic neurones induces changes in cell morphology via ephrin-B2 and Akt signalling. Cell Death Dis. 2013, 4, e669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, T.K.; Lamb, T.J. Emerging roles for Eph receptors and ephrin ligands in immunity. Front. Immunol. 2019, 10, 1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, Y.; Li, G.; Yu, H.; Xie, X. Down-regulation of nicotinamide N-methyltransferase induces apoptosis in human breast cancer cells via the mitochondria-mediated pathway. PLoS ONE 2014, 9, e89202. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer: Warburg addressed. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J. 2019, 38, e99435. [Google Scholar] [CrossRef]
- Kaboli, P.J.; Salimian, F.; Aghapour, S.; Xiang, S.; Zhao, Q.; Li, M.; Wu, X.; Du, F.; Zhao, Y.; Shen, J.; et al. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer—A comprehensive review from chemotherapy to immunotherapy. Pharmacol. Res. 2020, 156, 104806. [Google Scholar] [CrossRef]
- Yu, M.; Qi, B.; Wu, X.; Xu, J.; Liu, X. Baicalein increases cisplatin sensitivity of A549 lung adenocarcinoma cells via PI3K/Akt/NFκB pathway. Biomed. Pharmacother. 2017, 90, 677–685. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTor signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.-Q.; Zhai, X.-Q.; Ma, J.-W.; Fu, X.-Q.; Zhao, B.-S.; Zhang, P.; Fu, X.-Y. Natural borneol sensitizes human glioma cells to cisplatin-induced apoptosis by triggering ROS-mediated oxidative damage and regulation of MAPKs and PI3K/AKT pathway. Pharm. Biol. 2020, 58, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Xu, H.; Zhang, S.; Chen, J.; Yu, Z. SDC1 promotes cisplatin resistance in hepatic carcinoma cells via PI3K-AKT pathway. Hum. Cell 2020, 33, 721–729. [Google Scholar] [CrossRef]
- Milani, Z.H.; Ramsden, D.B.; Parsons, R.B. Neuroprotective effects of nicotinamide N-methyltransferase and its metabolite 1-methylnicotinamide. J. Biochem. Mol. Toxicol. 2013, 27, 451–456. [Google Scholar] [CrossRef]
- Thomas, M.G.; Sartini, D.; Emanuelli, M.; van Haren, M.J.; Martin, N.I.; Mountford, D.M.; Barlow, D.J.; Klamt, F.; Ramsden, D.B.; Reza, M.; et al. Nicotinamide N-methyltransferase catalyses the N-methylation of the endogenous β-carboline norharman: Evidence for a novel detoxification pathway. Biochem. J. 2016, 473, 3253–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Haren, M.J.; Thomas, M.G.; Sartini, D.; Barlow, D.J.; Ramsden, D.B.; Emanuelli, M.; Klamt, F.; Martin, N.I.; Parsons, R.B. The kinetic analysis of the N-methylation of 4-phenylpyridine by nicotinamide N-methyltransferase: Evidence for a novel mechanism of substrate inhibition. Int. J. Biochem. Cell Biol. 2018, 98, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Kong, B.; Tong, Q.; Li, Y.; Chen, L.; Zeng, J.; Yu, H.; Xie, X.; Zhang, J. Vanillin downregulates NNMT and attenuates NNMT-related resistance to 5-fluorouracil via ROS-induced cell apoptosis in colorectal cancer cells. Oncol. Rep. 2021, 45, 110. [Google Scholar] [CrossRef] [PubMed]
- Akar, S.; Harmankaya, I.; Ugras, S.; Celik, C. Expression and clinical significance of nicotinamide N-methyltransferase in cervical squamous cell carcinoma. Int. J. Gynecol. Pathol. 2019, 39, 289–295. [Google Scholar] [CrossRef]
- Bach, D.-H.; Kim, D.; Bae, S.Y.; Kim, W.K.; Hong, J.-Y.; Lee, H.-J.; Rajasekaran, N.; Kwon, S.; Fan, Y.; Luu, T.-T.-T.; et al. Targeting nicotinamide N-methyltransferase and miR-449a in EGFR-TKI-resistant non-small-cell lung cancer cells. Mol. Ther. Nucleic Acids 2018, 11, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Liu, H.; Wang, Y.; Zhou, Y.; Yu, H.; Li, G.; Ruan, Z.; Li, F.; Wang, X.; Zhang, J. Nicotinamide N-methyltransferase enhances resistance to 5-fluorouracil in colorectal cancer cells through inhibition of the ASK1-pMAPK pathway. Oncotarget 2016, 7, 45837–45848. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Poylak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The role of matrix metalloproteinases in the epithelial-mesenchymal transition of hepatocellular carcinoma. Anal. Cell Pathol. 2019, 2019, 9423907. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, R.B. Nicotinamide N-methyltransferase and metastasis: A new player in cancer therapeutics. Biotarget 2019, 3, 20. [Google Scholar] [CrossRef]
- Muller, T. Catechol-O-methyltransferase enzyme: Cofactor S-adenosyl-L-methionine and related mechanisms. Int. Rev. NeuroBiol. 2010, 95, 49–71. [Google Scholar]
- Yoshikawa, T.; Nakamura, T.; Yanai, K. Histamine N-methyltransferase in the brain. Int. J. Mol. Sci. 2019, 20, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesche, J.; Kuhn, S.; Kessler, B.M.; Salton, M.; Wolf, A. Protein arginine methylation: A prominent modification and its demethylation. Cell Mol. Life Sci. 2017, 74, 3305–3315. [Google Scholar] [CrossRef]
- Rui, L.; Wei, X.; Jiang, D.-S. Protein methylation functions as the posttranslational modification switch to regulate autophagy. Cell Mol. Life Sci. 2019, 76, 3711–3722. [Google Scholar]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodelling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmukh, D.; Qiu, Y. Role of PARP-1 in prostate cancer. J. Clin. Exp. Urol. 2015, 3, 1–12. [Google Scholar]
- Burkle, A. PARP-1: A regulator of genomic stability linked with mammalian longevity. ChemBiochem 2001, 2, 725–728. [Google Scholar] [CrossRef]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose)polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef]
- D’Andrea, F.P.; Horsman, M.R.; Kassem, M.; Overgaard, J.; Safwat, A. Tumourigenicity and radiation resistance of mesenchymal stem cells. Acta Oncol. 2012, 51, 669–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, F.P.; Safwat, A.; Kassem, M.; Gautier, L.; Overgaard, J.; Horsman, M.R. Cancer stem cell overexpression of nicotinamide N-methyltransferase enhances cellular radiation resistance. Radiother. Oncol. 2011, 99, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Kassem, H.S.; Sanger, V.; Cowan, R.; Clarke, N.; Margison, G.P. A potential role of heat shock proteins and nicotinamide N-methyltransferase in predicting response to radiation in bladder cancer. Int. J. Cancer 2002, 101, 454–460. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berrocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal binding of PARP-1 and histone H1 promoters specifies transcriptional outcomes. Science 2008, 319, 819–821. [Google Scholar] [CrossRef]
- Gebicki, J.; Sysa-Jedrzejowska, A.; Adamus, J.; Wozniacka, A.; Rybak, M.; Zielonka, J. 1-Methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated via a cyclooxygenase-2/prostacyclin pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar]
- Huang, Q.; Chen, Z.; Cheng, P.; Jiang, Z.; Wang, Z.; Huang, Y.; Yang, C.; Pan, J.; Qiu, F.; Huang, J. LYRM2 directly regulates complex I activity to support tumor growth in colorectal cancer by oxidative phosphorylation. Cancer Lett. 2019, 455, 36–47. [Google Scholar] [CrossRef]
- Marquez, J.; Kratchmarova, I.; Akimov, V.; Unda, F.; Ibarretxe, G.; Clerigue, A.S.; Osinalde, N.; Badiola, I. NADH dehydrogenase complex I is overexpressed in incipient metastatic murine colon cancer cells. Oncol. Rep. 2019, 41, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Xinyou, X.; Yu, H.; Wang, Y.; Zhou, Y.; Li, G.; Ruan, Z.; Li, F.; Wang, X.; Liu, H.; Zhang, J. Nicotinamide N-methyltransferase enhances the capacity of tumorigenesis associated with the promotion of cell cycle progression in human colorectal cancer cells. Ann. Biochem. Biophys. 2014, 564, 52–66. [Google Scholar]
- Yu, H.; Zhou, X.; Wang, Y.; Huang, X.; Yang, J.; Zeng, J.; Li, G.; Xie, X.; Zhang, J. Nicotinamide N-methyltransferase inhibits autophagy induced by oxidative stress through suppressing the AMPK pathway in breast cancer cells. Cancer Cell Int. 2020, 20, 191. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Na, N.; Lin, S. Diagnostic value of serum N1-methylnicotinamide in cervical cancer patients. Clin. Lab. 2021, 67. [Google Scholar] [CrossRef] [PubMed]
- Blazejczyk, A.; Switalska, M.; Chlopicki, S.; Marcinek, A.; Gebicki, J.; Nowak, M.; Nasulewicz-Goldeman, A.; Wietrzyk, J. 1-Methylnicotinamide and its structural analogue 1,4-dimethylpyridine for the prevention of cancer metastasis. J. Exp. Clin. Cancer Res. 2016, 35, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, Y.; Sasaki, T.; Bochimoto, H.; Kawabe, J.-I.; Endo, S.; Hira, Y.; Watanabe, T.; Okumura, S.; Hasebe, N.; Ohsaki, Y. Prostaglandin I2 analog suppresses lung metastasis by recruiting pericytes in tumor angiogenesis. Int. J. Oncol. 2015, 46, 548–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.-H.; Lee, K.-T.; Choi, Y.S.; Choi, J.-H. Iloprost, a prostacyclin analog, inhibits the invasion of ovarian cancer cells by downregulating matrix metallopeptidase (MMP-2) through the IP-dependent pathway. Prostaglandins Other Lipid Mediat. 2018, 134, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Tang, D.G.; Schirner, M.; Honn, K.V. Prostacyclin and its analogues: Antimetastatic effects and mechanisms of action. Cancer Metastasis Rev. 1994, 13, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Shang, F.-F.; Long, H.; Jiang, L.; Zhu, R.; Zhao, Q.; Gu, H.; Kong, J.; Xu, W.; Zhao, Y.; et al. Role of nicotinamide N-methyltransferase in dorsal striatum in cocaine place preference. Neuropsychopharmacology 2017, 42, 2333–2343. [Google Scholar] [CrossRef] [Green Version]
- Tomida, M.; Ohtake, H.; Yokota, T.; Kobayashi, Y.; Kurosomi, M. Statup-regulates expression of nicotinamide N-methyltransferease in human cancer cells. J. Cancer Res. Clin. Oncol. 2008, 134, 551–559. [Google Scholar] [CrossRef]
- Katiyar, S.; Jiao, X.; Addya, S.; Ertel, A.; Covarrubias, Y.; Rose, V.; Casimiro, M.C.; Zhou, J.; Lisanti, M.P.; Nasim, T.; et al. Mammary gland selective excision of c-jun identifies its role in mRNA splicing. Cancer Res. 2012, 72, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Nabokikh, A.; Ilhan, A.; Bilban, M.; Gartner, W.; Vila, G.; Niederle, B.; Nielsen, J.H.; Wagner, O.; Base, W.; Luger, A.; et al. Reduced TGF-beta expression and its target genes in human insulinomas. Exp. Clin. Endocrinol. Diabetes 2007, 115, 674–682. [Google Scholar] [CrossRef]
- Mantaj, J.; Rahman, S.M.A.; Bokshi, B.; Hasan, C.M.; Jackson, P.J.M.; Parsons, R.B.; Rahman, K.M. Crispene E, a cis-clerodane diterpene inhibits STAT3 dimerization in breast cancer cells. Org. Biomol. Chem. 2015, 13, 3882–3886. [Google Scholar] [CrossRef]
- Available online: http://jaspar.genereg.net/ (accessed on 27 July 2021).
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Ma, J.-H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal. 2020, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Rostami, N.; Nikkhoo, A.; Ajjoolabady, A.; Azizi, G.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Yousefi, B.; Yousefi, M.; Jadidi-Niaragh, F. S1PR1 as a novel promising therapeutic target in cancer therapy. Mol. Diagn. Ther. 2019, 23, 467–487. [Google Scholar] [CrossRef]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.-J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018, 27, 136–150. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yao, Y.; Yao, M.; Fu, P.; Wang, W. Interleukin-2 promotes triple negative breast cancer cells migration and paclitaxel resistance through JAK-STAT3/MAPKs/AKT signaling pathways. Biochem. Biophys. Res. Commun. 2018, 503, 1605–1609. [Google Scholar] [CrossRef]
- Liu, C.; Xing, H.; Guo, C.; Yang, Z.; Wang, Y.; Wang, Y. MiR-124 reversed the doxorubicin resistance of breast cancer stem cells through STAT3/HIF-signaling pathways. Cell Cycle 2019, 18, 2215–2217. [Google Scholar] [CrossRef]
- Lida, Y.; Okamoto, A.; Hollis, R.L.; Gourley, C.; Herrington, C.S. Clear cell carcinoma of the ovary: A clinical and molecular perspective. Int. J. Gynecol. Cancer 2021, 31, 605–616. [Google Scholar]
- Yu, D.-D.; Guo, S.-W.; Jing, Y.-Y.; Dong, Y.-L.; Wei, L.-X. A review on hepatocyte nuclear factor-1beta and tumor. Cell Biosci. 2015, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.; Liao, J.; Zhang, M.; Talamonti, M.L.; Bentrem, D.; Rao, S.; Yang, G.Y. Clear cell carcinoma of the pancreas: Histopathologic features and a unique biomarker: Hepatocyte nuclear factor-1beta. Mod. Pathol. 2008, 21, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.C.; Lin, M.C.; Lin, W.C.; Jeng, Y.M.; Mao, T.L. Utility of hepatocyte nuclear factor-1beta as a diagnostic marker in ovarian carcinomas with clear cells. Histopathology 2012, 61, 760–768. [Google Scholar] [CrossRef]
- Setiawan, V.W.; Haessler, J.; Schumacher, F.; Cote, M.L.; Deelman, E.; Fesinmeyer, M.D.; Hendersen, B.E.; Jackson, R.D.; Vockler, J.-S.; Wilkens, L.R.; et al. HNF1B and endometrial cancer risk: Results from the PAGE study. PLoS ONE 2012, 7, e303090. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Goncalves, L.G.; Nunes, C.; Faustino, I.; Silva, F.; Felix, A.; Pereira, S.A.; Serpa, J. HNF1β drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumor Biol. 2016, 37, 4813–4829. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Amano, Y.; Amano, Y.; Yamaguchi, K.; Matsumura, N.; Baba, T.; Konishi, I. Ovarian clear cell carcinoma meets metabolism; HNF-1β confers survival benefits through the Warburg effect and ROS reduction. Oncotarget 2015, 6, 30704–30714. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Madzharova, E.; Kastl, P.; Sabino, F.; auf dem Keller, U. Post-translational modification-dependent activity of matric metalloproteinases. Int. J. Mol. Sci. 2019, 20, 3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.-H.; Cho, B.-I.; Kim, Y.N.; Kim, J.W.; Park, S.-T.; Lee, C.-W. Overexpression of nicotinamide N-methyltransferase in gastric cancer tissues and its potential post-translational modification. Exp. Mol. Med. 2006, 38, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: httpps://www.phosphosite.org/proteinAction?id=14006&showAllSites=true#appletMsg (accessed on 26 July 2021).
- Nemmara, V.V.; Tilvawala, R.; Salinger, A.J.; Miller, L.; Bguyen, S.H.; Weerapana, E.; Thompson, P.R. Citrullination inactivates nicotinamide N-methyltransferase. ACS Chem. Biol. 2018, 13, 2663–2672. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Yuan, Y.; Liu, Z.; He, Y.; Pan, D. Casein kinase 2 inhibitor CX-4945 elicits an anti-Warburg effects through the downregulation of Tap73 and inhibits gastric tumorigenesis. Biochem. Biophys. Res. Commun. 2020, 530, 686–691. [Google Scholar] [CrossRef]
- Jung, M.; Park, K.H.; Kim, H.M.; Kim, T.S.; Zhang, X.; Park, S.-M.; Beom, S.-H.; Kim, H.S.; Cheong, J.-H.; Chung, H.C.; et al. Inhibiting casein kinase overcomes paclitaxel resistance in gastric cancer. Gastric Cancer 2019, 22, 1153–1163. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R. Regulatory subunits of PKA and breast cancer. Ann. N. Y. Acad. Sci. 2002, 968, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and associated signalling pathways involved in cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef] [PubMed]
- Tilvawala, R.; Nguyen, S.H.; Maurais, A.J.; Nemmara, V.V.; Nagar, M.; Salinger, A.J.; Nagpal, S.; Weerapana, E.; Thompson, P.R. The rheumatoid arthritis-associated citrullinome. Cell Chem. Biol. 2018, 25, 691–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witalison, W.E.; Thompson, P.R.; Hofseth, L.J. Protein arginine deiminases and associated citrullination: Physiological functions and diseases associated with dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Thompson, P.R. Protein arginine deiminases (PADs): Biochemistry and chemical biology of protein citrullination. Acc. Chem. Res. 2019, 52, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Liu, X.; Li, F.; Miao, L.; Li, T.; Xu, B.; An, X.; Muth, A.; Thompson, P.R.; Coonrod, S.A.; et al. PAD1 promotes epithelial-mesenchymal transition and metastasis in triple-negative breast cancer cells by regulating MEK1-ERK1/2-MMPsignaling. Cancer Lett. 2017, 409, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Balan, V.; Miller, G.S.; Kaplan, L.; Balan, K.; Chong, Z.-Z.; Li, F.; Kaplun, A.; Van Berkum, M.F.A.; Arking, R.; Freeman, D.C.; et al. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J. Biol. Chem. 2008, 283, 27810–27819. [Google Scholar] [CrossRef] [Green Version]
- Zhai, R.G.; Rizzi, M.; Garavaglia, S. Nicotinamide/nicotinic acid mononucleotide adenylyltransferase, new insights into an ancient enzyme. Cell. Mol. Life Sci. 2009, 66, 2805–2818. [Google Scholar]
- Enomoto, M.; Siow, C.; Igaki, T. Drosophila as a cancer model. Adv. Exp. Med. Biol. 2018, 1076, 173–194. [Google Scholar]
- Herranz, H.; Eichenlaub, T.; Cohen, S.M. Cancer in Drosophila: Imaginal discs as a model for epithelial tumor formation. Curr. Top. Dev. Biol. 2016, 116, 181–199. [Google Scholar]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.N.; Jackson, F.R. Tumors of the testis and midgut in aging flies. Fly 2008, 2, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Yang, D.; Wang, W.; Zhang, L.; Liu, H.; Ma, S.; Guo, W.; Yao, M.; Zhang, K.; Li, W.; et al. Nicotinamide N-methyltransferase decreases 5-fluorouracil sensitivity in human esophageal squamous cell carcinoma through metabolic reprogramming and promoting the Warburg effect. Mol. Carcinog. 2020, 59, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.K.; Webb, A.E. Regulation of FOXO factors in mammalian cells. Curr. Top. Dev. Biol. 2018, 127, 165–192. [Google Scholar]
- Frum, T.; Ralston, A. Cell signaling and transcription factors regulating cell fate during formation of the mouse blastocyst. Trends Genet. 2015, 31, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Martin, N.I.; van Haren, M.J. Nicotinamide N-methyl transferase (NNMT): An emerging therapeutic target. Drug Discov. Today 2021. [Google Scholar] [CrossRef]
- Cox, R. Nicotinamide methyltransferase inhibition by S-adenosylethionine. Cancer Lett. 1983, 17, 295–300. [Google Scholar] [CrossRef]
- Peng, Y.; Sartini, D.; Pozzi, V.; Wilk, D.; Emanuelli, M.; Yee, V.C. Structural basis of substrate recognition in human nicotinamide N-methyltransferase. Biochemistry 2011, 50, 7800–7808. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Chen, Y.; Wang, J.; Hao, L.; Huang, C.; Griffiths, A.; Sun, Z.; Zhou, Z.; Song, Z. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J. Hepatol. 2020, 73, 783–793. [Google Scholar] [CrossRef]
- Kannt, A.; Rajagopal, S.; Kadnur, S.V.; Suresh, J.; Bhamidipati, R.K.; Swaminathan, S.; Hallur, M.S.; Kristam, R.; Elvert, R.; Czech, J.; et al. Nicotinamide N-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 2018, 8, 3660. [Google Scholar] [CrossRef]
- Kannt, A.; Rajagopal, S.; Hallur, M.S.; Swamy, I.; Kristam, R.; Dhakshinamoorthy, S.; Czech, J.; Zech, G.; Schreuder, H.; Ruf, S. Novel inhibitors of nicotinamide N-methyltransferase for the treatment of metabolic disorders. Molecules 2021, 26, 991. [Google Scholar] [CrossRef]
- Sampson, C.M.; Dimet, A.L.; Neelakantan, H.; Ogunseye, K.O.; Stevenson, H.L.; Hommel, J.D.; Watowich, S.J. Combined nicotinamide N-methyltransferase inhibition and reduced-calorie diet normalises body composition and enhanced metabolic benefits in obese mice. Sci. Rep. 2021, 11, 5637. [Google Scholar] [CrossRef]
- Neelakantan, H.; Vance, V.; Wetzel, M.D.; Wang, H.-Y.L.; McHardy, S.F.; Finnerty, C.C.; Hommel, J.D.; Watowich, S.J. Selective and membrane-permeable small molecule inhibitors of nicotinamide N-methyltransferase reverse high-fat diet-induced obesity in mice. Biochem. Pharmacol. 2018, 147, 141–152. [Google Scholar] [CrossRef]
- Ziegler, M.G.; Bao, X.; Kennedy, B.P.; Joyner, A.; Enns, R. Location, development, control, and function of extraadrenal phenylethanolamine N-methyltransferase. Ann. N. Y. Acad. Sci. 2002, 971, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.A.; Moon, E.; Kim, U.-J.; Xu, J.; Siciliano, M.J.; Weinshilboum, R.M. Human indolethylamine N-methyltransferase: cDNA cloning and expression, gene cloning, and chromosomal localisation. Genomics 1999, 61, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; van Haren, M.J.; Moret, E.E.; Rood, J.J.M.; Sartini, D.; Salvucci, A.; Emanuelli, M.; Craveur, P.; Babault, N.; Jin, J.; et al. Bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT) with enhanced activity. J. Med. Chem. 2019, 62, 6597–6614. [Google Scholar] [CrossRef] [Green Version]
- Alson, T.A.; Abeles, R.H. Substrate specificity of nicotinamide methyltransferase isolated from porcine liver. Arch. Biochem. Biophys. 1988, 260, 601–608. [Google Scholar] [CrossRef]
- Akar, S.; Duran, T.; Azzawri, A.A.; Kocak, N.; Celik, C.; Yildirim, H.I. Small molecule inhibitor of nicotinamide N-methyltransferase shows anti-proliferative activity in HeLa cells. J. Obstet. Gynaecol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Chung, H.J.; Lee, H.J.; Park, H.J.; Lee, S.K. Growth inhibition of human lung cancer cells via down-regulation of epidermal growth factor receptor signaling by yuanhuadine, a daphnane diterpene from Dapne genkwa. J. Nat. Prod. 2011, 74, 2102–2108. [Google Scholar] [CrossRef]
- Rothe, M.L.; Li, J.; Garibay, E.; Moore, B.S.; McKinnie, S.M.K. Synthesis, bioactivity, and enzymatic modification of antibacterial thiotetromycin derivatives. Org. Biomol. Chem. 2019, 17, 3416–3423. [Google Scholar] [CrossRef]
- Ruf, S.; Hallur, M.S.; Anchan, N.K.; Swamy, I.N.; Murugesan, K.R.; Sarkar, S.; Narasimhulu, L.K.; Putta, V.P.P.R.; Shaik, S.; Chandrasekar, D.V.; et al. Novel nicotinamide analog as an inhibitor of nicotinamide N-methyltransferase. Bioorg. Med. Chem. Lett. 2018, 28, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, H.; Wang, H.-Y.; Vance, V.; Hommel, J.D.; McHardy, S.F.; Watowich, S.J. Structure-activity relationship for small molecule inhibitors of nicotinamide N-methyltransferase. J. Med. Chem. 2017, 60, 5015–5028. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Novel pyrimidine-5-carboxamide compounds as NNMT inhibitors for treating diabetes. ACS Med. Chem. Lett. 2021, 12, 538–539. [Google Scholar] [CrossRef]
- Horning, B.D.; Suciu, R.M.; Ghadiri, D.; Ulanovskaya, O.; Matthews, M.L.; Lum, K.M.; Backus, K.; Brown, S.J.; Rosen, H.; Cravatt, B.F. Chemical proteomic profiling of human methyltransferases. J. Am. Chem. Soc. 2016, 138, 13335–13343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-Y.; Suciu, R.M.; Horning, B.D.; Vinogradova, E.V.; Ulanovskaya, O.A.; Cravatt, B.F. Covalent inhibitors of nicotinamide N-methyltransferase (NNMT) provide evidence for target engagement challenges in situ. Bioorg. Med. Chem. Lett. 2018, 28, 2682–2687. [Google Scholar] [CrossRef]
- Sen, S.; Mondal, S.; Zheng, L.; Salinger, A.J.; Fast, W.; Weerapana, E.; Thompson, P.R. Development of a suicide inhibition based protein labelling (SIBLing) strategy for nicotinamide N-methyltransferase. ACS Chem. Biol. 2019, 14, 613–618. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef]
- van Haren, M.J.; Taig, R.; Kuppens, J.; Torano, J.S.; Moret, E.E.; Parsons, R.B.; Sartini, D.; Emanuelli, M.; Martin, N.I. Inhibitors of nicotinamide N-methyltransferase designed to mimic the methylation reaction transition state. Org. Biomol. Chem. 2017, 15, 6656–6667. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; van Haren, M.J.; Buis, N.; Innocenti, P.; Zhang, Y.; Sartini, D.; Campagna, R.; Emanuelli, M.; Parsons, R.B.; Jespers, W.; et al. Potent inhibition of nicotinamide N-methyltransferase by alkene-linked bisubstrate mimics bearing electron deficient aromatics. J. Med. Chem. 2021, 64, 12938–12963. [Google Scholar] [CrossRef]
- Babault, N.; Allali-Hassani, A.; Li, F.; Fan, J.; Yue, A.; Ju, K.; Liu, F.; Vedadi, M.; Liu, J.; Jin, J. Discovery of bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT). J. Med. Chem. 2018, 61, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Policarop, R.L.; Decultot, L.; May, E.; Kuzmic, P.; Carlson, S.; Huang, D.; Chu, V.; Wright, B.A.; Dhakshinamoorthy, S.; Kannt, A.; et al. High-affinity alkynyl bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT). J. Med. Chem. 2019, 62, 9837–9873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Li, L.; Diaz, K.; Iyamu, I.; Yadav, R.; Noinaj, N.; Huang, R. Novel propargyl-linked bisubstrate analogs as tight-binding inhibitors for nicotinamide N-methyltransferase. J. Med. Chem. 2019, 62, 10783–10797. [Google Scholar] [CrossRef] [PubMed]
- Tehlivets, O.; Malanovic, N.; Visram, M.; Pavkov-Keller, T.; Keller, W. S-Adenosyl-L-homocysteine hydrolase and methylation disorders: Yeast as a model system. Biochim. Biophys. Acta 2013, 1832, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlessinger, A.; Welch, M.A.; van Vlijmen, H.; Korzekwa, K.; Swaan, P.W.; Matsson, P. Molecular modelling of drug-transporter interactions—An international transporter consortium perspective. Clin. Pharmacol. Ther. 2018, 104, 818–835. [Google Scholar] [CrossRef]
- Varma, M.V.; Lai, Y.; El-Kattan, A.F. Molecular properties associated with transporter-mediated drug disposition. Adv. Drug Deliv. Rev. 2017, 116, 92–99. [Google Scholar] [CrossRef]
- Fathi, N.; Rashidi, G.; Khodadadi, A.; Shahi, S.; Sharifi, S. STAT3 and apoptosis challenges in cancer. Int. J. Biol. Macromol. 2018, 117, 993–1001. [Google Scholar] [CrossRef]
- Kim, M.; Morales, L.D.; Jang, I.-S.; Cho, Y.-Y.; Kim, D.J. Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef] [Green Version]
- Yong-Ming, H.; Ai-Jun, J.; Xiao-Yue, X.; Jian-Wei, J.; Chen, Y.; Ye, C. miR-449a: A potential therapeutic agent for cancer. Anticancer Drugs 2017, 28, 1067–1078. [Google Scholar] [CrossRef]
- Ellinger, P.; Coulson, R.A. The urinary elimination of nicotinamide methochloride by man. Biochem. J. 1944, 38, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Ellinger, P.; Abdel Kader, M.M. Nicotinamide metabolism in mammals. Biochem. J. 1949, 44, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, P.J.G.; Quastel, J.H. Nicotinamide, cozymase and tissue metabolism. Biochem. J. 1941, 35, 502–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenis, A.T.; Lec, P.M.; Chamie, K.; Mshs, M.D. Bladder cancer: A review. JAMA 2020, 324, 1980–1991. [Google Scholar] [CrossRef]
- DeGeorge, K.C.; Holt, H.R.; Hodges, S.C. Bladder cancer: Diagnosis and treatment. Am. Fam. Physician 2017, 96, 507–514. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intron a | Intron Coordinates b | Identified Transcription Factor Binding Motifs | |
|---|---|---|---|
| STAT3 | HNF-1β | ||
| 2 | 114,262,935–114,296,427 | CTTCCTGGAAT GTTCCTGGAAT CTGCTGGGAAC GTTCTGGAAAA GTGCTAGGAAG CTTTTGGGAAA TTCCTGGGAAA TTCCTGGGAAA GTTCCTGAAAA CTCCTTGGAAA CTGCTAGAAAA | GTCAATTATTTAC TTTAAAAATTAAT GTTATTAATTACC GTAAATCAATTAT TTGAATTATTAAT |
| 4 | 114,298,160–114,312,044 | TTTCTAGGAAT ATTCTGGAAAA GTTCCTGGAAC | TTAAATGATTGAT TTCAATGATTTAT |
| Residue | Modification a | Enzyme(s) b | Prediction/Confirmation |
|---|---|---|---|
| Serine-3 | Phosphorylation | CK1, GSK3 | PhosphoSite [178] (site), Lim et al. [177] (site & enzymes) |
| Serine-7 | Phosphorylation | CK1 | Nemmara et al. [179] |
| Lysine-8 | Acetylation | - | PhosphoSite |
| Ubiquitinylation | - | ||
| Methylation | - | ||
| Succinylation | - | ||
| Tyrosine-11 | Phosphorylation | - | PhosphoSite |
| Arginine-18 | Citrullination | PAD | Nemmara et al. |
| Lysine-23 | Ubiquitinylation | - | PhosphoSite |
| Succinylation | - | ||
| Tyrosine-24 | Phosphorylation | - | PhosphoSite |
| Tyrosine-25 | Phosphorylation | - | PhosphoSite |
| Lysine-26 | Ubiquitinylation | - | PhosphoSite |
| Serine-29 | Phosphorylation | CK1 | Lim et al. |
| Serine-32 | Phosphorylation | CK1, PKA2 | Lim et al. |
| Serine-35 | Phosphorylation | - | PhosphoSite |
| Lysine-39 | Acetylation | - | PhosphoSite |
| Ubiquitinylation | - | PhosphoSite | |
| Lysine-43 | Acetylation | - | PhosphoSite |
| Ubiquitinylation | - | PhosphoSite | |
| Lysine-47 | Ubiquitinylation | - | PhosphoSite |
| Serine-64 | Phosphorylation | CK1 | Lim et al. |
| Serine-73 | Phosphoroylation | CK2 c, GSK3 | Lim et al. |
| Serine-77 | Phosphorylation | CK2 c | Lim et al. |
| Lysine-96 | Ubiquitinylation | - | PhosphoSite |
| Lysine-99 | Sumoylation | - | Lim et al. |
| Lysine-100 | Ubiquitinylation | - | PhosphoSite |
| Serine-108 | Phosphorylation | GSK3, ProDKin | PhosphoSite (site), Lim et al. (site & enzymes) |
| Tyrosine-113 | Phosphorylation | - | PhosphoSite |
| Lysine-123 | Ubiquitinylation | - | PhosphoSite |
| Arginine-132 | Citrullination | PAD | Nemmara et al. |
| Lysine-136 | Ubiquitinylation | - | PhosphoSite |
| Arginine-181 | Citrullination | PAD | Nemmara et al. |
| Tyrosine-203 | Phosphorylation | - | PhosphoSite |
| Tyrosine-204 | Phosphorylation | - | PhosphoSite |
| Lysine-210 | Ubiquitinylation | - | PhosphoSite |
| Serine-239 | Phosphorylation | GSK3 | Lim et al. |
| Serine-241 | Phosphorylation | CK1, GSK3 | Lim et al. |
| Serine-261 | Phosphorylation | PKA1 | Lim et al. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parsons, R.B.; Facey, P.D. Nicotinamide N-Methyltransferase: An Emerging Protagonist in Cancer Macro(r)evolution. Biomolecules 2021, 11, 1418. https://doi.org/10.3390/biom11101418
Parsons RB, Facey PD. Nicotinamide N-Methyltransferase: An Emerging Protagonist in Cancer Macro(r)evolution. Biomolecules. 2021; 11(10):1418. https://doi.org/10.3390/biom11101418
Chicago/Turabian StyleParsons, Richard B., and Paul D. Facey. 2021. "Nicotinamide N-Methyltransferase: An Emerging Protagonist in Cancer Macro(r)evolution" Biomolecules 11, no. 10: 1418. https://doi.org/10.3390/biom11101418
APA StyleParsons, R. B., & Facey, P. D. (2021). Nicotinamide N-Methyltransferase: An Emerging Protagonist in Cancer Macro(r)evolution. Biomolecules, 11(10), 1418. https://doi.org/10.3390/biom11101418