
Lipid Profiles of RAS Nanoclusters Regulate RAS Function



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
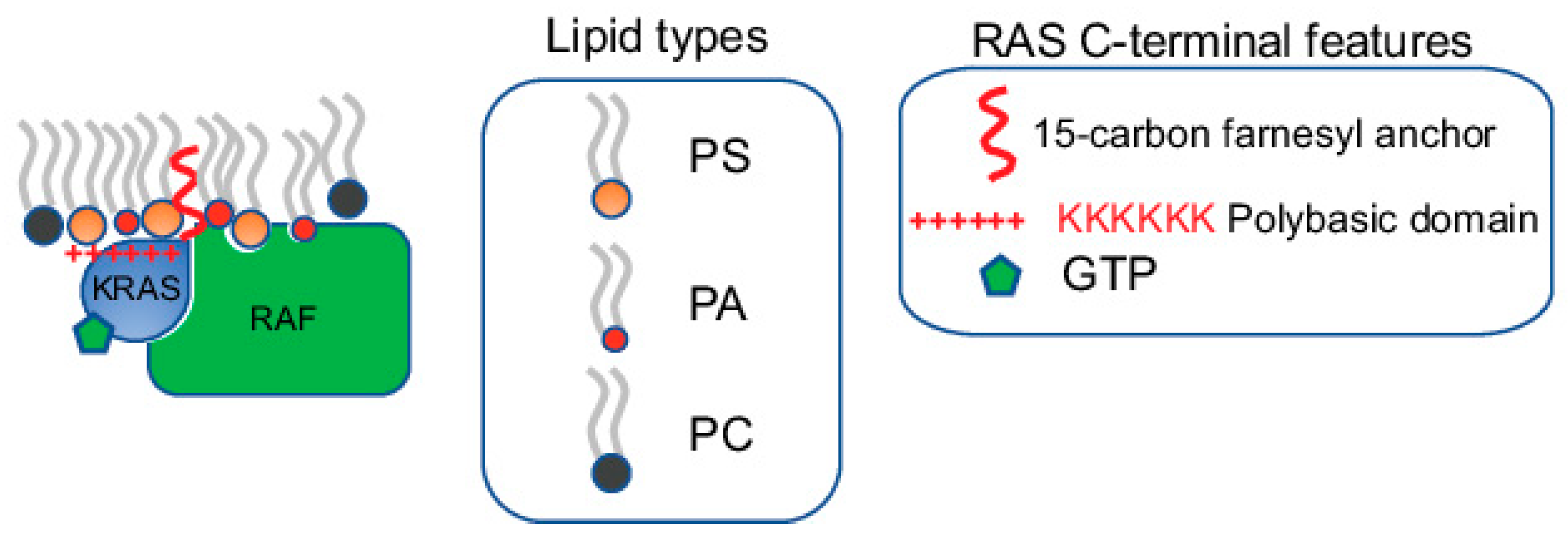
1.1. RAS Nanoclusters Selectively Sort Distinct Lipids in the Plasma Membrane
1.2. Electron Microscopy-Spatial Analysis Quantifies the Nanoclustering and Selective Lipid Sorting of RAS on Intact Plasma Membrane
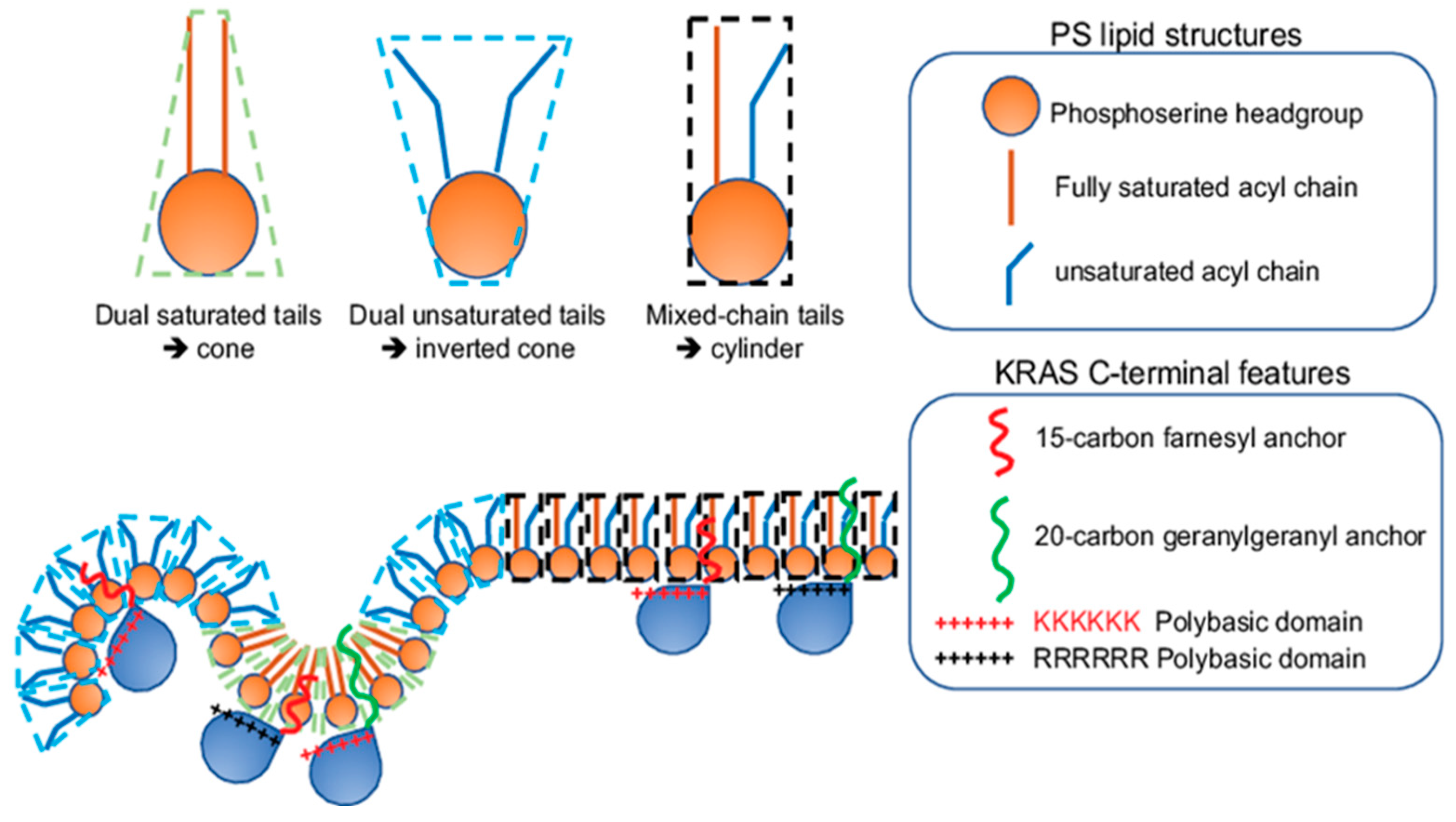
2. KRAS4B Nanoclusters Selectively Enrich Lipids with Distinct Headgroup and Acyl Chain Structures
2.1. KRAS4B Nanoclusters on the PM Contain Distinct and Precise Molecular Contents Important for Signal Propagation
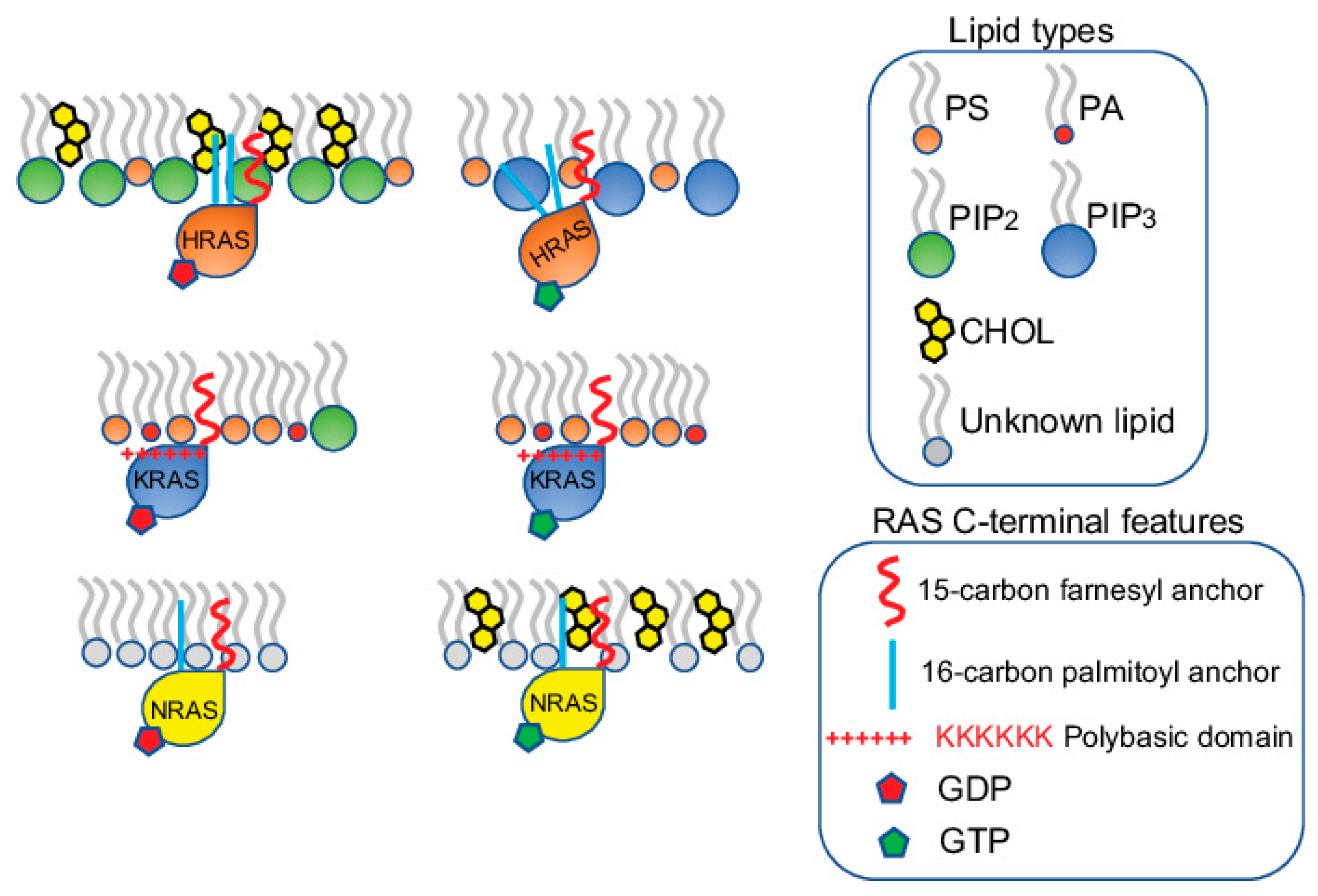
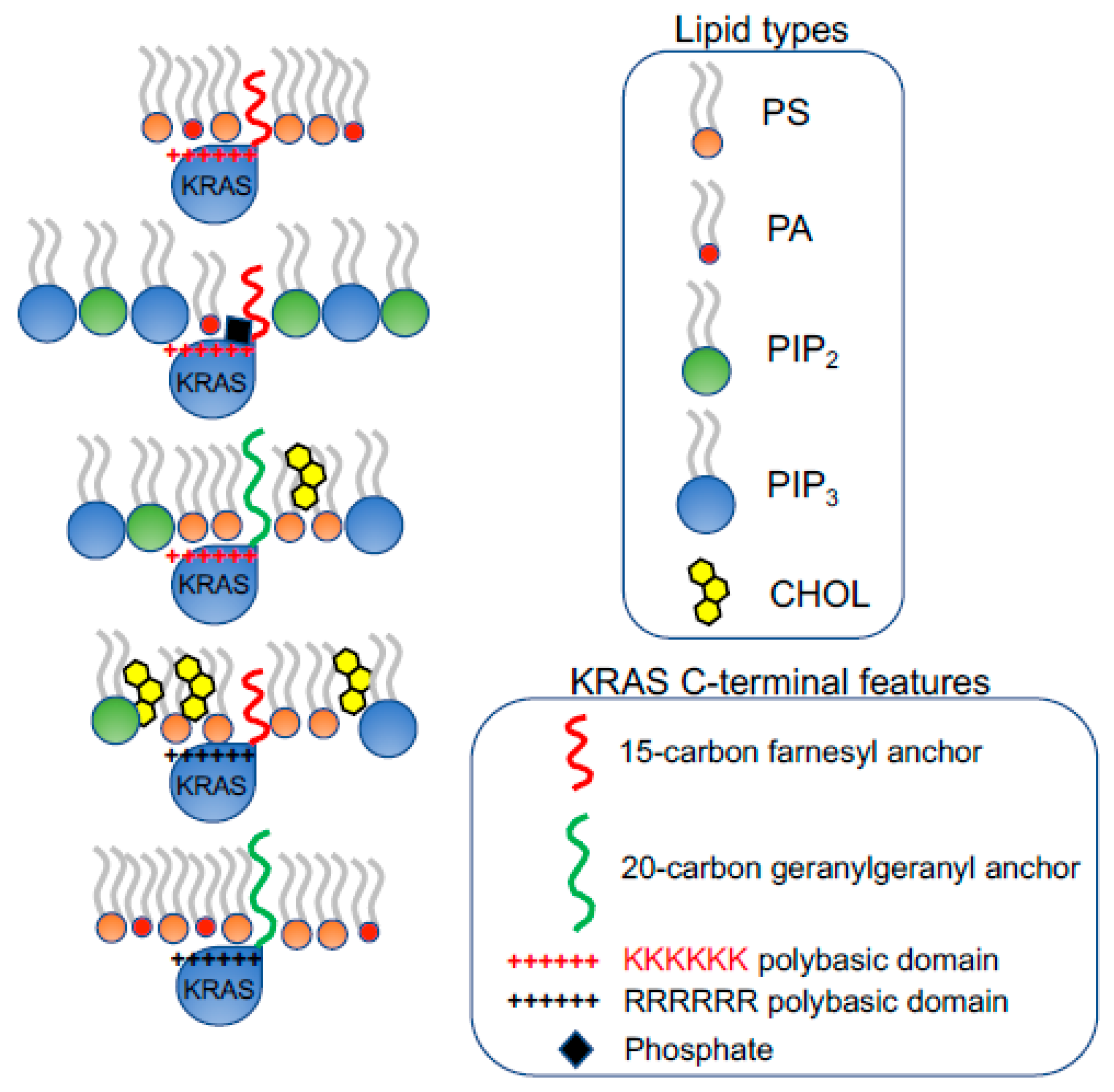
2.2. KRAS Nanoclusters Contain Distinct Lipid Types with Specific Headgroups, with a Selective Enrichment of PS Lipids
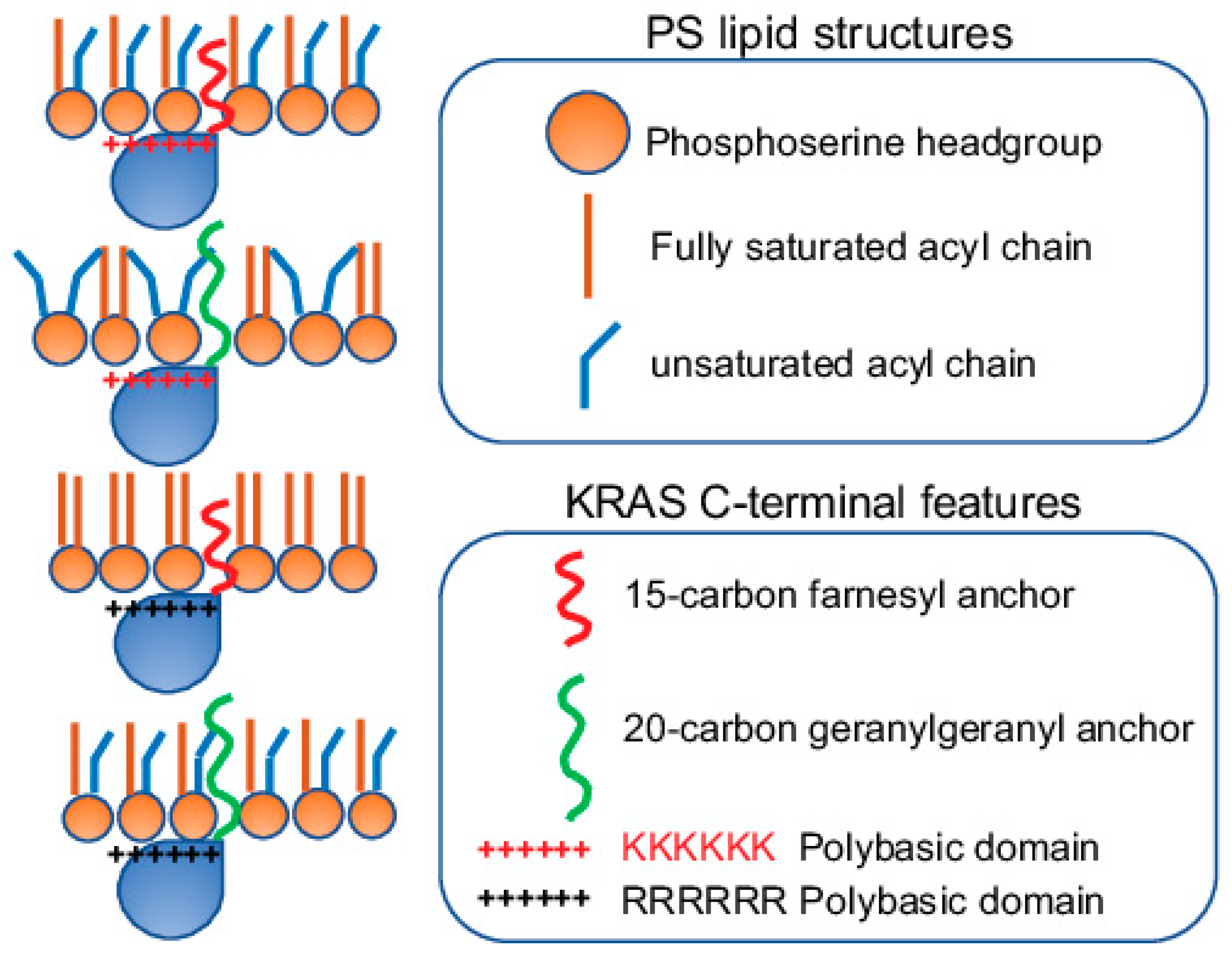
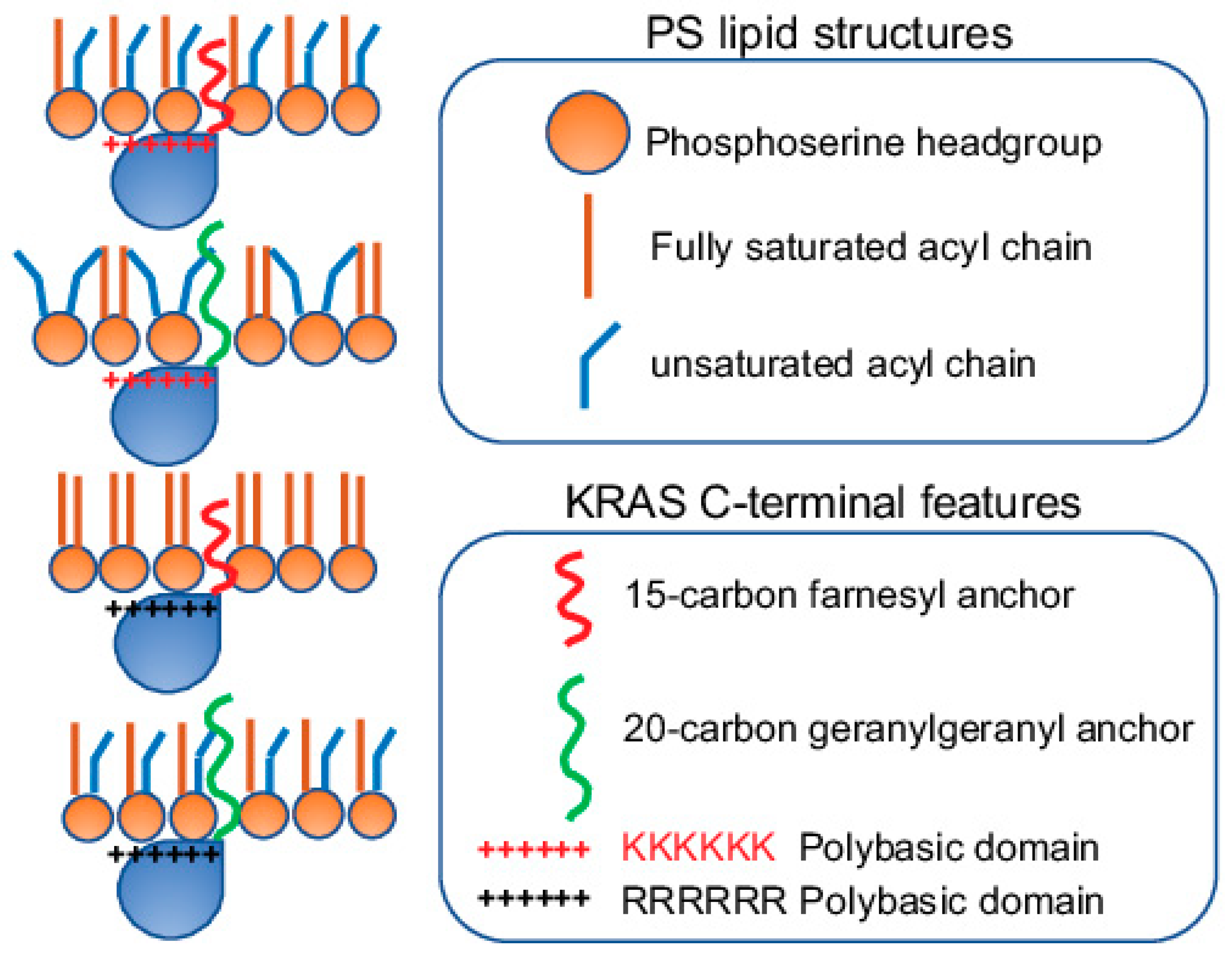
2.3. KRAS Nanoclusters Selectively Sort Distinct PS Species with Specific Acyl Chain Structures
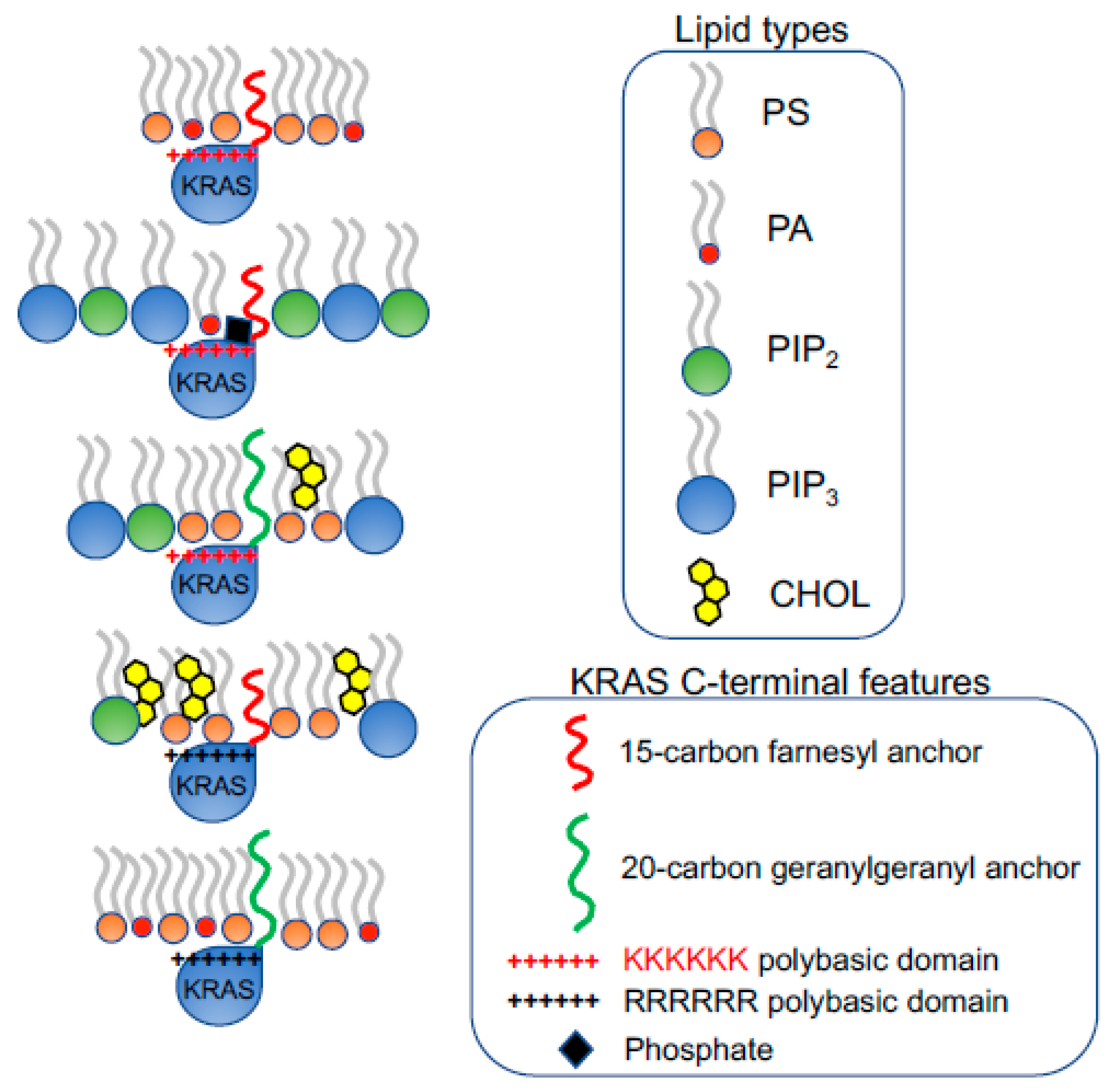
3. Conformational Sampling of Polybasic Domain Contributes to Selective Lipid Sorting of KRAS
4. Selective Lipid Sorting Capability Mediates Distinct Responses of KRAS Nanoclustering and Signaling to Perturbations of Membrane Properties
4.1. PM Depolarization Enhances the Nanoclustering and Signaling of Oncogenic Mutant KRAS
4.2. PM Curvature Disrupts the Nanoclustering and Signaling of KRAS
4.3. KRAS Polybasic Domain Mutants Possess Distinct Dependence on Cholesterol in the PM
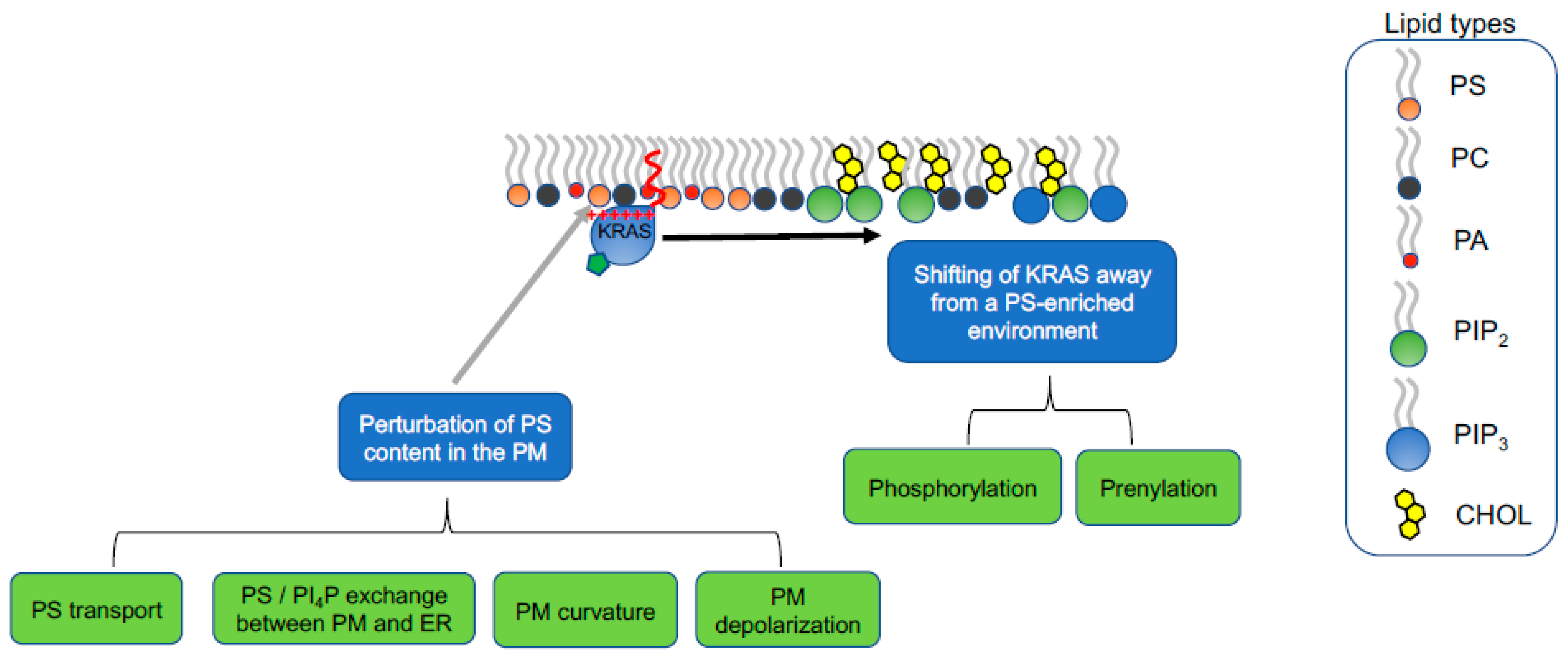
5. Strategies of Therapeutically Perturbing Lipid Profiles of RAS Nanoclusters
5.1. Interfering with PS Trafficking through Recycling Endosomes
5.2. Perturbation of PI4P/PS Exchange at the PM/ER Contact Sites
5.3. Perturbation of Lipid Sorting Specificity of KRAS Polybasic Domain
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Papke, B.; Der, C.J. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Veatch, S.L. Lipids out of order. Nat. Chem. Biol. 2008, 4, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Veatch, S.L.; Keller, S.L. Organization in lipid membranes containing cholesterol. Phys. Rev. Lett. 2002, 89, 268101. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Gerl, M.J. Revitalizing membrane rafts: New tools and insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688–699. [Google Scholar] [CrossRef]
- Baumgart, T.; Hess, S.T.; Webb, W.W. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature 2003, 425, 821–824. [Google Scholar] [CrossRef]
- Prior, I.A.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plowman, S.J.; Muncke, C.; Parton, R.G.; Hancock, J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 102, 15500–15505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, C.; Baranski, J.; Schlummer, S.; Palomo, J.; Lumbierres-Burgues, M.; Kahms, M.; Kuhlmann, J.; Sanchez, S.; Gratton, E.; Waldmann, H.; et al. Visualizing Association of N-Ras in Lipid Microdomains: Influence of Domain Structure and Interfacial Adsorption. J. Am. Chem. Soc. 2006, 128, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Harding, A.; Inder, K.; Plowman, S.; Parton, R.G.; Hancock, J.F. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007, 9, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Weise, K.; Triola, G.; Brunsveld, L.; Waldmann, H.; Winter, R. Influence of the lipidation motif on the partitioning and association of N-Ras in model membrane subdomains. J. Am. Chem. Soc. 2009, 131, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Weise, K.; Kapoor, S.; Denter, C.; Nikolaus, J.; Opitz, N.; Koch, S.; Triola, G.; Herrmann, A.; Waldmann, H.; Winter, R. Membrane-mediated induction and sorting of K-Ras microdomain signaling platforms. J. Am. Chem. Soc. 2011, 133, 880–887. [Google Scholar] [CrossRef]
- Janosi, L.; Li, Z.; Hancock, J.F.; Gorfe, A.A. Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc. Natl. Acad. Sci. USA 2012, 109, 8097–8102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, S.; Triola, G.; Vetter, I.R.; Erlkamp, M.; Waldmann, H.; Winter, R. Revealing conformational substates of lipidated N-Ras protein by pressure modulation. Proc. Natl. Acad. Sci. USA 2012, 109, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Janosi, L.; Gorfe, A.A. Formation and domain partitioning of H-ras peptide nanoclusters: Effects of peptide concentration and lipid composition. J. Am. Chem. Soc. 2012, 134, 17278–17285. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, S.; Weise, K.; Erlkamp, M.; Triola, G.; Waldmann, H.; Winter, R. The role of G-domain orientation and nucleotide state on the Ras isoform-specific membrane interaction. Eur. Biophys. J. 2012, 41, 801–813. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, H.; Rodkey, T.; Ariotti, N.; Parton, R.G.; Hancock, J.F. Signal Integration by Lipid-Mediated Spatial Cross Talk between Ras Nanoclusters. Mol. Cell Biol. 2014, 34, 862–876. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wong, C.O.; Cho, K.J.; van der Hoeven, D.; Liang, H.; Thakur, D.P.; Luo, J.; Babic, M.; Zinsmaier, K.E.; Zhu, M.X.; et al. SIGNAL TRANSDUCTION. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 2015, 349, 873–876. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Prakash, P.; Liang, H.; Cho, K.J.; Gorfe, A.A.; Hancock, J.F. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell 2017, 168, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Mu, H.; Jean-Francois, F.; Lakshman, B.; Sarkar-Banerjee, S.; Zhuang, Y.; Zeng, Y.; Gao, W.; Zaske, A.M.; Nissley, D.V.; et al. Membrane curvature sensing of the lipid-anchored K-Ras small GTPase. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Prakash, P.S.; Liang, H.; Gorfe, A.A.; Hancock, J.F. The KRAS and other prenylated polybasic domain membrane anchors recognize phosphatidylserine acyl chain structure. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Zhou, Y.; Ariotti, N.; Rae, J.; Liang, H.; Tillu, V.; Tee, S.; Bastiani, M.; Bademosi, A.T.; Collins, B.M.; Meunier, F.A.; et al. Caveolin-1 and cavin1 act synergistically to generate a unique lipid environment in caveolae. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Murakoshi, H.; Iino, R.; Kobayashi, T.; Fujiwara, T.; Ohshima, C.; Yoshimura, A.; Kusumi, A. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7317–7322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inder, K.; Hancock, J.F. System output of the MAPK module is spatially regulated. Commun. Integr. Biol. 2008, 1, 178–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inder, K.; Harding, A.; Plowman, S.J.; Philips, M.R.; Parton, R.G.; Hancock, J.F. Activation of the MAPK module from different spatial locations generates distinct system outputs. Mol. Biol. Cell. 2008, 19, 4776–4784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belanis, L.; Plowman, S.J.; Rotblat, B.; Hancock, J.F.; Kloog, Y. Galectin-1 is a novel structural component and a major regulator of h-ras nanoclusters. Mol. Biol. Cell 2008, 19, 1404–1414. [Google Scholar] [CrossRef] [Green Version]
- Blazevits, O.; Mideksa, Y.G.; Solman, M.; Ligabue, A.; Ariotti, N.; Nakhaeizadeh, H.; Fansa, E.K.; Papageorgiou, A.C.; Wittinghofer, A.; Ahmadian, M.R.; et al. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci. Rep. 2016, 6, 24165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, I.M.D.; Lectez, B.; Sharma, M.; Oetken-Lindholm, C.; Yetukuri, L.; Zhou, Y.; Aittokallio, T.; Abankwa, D. Rapalogs can promote cancer cell stemness in vitro in a Galectin-1 and H-ras-dependent manner. Oncotarget 2017, 8, 44550–44566. [Google Scholar] [CrossRef] [PubMed]
- Rotblat, B.; Niv, H.; Andre, S.; Kaltner, H.; Gabius, H.J.; Kloog, Y. Galectin-1(L11A) predicted from a computed galectin-1 farnesyl-binding pocket selectively inhibits Ras-GTP. Cancer Res. 2004, 64, 3112–3118. [Google Scholar] [CrossRef] [Green Version]
- Elad-Sfadia, G.; Haklai, R.; Balan, E.; Kloog, Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J. Biol. Chem. 2004, 279, 34922–34930. [Google Scholar] [CrossRef] [Green Version]
- Shalom-Feuerstein, R.; Plowman, S.J.; Rotblat, B.; Ariotti, N.; Tian, T.; Hancock, J.F.; Kloog, Y. K-ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008, 68, 6608–6616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, I.M.; Serulla, M.; Zhou, Y.; Oetken-Lindholm, C.; Abankwa, D.; Lectez, B. ASPP2 Is a Novel Pan-Ras Nanocluster Scaffold. PLoS ONE 2016, 11, e0159677. [Google Scholar] [CrossRef]
- Inder, K.L.; Hill, M.M.; Hancock, J.F. Nucleophosmin and nucleolin regulate K-Ras signaling. Commun. Integr. Biol. 2010, 3, 188–190. [Google Scholar] [CrossRef]
- Inder, K.L.; Lau, C.; Loo, D.; Chaudhary, N.; Goodall, A.; Martin, S.; Jones, A.; van der Hoeven, D.; Parton, R.G.; Hill, M.M.; et al. Nucleophosmin and nucleolin regulate K-Ras plasma membrane interactions and MAPK signal transduction. J. Biol. Chem. 2009, 284, 28410–28419. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hancock, J.F. Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta 2015, 1853, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hancock, J.F. Deciphering lipid codes: K-Ras as a paradigm. Traffic 2018, 19, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hancock, J.F. A novel prenyl-polybasic domain code determines lipid-binding specificity of the K-Ras membrane anchor. Small GTPases 2020, 11, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Strum, J.C.; Sciorra, V.A.; Daniel, L.; Bell, R.M. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem. 1996, 271, 8472–8480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Xie, W.Q.; Quest, A.F.; Mabrouk, G.M.; Strum, J.C.; Bell, R.M. The cysteine-rich region of raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-ras. J. Biol. Chem. 1994, 269, 10000–10007. [Google Scholar] [CrossRef]
- Li, Z.L.; Prakash, P.; Buck, M. A “Tug of War” Maintains a Dynamic Protein-Membrane Complex: Molecular Dynamics Simulations of C-Raf RBD-CRD Bound to K-Ras4B at an Anionic Membrane. ACS Cent. Sci. 2018, 4, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.R.; Parker, J.A.; Chung, J.K.; Li, Z.; Lee, Y.K.; Cookis, T.; Guterres, H.; Alvarez, S.; Hossain, M.A.; Donnelly, D.P.; et al. Raf promotes dimerization of the Ras G-domain with increased allosteric connections. Proc. Natl. Acad. Sci. USA 2021, 118, e2015648118. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Miao, B.; Skidan, I.; Yang, J.; Lugovskoy, A.; Reibarkh, M.; Long, K.; Brazell, T.; Durugkar, K.A.; Maki, J.; Ramana, C.V.; et al. Small molecule inhibition of phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains. Proc. Natl. Acad. Sci. USA 2010, 107, 20126–20131. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hancock, J.F. Super-Resolution Imaging and Spatial Analysis of RAS on Intact Plasma Membrane Sheets. Methods Mol. Biol. 2021, 2262, 217–232. [Google Scholar] [CrossRef]
- Zhou, Y.; Hancock, J.F. Electron microscopy combined with spatial analysis: Quantitative mapping of the nano-assemblies of plasma membrane-associating proteins and lipids. Biophys. Rep. 2018, 4, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Sarkar-Banerjee, S.; Sayyed-Ahmad, A.; Prakash, P.; Cho, K.J.; Waxham, M.N.; Hancock, J.F.; Gorfe, A.A. Spatiotemporal Analysis of K-Ras Plasma Membrane Interactions Reveals Multiple High Order Homo-oligomeric Complexes. J. Am. Chem. Soc. 2017, 139, 13466–13475. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Paterson, H.; Marshall, C.J. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990, 63, 133–139. [Google Scholar] [CrossRef]
- Hancock, J.F.; Cadwallader, K.; Paterson, H.; Marshall, C.J. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991, 10, 4033–4039. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Uchida, Y.; Emoto, K.; Umeda, M.; Kuge, O.; Taguchi, T.; Arai, H. Impaired retrograde membrane traffic through endosomes in a mutant CHO cell defective in phosphatidylserine synthesis. Genes Cells 2012, 17, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; van der Hoeven, D.; Zhou, Y.; Maekawa, M.; Ma, X.; Chen, W.; Fairn, G.D.; Hancock, J.F. Inhibition of Acid Sphingomyelinase Depletes Cellular Phosphatidylserine and Mislocalizes K-Ras from the Plasma Membrane. Mol. Cell Biol. 2015, 36, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.J.; Park, J.H.; Piggott, A.M.; Salim, A.A.; Gorfe, A.A.; Parton, R.G.; Capon, R.J.; Lacey, E.; Hancock, J.F. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J. Biol. Chem. 2012, 287, 43573–43584. [Google Scholar] [CrossRef] [Green Version]
- van der Hoeven, D.; Cho, K.J.; Ma, X.; Chigurupati, S.; Parton, R.G.; Hancock, J.F. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol. Cell Biol. 2013, 33, 237–251. [Google Scholar] [CrossRef] [Green Version]
- van der Hoeven, D.; Cho, K.J.; Zhou, Y.; Ma, X.; Chen, W.; Naji, A.; Montufar-Solis, D.; Zuo, Y.; Kovar, S.E.; Levental, K.R.; et al. Sphingomyelin metabolism is a regulator of KRAS function. Mol. Cell Biol. 2017, 38, e00373-17. [Google Scholar] [CrossRef] [Green Version]
- Lakshman, B.; Messing, S.; Schmid, E.M.; Clogston, J.D.; Gillette, W.K.; Esposito, D.; Kessing, B.; Fletcher, D.A.; Nissley, D.V.; McCormick, F.; et al. Quantitative biophysical analysis defines key components modulating recruitment of the GTPase KRAS to the plasma membrane. J. Biol. Chem. 2019, 294, 2193–2207. [Google Scholar] [CrossRef] [Green Version]
- Janosi, L.; Gorfe, A.A. Segregation of negatively charged phospholipids by the polycationic and farnesylated membrane anchor of Kras. Biophys. J. 2010, 99, 3666–3674. [Google Scholar] [CrossRef] [Green Version]
- Rawicz, W.; Olbrich, K.C.; McIntosh, T.; Needham, D.; Evans, E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 2000, 79, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric controls of cell proliferation: Ion channels, membrane voltage and the cell cycle. Cell Cycle 2009, 8, 3519–3528. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Foller, M.; Lang, K.S.; Lang, P.A.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion channels in cell proliferation and apoptotic cell death. J. Membr. Biol. 2005, 205, 147–157. [Google Scholar] [CrossRef]
- Pardo, L.A. Voltage-gated potassium channels in cell proliferation. Physiology 2004, 19, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Szabo, I.; Zoratti, M.; Gulbins, E. Contribution of voltage-gated potassium channels to the regulation of apoptosis. FEBS Lett. 2010, 584, 2049–2056. [Google Scholar] [CrossRef] [Green Version]
- Miroshnikova, Y.A.; Le, H.Q.; Schneider, D.; Thalheim, T.; Rubsam, M.; Bremicker, N.; Polleux, J.; Kamprad, N.; Tarantola, M.; Wang, I.; et al. Adhesion forces and cortical tension couple cell proliferation and differentiation to drive epidermal stratification. Nat. Cell Biol. 2018, 20, 69–80. [Google Scholar] [CrossRef]
- Hirama, T.; Lu, S.M.; Kay, J.G.; Maekawa, M.; Kozlov, M.M.; Grinstein, S.; Fairn, G.D. Membrane curvature induced by proximity of anionic phospholipids can initiate endocytosis. Nat. Commun. 2017, 8, 1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakar, R.G.; Cheng, Q.; Patel, S.; Chu, J.; Nasir, M.; Liepmann, D.; Komvopoulos, K.; Li, S. Cell-shape regulation of smooth muscle cell proliferation. Biophys. J. 2009, 96, 3423–3432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Plowman, S.; Rotblat, B.; Prior, I.A.; Muncke, C.; Grainger, S.; Parton, R.G.; Henis, Y.I.; Kloog, Y.; Hancock, J.F. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol. Cell Biol. 2005, 25, 6722–6733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmick, M.; Vartak, N.; Papke, B.; Kovacevic, M.; Truxius, D.C.; Rossmannek, L.; Bastiaens, P.I. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014, 157, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.; et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2011, 14, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Moser von Filseck, J.; Copic, A.; Delfosse, V.; Vanni, S.; Jackson, C.L.; Bourguet, W.; Drin, G. INTRACELLULAR TRANSPORT. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science 2015, 349, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Moser von Filseck, J.; Vanni, S.; Mesmin, B.; Antonny, B.; Drin, G. A phosphatidylinositol-4-phosphate powered exchange mechanism to create a lipid gradient between membranes. Nat. Commun. 2015, 6, 6671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galmes, R.; Houcine, A.; van Vliet, A.R.; Agostinis, P.; Jackson, C.L.; Giordano, F. ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016, 17, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Sohn, M.; Ivanova, P.; Brown, H.A.; Toth, D.J.; Varnai, P.; Kim, Y.J.; Balla, T. Lenz-Majewski mutations in PTDSS1 affect phosphatidylinositol 4-phosphate metabolism at ER-PM and ER-Golgi junctions. Proc. Natl. Acad. Sci. USA 2016, 113, 4314–4319. [Google Scholar] [CrossRef] [Green Version]
- Gulbins, E.; Palmada, M.; Reichel, M.; Luth, A.; Bohmer, C.; Amato, D.; Muller, C.P.; Tischbirek, C.H.; Groemer, T.W.; Tabatabai, G.; et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat. Med. 2013, 19, 934–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhle, C.; Huttner, H.B.; Walter, S.; Reichel, M.; Canneva, F.; Lewczuk, P.; Gulbins, E.; Kornhuber, J. Characterization of acid sphingomyelinase activity in human cerebrospinal fluid. PLoS ONE 2013, 8, e62912. [Google Scholar] [CrossRef] [Green Version]
- Munzer, P.; Borst, O.; Walker, B.; Schmid, E.; Feijge, M.A.; Cosemans, J.M.; Chatterjee, M.; Schmidt, E.M.; Schmidt, S.; Towhid, S.T.; et al. Acid sphingomyelinase regulates platelet cell membrane scrambling, secretion, and thrombus formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Kattan, W.E.; Chen, W.; Ma, X.; Lan, T.H.; van der Hoeven, D.; van der Hoeven, R.; Hancock, J.F. Targeting plasma membrane phosphatidylserine content to inhibit oncogenic KRAS function. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; Casteel, D.E.; Prakash, P.; Tan, L.; van der Hoeven, D.; Salim, A.A.; Kim, C.; Capon, R.J.; Lacey, E.; Cunha, S.R.; et al. AMPK and Endothelial Nitric Oxide Synthase Signaling Regulates K-Ras Plasma Membrane Interactions via Cyclic GMP-Dependent Protein Kinase 2. Mol. Cell Biol. 2016, 36, 3086–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Hancock, J.F. Lipid Profiles of RAS Nanoclusters Regulate RAS Function. Biomolecules 2021, 11, 1439. https://doi.org/10.3390/biom11101439
Zhou Y, Hancock JF. Lipid Profiles of RAS Nanoclusters Regulate RAS Function. Biomolecules. 2021; 11(10):1439. https://doi.org/10.3390/biom11101439
Chicago/Turabian StyleZhou, Yong, and John F. Hancock. 2021. "Lipid Profiles of RAS Nanoclusters Regulate RAS Function" Biomolecules 11, no. 10: 1439. https://doi.org/10.3390/biom11101439
APA StyleZhou, Y., & Hancock, J. F. (2021). Lipid Profiles of RAS Nanoclusters Regulate RAS Function. Biomolecules, 11(10), 1439. https://doi.org/10.3390/biom11101439