
Crystal Structure Reveals the Full Ras–Raf Interface and Advances Mechanistic Understanding of Raf Activation

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Expression/Purification
2.2. Protein Crystallization, Data Collection, and Refinement
2.3. Model Preparation for Molecular Dynamics Simulations
2.4. Molecular Dynamics Simulations
2.5. Trajectory Analysis
2.6. Protein Interfaces Surfaces and Assemblies (PISA)
3. Results
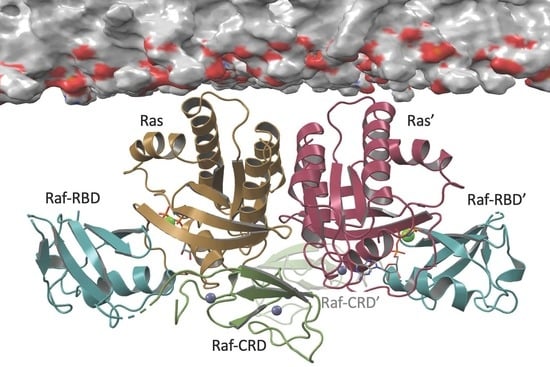
3.1. Overall Structure of Ras–Raf-RBD_CRD Complex
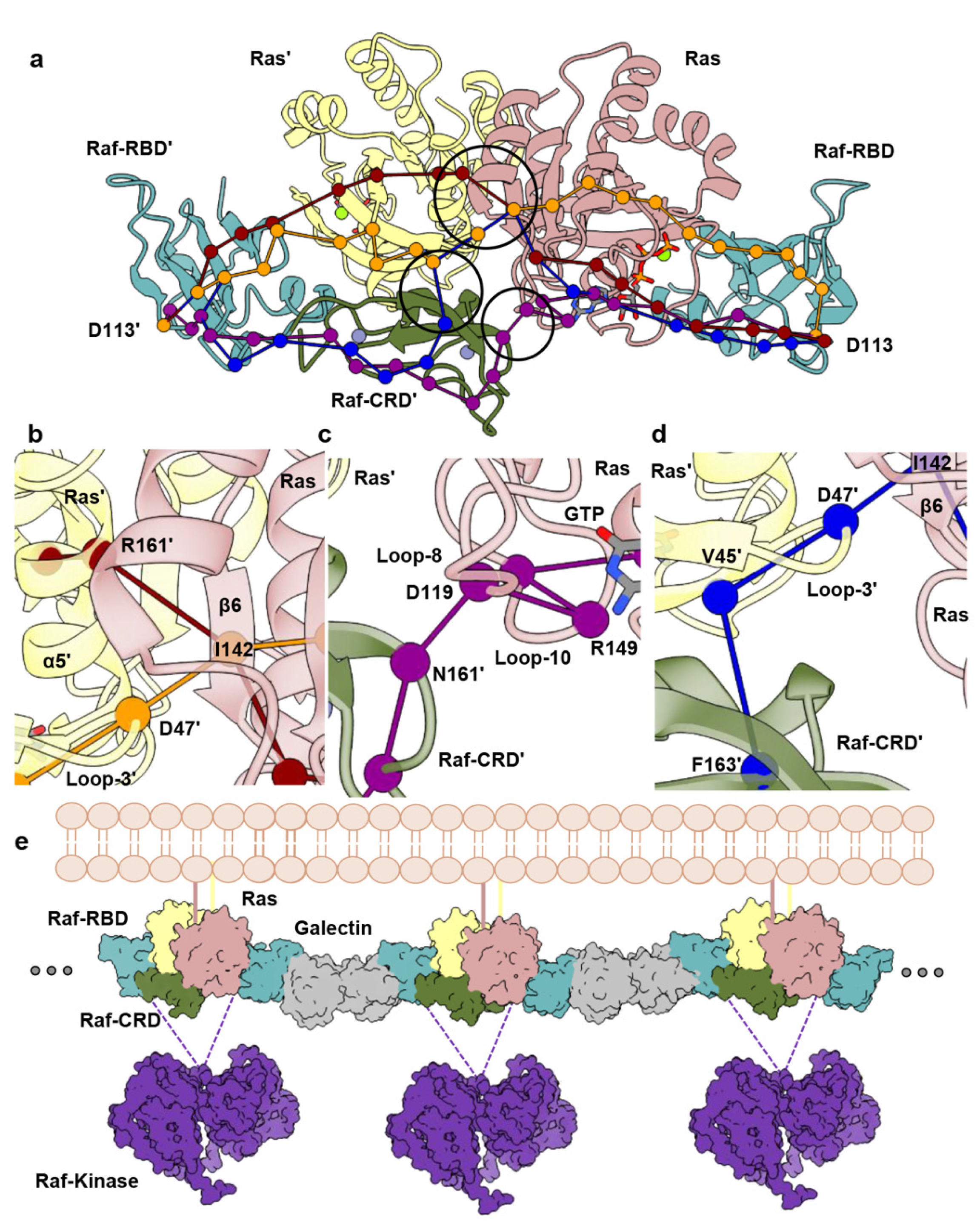
3.2. Raf-CRD Links to the Active Site through Loop 8 across the Dimer Interface
3.3. Allosteric Communication across the Ras–Raf-RBD_CRD Dimer Complex
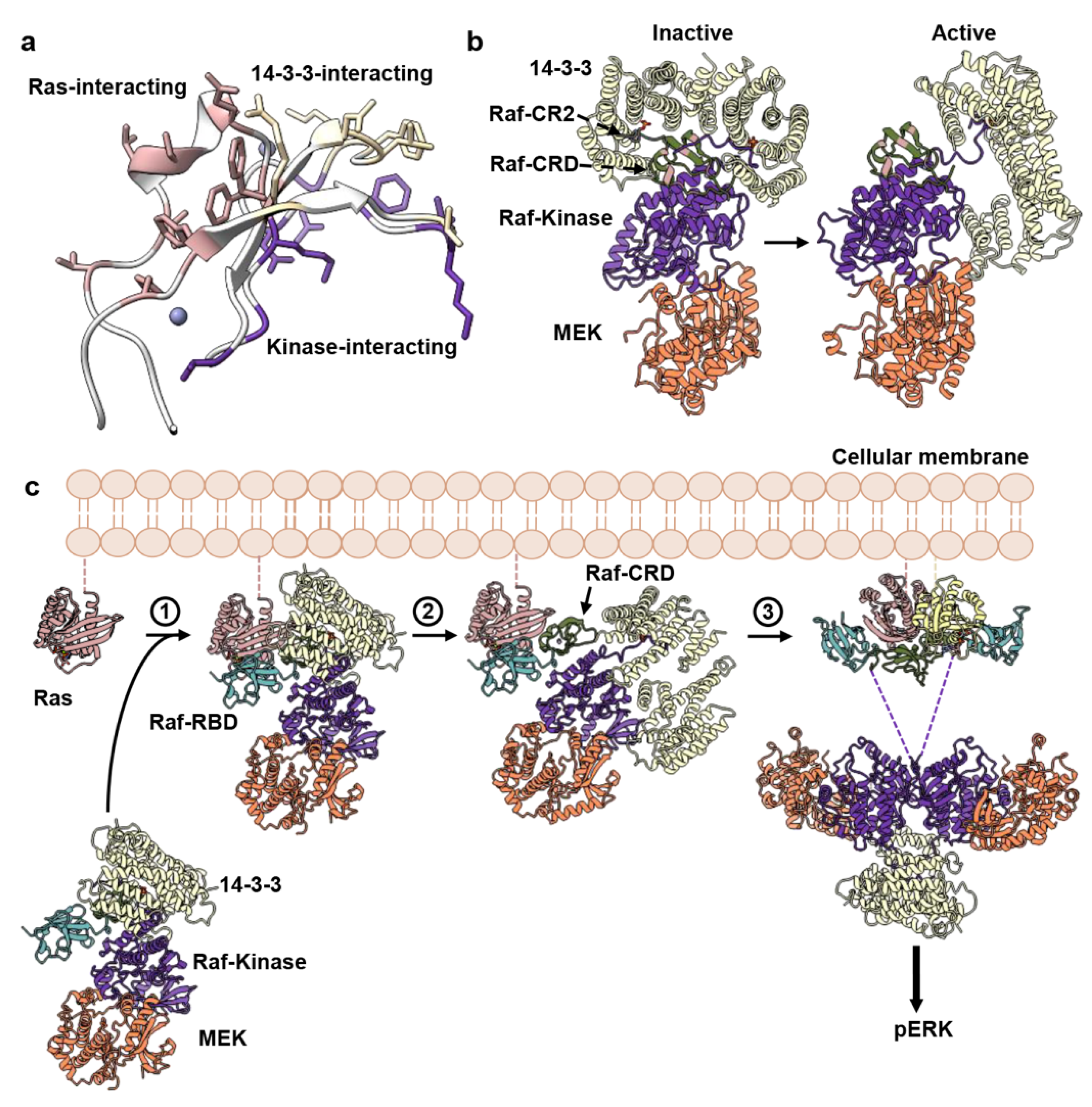
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winkler, D.G.; Cutler, R.E., Jr.; Drugan, J.K.; Campbell, S.; Morrison, D.K.; Cooper, J.A. Identification of Residues in the Cysteine-rich Domain of Raf-1 That Control Ras Binding and Raf-1 Activity. J. Biol. Chem. 1998, 273, 21578–21584. [Google Scholar] [CrossRef]
- Williams, J.G.; Drugan, J.K.; Yi, G.-S.; Clark, G.J.; Der, C.J.; Campbell, S. Elucidation of Binding Determinants and Functional Consequences of Ras/Raf-Cysteine-rich Domain Interactions. J. Biol. Chem. 2000, 275, 22172–22179. [Google Scholar] [CrossRef]
- Mott, H.; Carpenter, J.W.; Zhong, S.; Ghosh, S.; Bell, R.M.; Campbell, S. The solution structure of the Raf-1 cysteine-rich domain: A novel ras and phospholipid binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8312–8317. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Roskoski, R. RAF protein-serine/threonine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2010, 399, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, G.; Wink, G.; Mattos, C. Transformation Efficiency of RasQ61 Mutants Linked to Structural Features of the Switch Regions in the Presence of Raf. Structure 2007, 15, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Fetics, S.K.; Guterres, H.; Kearney, B.M.; Buhrman, G.; Ma, B.; Nussinov, R.; Mattos, C. Allosteric Effects of the Oncogenic RasQ61L Mutant on Raf-RBD. Structure 2015, 23, 505–516. [Google Scholar] [CrossRef]
- Packer, M.R.; Parker, J.A.; Chung, J.K.; Li, Z.; Lee, Y.K.; Cookis, T.; Guterres, H.; Alvarez, S.; Hossain, A.; Donnelly, D.P.; et al. Raf promotes dimerization of the Ras G-domain with increased allosteric connections. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Köhler, J.; Zhou, Z.-W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Spencer-Smith, R.; O’Bryan, J.P. Targeting the α4–α5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene 2019, 38, 2984–2993. [Google Scholar] [CrossRef]
- Inouye, K.; Mizutani, S.; Koide, H.; Kaziro, Y. Formation of the Ras Dimer Is Essential for Raf-1 Activation. J. Biol. Chem. 2000, 275, 3737–3740. [Google Scholar] [CrossRef]
- Nan, X.; Tamgüney, T.M.; Collisson, E.A.; Lin, L.-J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef]
- Blaževitš, O.; Mideksa, Y.G.; Šolman, M.; Ligabue, A.; Ariotti, N.; Nakhaeizadeh, H.; Fansa, E.K.; Papageorgiou, A.; Wittinghofer, A.; Ahmadian, M.R.; et al. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci. Rep. 2016, 6, 24165. [Google Scholar] [CrossRef]
- Caron, M.; Bladier, D.; Joubert, R. Soluble galactoside-binding vertebrate lectins: A protein family with common properties. Int. J. Biochem. 1990, 22, 1379–1385. [Google Scholar] [CrossRef]
- Paz, A.; Haklai, R.; Elad-Sfadia, G.; Ballan, E.; Kloog, Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 2001, 20, 7486–7493. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Feuerstein, R.; Plowman, S.J.; Rotblat, B.; Ariotti, N.; Tian, T.; Hancock, J.F.; Kloog, Y. K-Ras Nanoclustering Is Subverted by Overexpression of the Scaffold Protein Galectin-3. Cancer Res. 2008, 68, 6608–6616. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.-Y.; Hill, P.N.; Hsu, A.D.K.; Liu, F.-T. Role of the Carboxyl-Terminal Lectin Domain in Self-Association of Galectin-3. Biochemistry 1998, 37, 4086–4092. [Google Scholar] [CrossRef]
- Drugan, J.K.; Khosravi-Far, R.; White, M.A.; Der, C.J.; Sung, Y.-J.; Hwang, Y.-W.; Campbell, S. Ras Interaction with Two Distinct Binding Domains in Raf-1 5 Be Required for Ras Transformation. J. Biol. Chem. 1996, 271, 233–237. [Google Scholar] [CrossRef]
- Hu, C.-D.; Kariya, K.-I.; Tamada, M.; Akasaka, K.; Shirouzu, M.; Yokoyama, S.; Kataoka, T. Cysteine-rich Region of Raf-1 Interacts with Activator Domain of Post-translationally Modified Ha-Ras. J. Biol. Chem. 1995, 270, 30274–30277. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Chan, A.H.; Young, L.C.; Bindu, L.; Neale, C.; Messing, S.; Dharmaiah, S.; Taylor, T.; Denson, J.-P.; Esposito, D.; et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Xie, W.Q.; Quest, A.F.; Mabrouk, G.M.; Strum, J.C.; Bell, R.M. The cysteine-rich region of raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-ras. J. Biol. Chem. 1994, 269, 10000–10007. [Google Scholar] [CrossRef]
- Lakshman, B.; Messing, S.; Schmid, E.M.; Clogston, J.D.; Gillette, W.K.; Esposito, D.; Kessing, B.; Fletcher, D.A.; Nissley, D.V.; McCormick, F.; et al. Quantitative biophysical analysis defines key components modulating recruitment of the GTPase KRAS to the plasma membrane. J. Biol. Chem. 2019, 294, 2193–2207. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Lane, A.; Yan, J.; McPherson, R.; Hancock, J.F. Activity of Plasma Membrane-recruited Raf-1 Is Regulated by Ras via the Raf Zinc Finger. J. Biol. Chem. 1997, 272, 20139–20145. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Lee, K.-Y.; Huo, K.-G.; Gasmi-Seabrook, G.; Zheng, L.; Moghal, N.; Tsao, M.-S.; Ikura, M.; Marshall, C.B. Multivalent assembly of KRAS with the RAS-binding and cysteine-rich domains of CRAF on the membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 12101–12108. [Google Scholar] [CrossRef]
- Jang, H.; Zhang, M.; Nussinov, R. The quaternary assembly of KRas4B with Raf-1 at the membrane. Comput. Struct. Biotechnol. J. 2020, 18, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jang, H.; Zhang, J.; Nussinov, R. Raf-1 Cysteine-Rich Domain Increases the Affinity of K-Ras/Raf at the Membrane, Promoting MAPK Signaling. Structure 2018, 26, 513–525.e2. [Google Scholar] [CrossRef]
- Li, Z.-L.; Prakash, P.; Buck, M. A “Tug of War” Maintains a Dynamic Protein–Membrane Complex: Molecular Dynamics Simulations of C-Raf RBD-CRD Bound to K-Ras4B at an Anionic Membrane. ACS Central Sci. 2018, 4, 298–305. [Google Scholar] [CrossRef]
- Travers, T.; López, C.A.; Agamasu, C.; Hettige, J.J.; Messing, S.; García, A.E.; Stephen, A.G.; Gnanakaran, S. Anionic Lipids Impact RAS-Binding Site Accessibility and Membrane Binding Affinity of CRAF RBD-CRD. Biophys. J. 2020, 119, 525–538. [Google Scholar] [CrossRef]
- Hekman, M.; Wiese, S.; Metz, R.; Albert, S.; Troppmair, J.; Nickel, J.; Sendtner, M.; Rapp, U.R. Dynamic Changes in C-Raf Phosphorylation and 14-3-3 Protein Binding in Response to Growth Factor Stimulation: DIFFERENTIAL ROLES OF 14-3-3 PROTEIN BINDING SITES. J. Biol. Chem. 2004, 279, 14074–14086. [Google Scholar] [CrossRef]
- Muslin, A.J.; Tanner, J.; Allen, P.M.; Shaw, A.S. Interaction of 14-3-3 with Signaling Proteins Is Mediated by the Recognition of Phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef]
- Morrison, D.K.; Heidecker, G.; Rapp, U.R.; Copeland, T.D. Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem. 1993, 268, 17309–17316. [Google Scholar] [CrossRef]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Gonzalez-Del Pino, G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF–MEK1–14-3-3 complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Rommel, C.; Radziwill, G.; Moelling, K.; Hafen, E. Negative regulation of Raf activity by binding of 14-3-3 to the amino terminus of Raf in vivo. Mech. Dev. 1997, 64, 95–104. [Google Scholar] [CrossRef]
- Light, Y.; Paterson, H.; Marais, R. 14-3-3 Antagonizes Ras-Mediated Raf-1 Recruitment to the Plasma Membrane to Maintain Signaling Fidelity. Mol. Cell. Biol. 2002, 22, 4984–4996. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and Role of Raf-1/B-Raf Heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Stites, E.C.; Yu, H.; Germino, E.A.; Meharena, H.S.; Stork, P.J.; Kornev, A.P.; Taylor, S.S.; Shaw, A.S. Allosteric Activation of Functionally Asymmetric RAF Kinase Dimers. Cell 2013, 154, 1036–1046. [Google Scholar] [CrossRef]
- Clark, G.J.; Drugan, J.K.; Rossman, K.L.; Carpenter, J.W.; Rogers-Graham, K.; Fu, H.; Der, C.J.; Campbell, S.L. 14-3-3 zeta negatively regulates raf-1 activity by interactions with the Raf-1 cysteine-rich domain. J. Biol. Chem. 1997, 272, 20990–20993. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Ognjenović, J.; Banerjee, S.; Karandur, D.; Merk, A.; Kulhanek, K.; Wong, K.; Roose, J.P.; Subramaniam, S.; Kuriyan, J. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 2019, 366, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.W.; Buhrman, G.; Ting, P.Y.; Colicelli, J.; Mattos, C. Expression, purification, crystallization and X-ray data collection for RAS and its mutants. Data Brief 2016, 6, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 859–866. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K.D. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Glykos, N.M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Güldenhaupt, J.; Rudack, T.; Bachler, P.; Mann, D.; Triola, G.; Waldmann, H.; Kötting, C.; Gerwert, K. N-Ras Forms Dimers at POPC Membranes. Biophys. J. 2012, 103, 1585–1593. [Google Scholar] [CrossRef]
- Spencer-Smith, R.; Koide, A.; Zhou, Y.; Eguchi, R.R.; Sha, F.; Gajwani, P.; Santana, D.; Gupta, A.; Jacobs, M.; Herrero-Garcia, E.; et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. [Google Scholar] [CrossRef]
- Johnson, C.W.; Reid, D.; Parker, J.A.; Salter, S.; Knihtila, R.; Kuzmic, P.; Mattos, C. The small GTPases K-Ras, N-Ras, and H-Ras have distinct biochemical properties determined by allosteric effects. J. Biol. Chem. 2017, 292, 12981–12993. [Google Scholar] [CrossRef]
- Terrell, E.M.; Durrant, D.E.; Ritt, D.A.; Sealover, N.E.; Sheffels, E.; Spencer-Smith, R.; Esposito, D.; Zhou, Y.; Hancock, J.F.; Kortum, R.L.; et al. Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol. Cell 2019, 76, 872–884.e5. [Google Scholar] [CrossRef]
- Weber, C.K.; Slupsky, J.R.; Kalmes, H.A.; Rapp, U.R. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61, 3595–3598. [Google Scholar]
- Marcus, K.; Mattos, C. Direct Attack on RAS: Intramolecular Communication and Mutation-Specific Effects. Clin. Cancer Res. 2015, 21, 1810–1818. [Google Scholar] [CrossRef]
- Tiberti, M.; Invernizzi, G.; Papaleo, E. (Dis)similarity Index to Compare Correlated Motions in Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 4404–4414. [Google Scholar] [CrossRef] [PubMed]
- Botello-Smith, W.M.; Luo, Y. Robust Determination of Protein Allosteric Signaling Pathways. J. Chem. Theory Comput. 2019, 15, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.C.; Alvarez, S.; Kondo, Y.; Lee, Y.; Chung, J.K.; Lam, H.Y.M.; Biswas, K.H.; Kuriyan, J.; Groves, J.T. A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science 2019, 363, 1098–1103. [Google Scholar] [CrossRef]
- Belanis, L.; Plowman, S.J.; Rotblat, B.; Hancock, J.F.; Kloog, Y. Galectin-1 Is a Novel Structural Component and a Major Regulator of H-Ras Nanoclusters. Mol. Biol. Cell 2008, 19, 1404–1414. [Google Scholar] [CrossRef]
- Elad-Sfadia, G.; Haklai, R.; Ballan, E.; Gabius, H.-J.; Kloog, Y. Galectin-1 Augments Ras Activation and Diverts Ras Signals to Raf-1 at the Expense of Phosphoinositide 3-Kinase. J. Biol. Chem. 2002, 277, 37169–37175. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Phelps, C.; Huang, T.; Mostofian, B.; Wu, L.; Zhang, Y.; Tao, K.; Chang, Y.H.; Stork, P.J.; Gray, J.W.; et al. High-throughput, single-particle tracking reveals nested membrane domains that dictate KRasG12D diffusion and trafficking. eLife 2019, 8, 46393. [Google Scholar] [CrossRef]
- Murakoshi, H.; Iino, R.; Kobayashi, T.; Fujiwara, T.; Ohshima, C.; Yoshimura, A.; Kusumi, A. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7317–7322. [Google Scholar] [CrossRef] [PubMed]
- Goswami, D.; Chen, D.; Yang, Y.; Gudla, P.R.; Columbus, J.; Worthy, K.; Rigby, M.; Wheeler, M.; Mukhopadhyay, S.; Powell, K.; et al. Membrane interactions of the globular domain and the hypervariable region of KRAS4b define its unique diffusion behavior. eLife 2020, 9, 47654. [Google Scholar] [CrossRef]
- Van, Q.; Prakash, P.; Shrestha, R.; Balius, T.; Turbyville, T.; Stephen, A. RAS Nanoclusters: Dynamic Signaling Platforms Amenable to Therapeutic Intervention. Biomolecules 2021, 11, 377. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Chavan, T.S.; Freed, B.C.; Jang, H.; Khavrutskii, L.; Freed, R.N.; Dyba, M.A.; Stefanisko, K.; Tarasov, S.G.; Gursoy, A.; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Prakash, P.; Gorfe, A.A. Membrane orientation dynamics of lipid-modified small GTPases. Small GTPases 2016, 8, 129–138. [Google Scholar] [CrossRef]
- Sarkar-Banerjee, S.; Sayyed-Ahmad, A.; Prakash, P.; Cho, K.-J.; Waxham, M.N.; Hancock, J.F.; Gorfe, A.A. Spatiotemporal Analysis of K-Ras Plasma Membrane Interactions Reveals Multiple High Order Homo-oligomeric Complexes. J. Am. Chem. Soc. 2017, 139, 13466–13475. [Google Scholar] [CrossRef]
- Chung, J.; Lee, Y.; Denson, J.-P.; Gillette, W.K.; Alvarez, S.; Stephen, A.G.; Groves, J.T. K-Ras4B Remains Monomeric on Membranes over a Wide Range of Surface Densities and Lipid Compositions. Biophys. J. 2018, 114, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Terrell, E.M.; Morrison, D.K. Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, S.; Gimeno, A.; Núñez-Franco, R.; Bernardo-Seisdedos, G.; Delgado, S.; Jiménez-Osés, G.; Millet, O.; Jiménez-Barbero, J.; Ardá, A. Unravelling the Time Scale of Conformational Plasticity and Allostery in Glycan Recognition by Human Galectin-1. Chemistry 2020, 26, 15643–15653. [Google Scholar] [CrossRef]
- Lee, K.; Fang, Z.; Enomoto, M.; Gasmi-Seabrook, G.; Zheng, L.; Koide, S.; Ikura, M.; Marshall, C.B. Two Distinct Structures of Membrane-Associated Homodimers of GTP- and GDP-Bound KRAS4B Revealed by Paramagnetic Relaxation Enhancement. Angew. Chem. Int. Ed. 2020, 59, 11037–11045. [Google Scholar] [CrossRef] [PubMed]
- Bondeva, T.; Balla, A.; Várnai, P.; Balla, T. Structural Determinants of Ras-Raf Interaction Analyzed in Live Cells. Mol. Biol. Cell 2002, 13, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Thapar, R.; Williams, J.G.; Campbell, S.L. NMR Characterization of Full-length Farnesylated and Non-farnesylated H-Ras and its Implications for Raf Activation. J. Mol. Biol. 2004, 343, 1391–1408. [Google Scholar] [CrossRef]
- Daub, M.; Jöckel, J.; Quack, T.; Weber, C.K.; Schmitz, F.; Rapp, U.R.; Wittinghofer, A.; Block, C. The RafC1 Cysteine-Rich Domain Contains Multiple Distinct Regulatory Epitopes Which Control Ras-Dependent Raf Activation. Mol. Cell. Biol. 1998, 18, 6698–6710. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Improta-Brears, T.; Ghosh, S.; Bell, R.M. Mutational analysis of Raf-1 cysteine rich domain: Requirement for a cluster of basic aminoacids for interaction with phosphatidylserine. Mol. Cell. Biochem. 1999, 198, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Travers, T.; Lopez, C.A.; Van, Q.N.; Neale, C.; Tonelli, M.; Stephen, A.G.; Gnanakaran, S. Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci. Rep. 2018, 8, 8461. [Google Scholar] [CrossRef]
- Knihtila, R.; Holzapfel, G.; Weiss, K.; Meilleur, F.; Mattos, C. Neutron Crystal Structure of RAS GTPase Puts in Question the Protonation State of the GTP gamma-Phosphate. J. Biol. Chem. 2015, 290, 31025–31036. [Google Scholar] [CrossRef]
- Sarkozy, A.; Carta, C.; Moretti, S.; Zampino, G.; Digilio, M.C.; Pantaleoni, F.; Scioletti, A.P.; Esposito, G.; Cordeddu, V.; Lepri, F.R.; et al. GermlineBRAFmutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum. Hum. Mutat. 2009, 30, 695–702. [Google Scholar] [CrossRef]
- Anastasaki, C.; Estep, A.L.; Marais, R.; Rauen, K.A.; Patton, E.E. Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum. Mol. Genet. 2009, 18, 2543–2554. [Google Scholar] [CrossRef]
- Zhang, M.; Jang, H.; Li, Z.; Sacks, D.B.; Nussinov, R. B-Raf autoinhibition in the presence and absence of 14-3-3. Structure 2021. [Google Scholar] [CrossRef]
- Mysore, V.P.; Zhou, Z.W.; Ambrogio, C.; Li, L.; Kapp, J.N.; Lu, C.; Wang, Q.; Tucker, M.R.; Okoro, J.J.; Nagy-Davidescu, G.; et al. A structural model of a Ras-Raf signalosome. BioRxiv 2020. [Google Scholar] [CrossRef]
- Fuglebakk, E.; Reuter, N. A model for hydrophobic protrusions on peripheral membrane proteins. PLoS Comput. Biol. 2018, 14, e1006325. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ongusaha, P.P.; Miles, P.D.; Havstad, J.C.; Zhang, F.; So, W.V.; Kudlow, J.E.; Michell, R.H.; Olefsky, J.M.; Field, S.J.; et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008, 451, 964–969. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cookis, T.; Mattos, C. Crystal Structure Reveals the Full Ras–Raf Interface and Advances Mechanistic Understanding of Raf Activation. Biomolecules 2021, 11, 996. https://doi.org/10.3390/biom11070996
Cookis T, Mattos C. Crystal Structure Reveals the Full Ras–Raf Interface and Advances Mechanistic Understanding of Raf Activation. Biomolecules. 2021; 11(7):996. https://doi.org/10.3390/biom11070996
Chicago/Turabian StyleCookis, Trinity, and Carla Mattos. 2021. "Crystal Structure Reveals the Full Ras–Raf Interface and Advances Mechanistic Understanding of Raf Activation" Biomolecules 11, no. 7: 996. https://doi.org/10.3390/biom11070996
APA StyleCookis, T., & Mattos, C. (2021). Crystal Structure Reveals the Full Ras–Raf Interface and Advances Mechanistic Understanding of Raf Activation. Biomolecules, 11(7), 996. https://doi.org/10.3390/biom11070996