
Integrative Analysis of Multi-Omics and Genetic Approaches—A New Level in Atherosclerotic Cardiovascular Risk Prediction

 ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Need of in Depth Cardiovascular Risk Prevention
2. How to Correctly Identify High ACVD Risk Patients? Lessons from Genetics
3. Multi-Omics Tools for Cardiovascular Risk Prediction Tools: Transcriptomic and Epigenetic Markers
4. Multi-Omics Tools for Cardiovascular Risk Prediction Tools: Proteomics
5. Cardiovascular Risk Prediction Tools: The Opportunity to Use Artificial Intelligence/Machine Learning Approaches
6. Cardiovascular Risk Prediction Tools: A Joint Research Project
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACS | Acute Coronary Syndrome |
| ACVD | Atherosclerotic Cardiovascular Disease |
| AI | Artificial Intelligence |
| ANGPLT4 | Angiopoietin-Like 4 |
| APOB | Apolipoprotein B |
| APOE | Apolipoprotein E |
| CAD | Coronary Artery Disease |
| CAG | Coronary Angiography |
| CHD | Coronary Heart Disease |
| CRP | C-Reactive Protein |
| DLCN | Dutch Lipid Clinic Network |
| eQTL | Expression quantitative trait loci |
| ECG | Electrocardiogram |
| FH | Familial Hypercholesterolemia |
| GWAS | Genome-Wide Association Studies |
| HDL-C | High-Density Lipoprotein cholesterol content |
| LDLR | Low-Density Lipoprotein Receptor |
| LDL-C | Low-Density Lipoprotein cholesterol content |
| LDLRAP1 | Low Density Lipoprotein Receptor Adaptor Protein 1 |
| MI | Myocardial Infarction |
| ML | Machine Learning |
| MR | Mendelian Randomization |
| NGS | Next-Generation Sequencing |
| PCSK9 | Proprotein Convertase Subtilisin/Kexin type 9 |
| PhWAS | Phenome-wide association study |
| SNPs | Single Nucleotides Polymorphisms |
| STEMI | ST-elevation myocardial infarction (STEMI) |
| TWAS | Transcriptome Wide Association Study |
References
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Yusuf, S.; Joseph, P.; Rangarajan, S.; Islam, S.; Mente, A.; Hystad, P.; Brauer, M.; Kutty, V.R.; Gupta, R.; Wielgosz, A.; et al. Modifiable risk factors, cardiovascular disease, and mortality in 155722 individuals from 21 high-income, middle-income, and low-income countries (PURE): A prospective cohort study. Lancet 2019, 395, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, R.; Zekavat, S.M.; Uddin, M.; Pirruccello, J.; Niroula, A.; Gibson, C.; Griffin, G.K.; Libby, P.; Ebert, B.L.; Bick, A.; et al. Association of Diet Quality With Prevalence of Clonal Hematopoiesis and Adverse Cardiovascular Events. JAMA Cardiol. 2021, 6, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Baragetti, A.; Severgnini, M.; Olmastroni, E.; Dioguardi, C.; Mattavelli, E.; Angius, A.; Rotta, L.; Cibella, J.; Caredda, G.; Consolandi, C.; et al. Gut Microbiota Functional Dysbiosis Relates to Individual Diet in Subclinical Carotid Atherosclerosis. Nutrients 2021, 13, 304. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [Green Version]
- Berry, S.E.; Valdes, A.M.; Drew, D.A.; Asnicar, F.; Mazidi, M.; Wolf, J.; Capdevila, J.; Hadjigeorgiou, G.; Davies, R.; Al Khatib, H.; et al. Human postprandial responses to food and potential for precision nutrition. Nat. Med. 2020, 26, 964–973. [Google Scholar] [CrossRef]
- Joshi, A.; Rienks, M.; Theofilatos, K.; Mayr, M. Systems biology in cardiovascular disease: A multiomics approach. Nat. Rev. Cardiol. 2021, 18, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Borén, J.; Ginsberg, H.N.; Arca, M.; Averna, M.; Binder, C.J.; Calabresi, L.; Chapman, M.J.; Cuchel, M.; von Eckardstein, A.; et al. Rare dyslipidaemias, from phenotype to genotype to management: A European Atherosclerosis Society task force consensus statement. Lancet Diabetes Endocrinol. 2020, 8, 50–67. [Google Scholar] [CrossRef] [Green Version]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478-90a. [Google Scholar] [CrossRef] [Green Version]
- Austin, M.A.; Hutter, C.M.; Zimmern, R.L.; Humphries, S.E. Genetic causes of monogenic heterozygous familial hypercholesterolemia: A HuGE prevalence review. Am. J. Epidemiol. 2004, 160, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Garlaschelli, K.; Arca, M.; Averna, M.; Bertolini, S.; Calandra, S.; Tarugi, P.; Catapano, A.L.; Pellegatta, F.; Angelico, F.; et al. Spectrum of mutations in Italian patients with familial hypercholesterolemia: New results from the LIPIGEN study. Atheroscler. Suppl. 2017, 29, 17–24. [Google Scholar] [CrossRef]
- Gazzotti, M.; Casula, M.; Olmastroni, E.; Averna, M.; Arca, M.; Catapano, A.L. How registers could enhance knowledge and characterization of genetic dyslipidaemias: The experience of the LIPIGEN in Italy and of other networks for familial hypercholesterolemia. Atheroscler. Suppl. 2020, 42, e35–e40. [Google Scholar] [CrossRef]
- Musunuru, K.; Kathiresan, S. Genetics of Common, Complex Coronary Artery Disease. Cell 2019, 177, 132–145. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, M.; Dlouha, D.; Todorovova, V.; Stefler, D.; Hubacek, J. Genetics of Cardiovascular Disease: How Far Are We from Personalized CVD Risk Prediction and Management? Int. J. Mol. Sci. 2021, 22, 4182. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A. How to use Mendelian randomization to anticipate the results of randomized trials. Eur. Heart J. 2018, 39, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Bhatt, D.L.; Catapano, A.L.; Packard, C.J.; Graham, I.; Kaptoge, S.; Ference, T.B.; Guo, Q.; Laufs, U.; Ruff, C.T.; et al. Association of Genetic Variants Related to Combined Exposure to Lower Low-Density Lipoproteins and Lower Systolic Blood Pressure With Lifetime Risk of Cardiovascular Disease. JAMA 2019, 322, 1381–1391. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.D.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef] [Green Version]
- Mehta, N.N. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Circ. Cardiovasc. Genet. 2011, 4, 327–329. [Google Scholar] [CrossRef]
- Peden, J.F.; Hopewell, J.C.; Saleheen, D.; Chambers, J.C.; Hager, J.; Soranzo, N.; Collins, R.; Danesh, J.; Elliott, P.; Farrall, M.; et al. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat. Genet. 2011, 43, 339–346. [Google Scholar] [CrossRef]
- van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef]
- Verweij, N.; Eppinga, R.N.; Hagemeijer, Y.; van der Harst, P. Identification of 15 novel risk loci for coronary artery disease and genetic risk of recurrent events, atrial fibrillation and heart failure. Sci. Rep. 2017, 7, 2761. [Google Scholar] [CrossRef] [PubMed]
- Brænne, I.; Civelek, M.; Vilne, B.; Di Narzo, A.; Johnson, A.D.; Zhao, Y.; Reiz, B.; Codoni, V.; Webb, T.R.; Asl, H.F.; et al. Prediction of Causal Candidate Genes in Coronary Artery Disease Loci. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2207–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samani, N.J.; Erdmann, J.; Hall, A.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.S.; Wichmann, H.-E.; et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emilsson, V.; Thorleifsson, G.; Zhang, B.; Leonardson, A.S.; Zink, F.; Zhu, J.; Carlson, S.; Helgason, A.; Walters, G.B.; Gunnarsdottir, S.; et al. Genetics of gene expression and its effect on disease. Nature 2008, 452, 423–428. [Google Scholar] [CrossRef]
- Klarin, D.; Damrauer, S.M.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; DuVall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Inouye, M.; Silander, K.; Hämäläinen, E.; Salomaa, V.; Harald, K.; Jousilahti, P.; Mannisto, S.; Eriksson, J.G.; Saarela, J.; Ripatti, S.; et al. An immune response network associated with blood lipid levels. PLoS Genet. 2010, 6, e1001113. [Google Scholar] [CrossRef] [Green Version]
- Rockman, M.V.; Kruglyak, L. Genetics of global gene expression. Nat. Rev. Genet. 2006, 7, 862–872. [Google Scholar] [CrossRef]
- Alieva, A.S.; Olmastroni, E.; Reutova, O.V.; Rotar, O.P.; Konradi, A.O.; Shlyakhto, E.V.; Baragetti, A.; Grigore, L.; Pellegatta, F.; Casula, M.; et al. Prevalence and relationship between metabolic syndrome and risk of cardiovascular disease: Evidence from two population-based studies. Atheroscler. Suppl. 2020, 42, e41–e48. [Google Scholar] [CrossRef]
- Boardman-Pretty, F.; Smith, A.J.P.; Cooper, J.; Palmen, J.; Folkersen, L.; Hamsten, A.; Catapano, A.L.; Melander, O.; Price, J.F.; Kumari, M.; et al. Functional Analysis of a Carotid Intima-Media Thickness Locus Implicates BCAR1 and Suggests a Causal Variant. Circ. Cardiovasc. Genet. 2015, 8, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.S.; Krishnan, A.; Wong, A.; Ricciotti, E.; Zelaya, R.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.; Zaslavsky, E.; Sealfon, S.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santolini, M.; Romay, M.C.; Yukhtman, C.L.; Rau, C.D.; Ren, S.; Saucerman, J.J.; Wang, J.J.; Weiss, J.N.; Wang, Y.; Lusis, A.J.; et al. A personalized, multiomics approach identifies genes involved in cardiac hypertrophy and heart failure. NPJ Syst. Biol. Appl. 2018, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Winkels, H.; Cochain, C.; Williams, J.W.; Wolf, D.; Soehnlein, O.; Robbins, C.S.; Monaco, C.; Park, I.; McNamara, C.A.; et al. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ. Res. 2020, 127, 402–426. [Google Scholar] [CrossRef]
- Veltman, D.; Wu, M.; Pokreisz, P.; Claus, P.; Gillijns, H.; Caluwé, E.; Vanhaverbeke, M.; Gsell, W.; Himmelreich, U.; Sinnaeve, P.R.; et al. Clec4e-Receptor Signaling in Myocardial Repair After Ischemia-Reperfusion Injury. JACC Basic Transl. Sci. 2021, 6, 631–646. [Google Scholar] [CrossRef]
- Asl, H.F.; Talukdar, H.A.; Kindt, A.S.; Jain, R.K.; Ermel, R.; Ruusalepp, A.; Nguyen, K.-D.H.; Dobrin, R.; Reilly, D.F.; Schunkert, H.; et al. Expression quantitative trait Loci acting across multiple tissues are enriched in inherited risk for coronary artery disease. Circ. Cardiovasc. Genet. 2015, 8, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, H.A.; Asl, H.F.; Jain, R.K.; Ermel, R.; Ruusalepp, A.; Franzén, O.; Kidd, B.A.; Readhead, B.; Giannarelli, C.; Kovacic, J.C.; et al. Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst. 2016, 2, 196–208. [Google Scholar] [CrossRef] [Green Version]
- Abstract 20541: Multi-Omics Modeling of Carotid Atherosclerotic Plaques Reveals Molecular Networks and Regulators of Stroke | Circulation. Available online: https://www.ahajournals.org/doi/10.1161/circ.136.suppl_1.20541 (accessed on 26 September 2021).
- Georgakis, M.K.; van der Laan, S.W.; Asare, Y.; Mekke, J.M.; Haitjema, S.; Schoneveld, A.H.; de Jager, S.C.; Nurmohamed, N.S.; Kroon, J.; Stroes, E.S.; et al. Monocyte-Chemoattractant Protein-1 Levels in Human Atherosclerotic Lesions Associate With Plaque Vulnerability. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- Jarinova, O.; Stewart, A.F.; Roberts, R.; Wells, G.; Lau, P.; Naing, T.; Buerki, C.; McLean, B.W.; Cook, R.C.; Parker, J.S.; et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1671–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holdt, L.M.; Stahringer, A.; Sass, K.; Pichler, G.; Kulak, N.A.; Wilfert, W.; Kohlmaier, A.; Herbst, A.; Northoff, B.H.; Nicolaou, A.; et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat. Commun. 2016, 7, 12429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Shen, G.-Q.; Wang, X.; Wang, F.; Archacki, S.; Li, Y.; Yu, G.; Chakrabarti, S.; Chen, Q.; Wang, Q.K. Long noncoding RNA ANRIL regulates endothelial cell activities associated with coronary artery disease by up-regulating CLIP1, EZR, and LYVE1 genes. J. Biol. Chem. 2019, 294, 3881–3898. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis Is an Epigenetic Disease. Trends Endocrinol. Metab. 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Wang, P.; Shen, C.; Diao, L.; Yang, Z.; Fan, F.; Wang, C.; Liu, X.; Sun, X.; Dong, Z.; Zhu, H.; et al. Aberrant hypermethylation of aldehyde dehydrogenase 2 promoter upstream sequence in rats with experimental myocardial infarction. Biomed. Res. Int. 2015, 2015, 503692. [Google Scholar] [CrossRef] [PubMed]
- Ek, W.E.; Hedman, Å.K.; Enroth, S.; Morris, A.P.; Lindgren, C.M.; Mahajan, A.; Gustafsson, S.; Gyllensten, U.; Lind, L.; Johansson, Å. Genome-wide DNA methylation study identifies genes associated with the cardiovascular biomarker GDF-15. Hum. Mol. Genet. 2016, 25, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
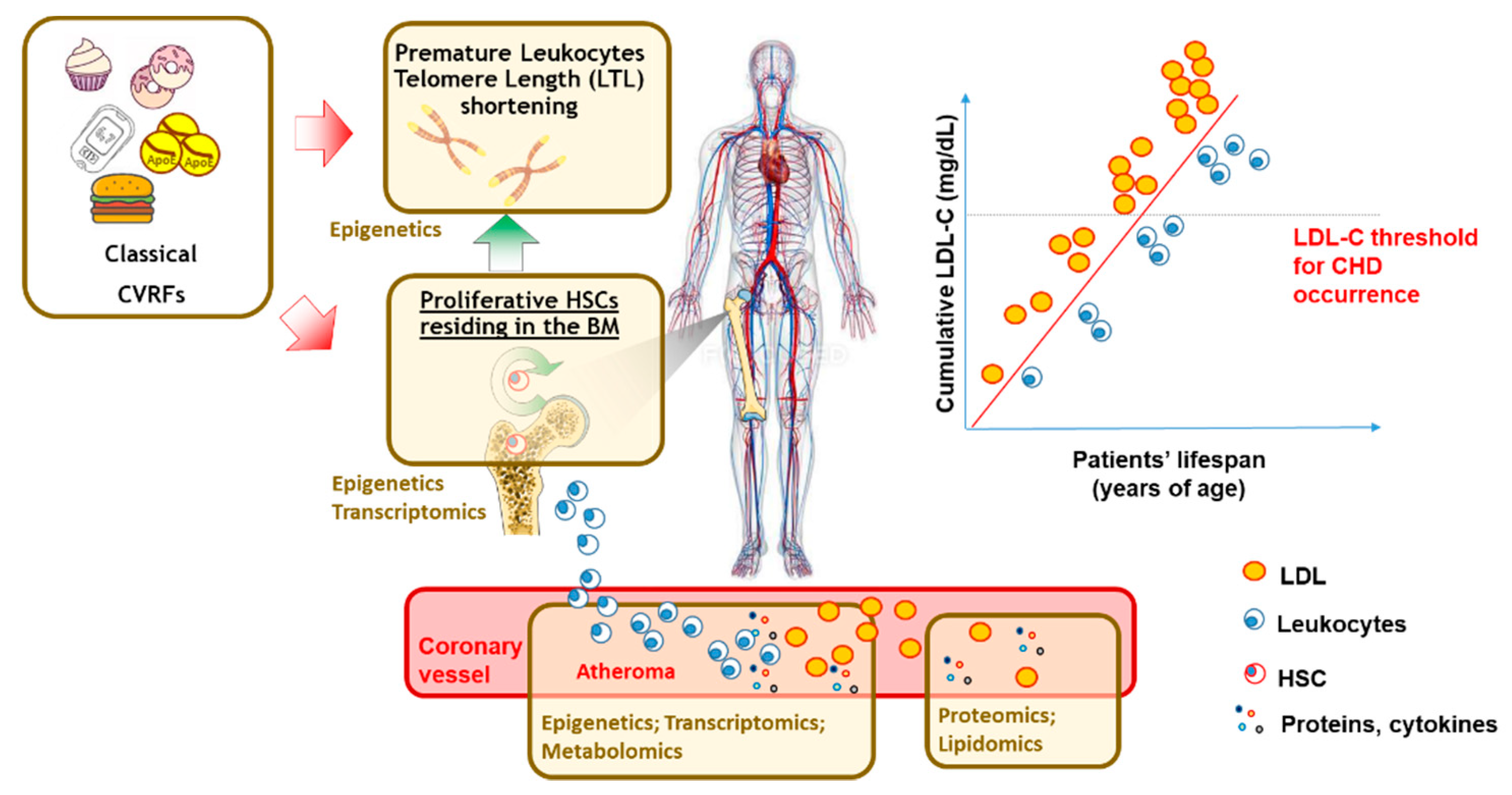
- Baragetti, A.; Bonacina, F.; Catapano, A.L.; Norata, G.D. Effect of Lipids and Lipoproteins on Hematopoietic Cell Metabolism and Commitment in Atherosclerosis. Immunometabolism 2021, 3, e210014. [Google Scholar] [CrossRef]
- Huzen, J.; Wong, L.S.M.; van Veldhuisen, D.J.; Samani, N.J.; Zwinderman, A.H.; Codd, V.; Cawthon, R.M.; Benus, G.F.J.D.; van der Horst, I.C.C.; Navis, G.; et al. Telomere length loss due to smoking and metabolic traits. J. Intern. Med. 2014, 275, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Mazidi, M.; Rezaie, P.; Covic, A.; Malyszko, J.; Rysz, J.; Kengne, A.P.; Banach, M. Telomere attrition, kidney function, and prevalent chronic kidney disease in the United States. Oncotarget 2017, 8, 80175–80181. [Google Scholar] [CrossRef] [Green Version]
- Codd, V.; Nelson, C.; Albrecht, E.; Mangino, M.; Deelen, J.; Buxton, J.L.; Hottenga, J.J.; Fischer, K.; Esko, T.; Surakka, I.; et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet. 2013, 45, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Baragetti, A.; Palmen, J.; Garlaschelli, K.; Grigore, L.; Pellegatta, F.; Tragni, E.; Catapano, A.L.; Humphries, S.E.; Norata, G.D.; Talmud, P.J. Telomere shortening over 6 years is associated with increased subclinical carotid vascular damage and worse cardiovascular prognosis in the general population. J. Intern. Med. 2015, 277, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Brümmendorf, T.H.; Balabanov, S. Telomere length dynamics in normal hematopoiesis and in disease states characterized by increased stem cell turnover. Leukemia 2006, 20, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
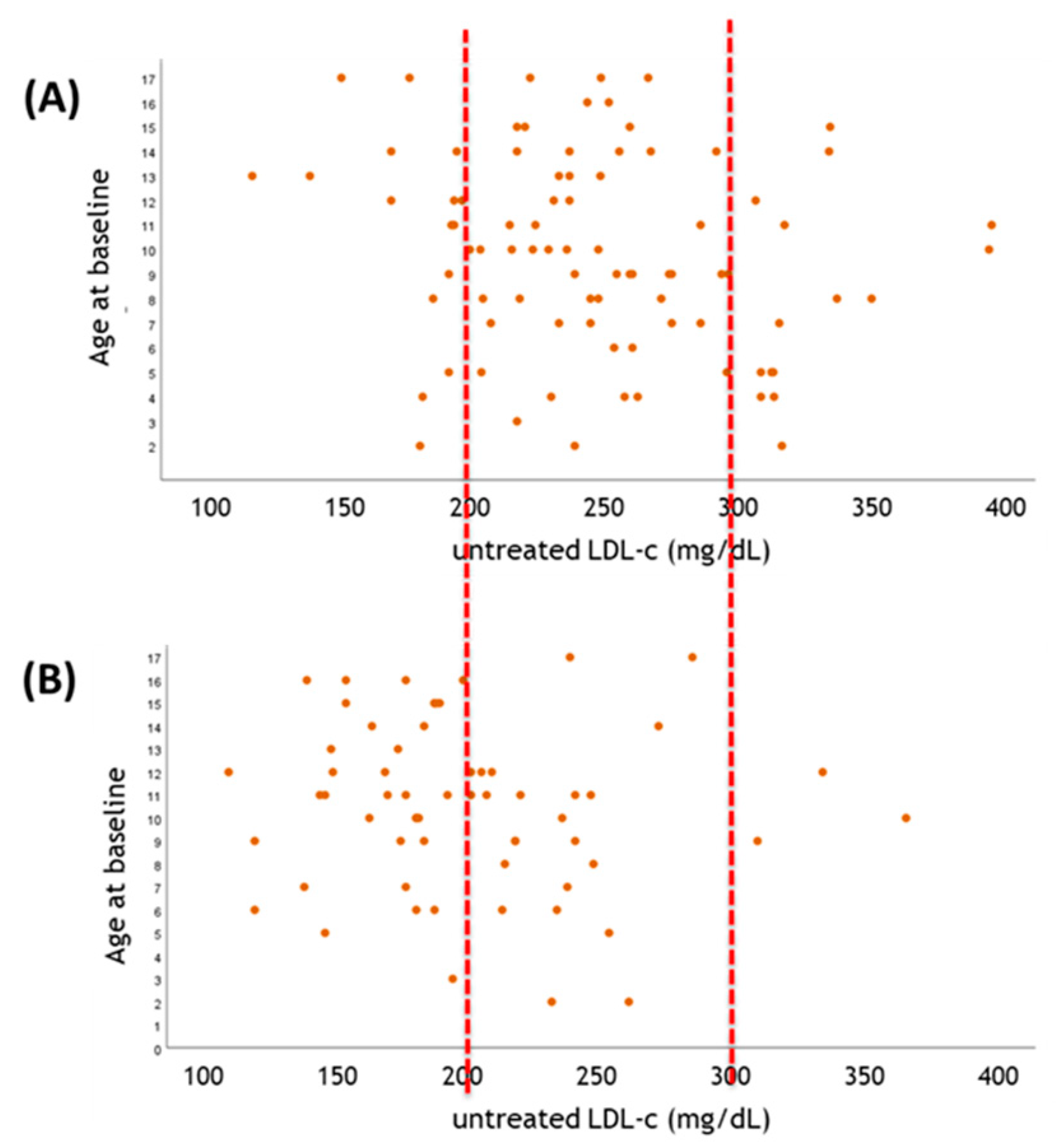
- Baragetti, A.; Bonacina, F.; Da Dalt, L.; Moregola, A.; Zampoleri, V.; Pellegatta, F.; Grigore, L.; Pirillo, A.; Spina, R.; Cefalù, A.B.; et al. Genetically determined hypercholesterolaemia results into premature leucocyte telomere length shortening and reduced haematopoietic precursors. Eur. J. Prev. Cardiol. 2020, zwaa115. [Google Scholar] [CrossRef]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Thum, T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 661–674. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Mayr, M.; Gomes, A.V.; Delles, C.; Arrell, D.K.; Murphy, A.M.; Lange, R.A.; Costello, C.E.; Jin, Y.-F.; Laskowitz, D.T.; et al. Transformative Impact of Proteomics on Cardiovascular Health and Disease: A Scientific Statement From the American Heart Association. Circulation 2015, 132, 852–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Canela, M.; Hruby, A.; Clish, C.; Liang, L.; Martínez-González, M.A.; Hu, F.B. Comprehensive Metabolomic Profiling and Incident Cardiovascular Disease: A Systematic Review. J. Am. Heart Assoc. 2017, 6, e005705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarrah, R.W.; Crown, S.B.; Zhang, G.-F.; Shah, S.H.; Newgard, C.B. Cardiovascular Metabolomics. Circ. Res. 2018, 122, 1238–1258. [Google Scholar] [CrossRef]
- Stegemann, C.; Pechlaner, R.; Willeit, P.; Langley, S.R.; Mangino, M.; Mayr, U.; Menni, C.; Moayyeri, A.; Santer, P.; Rungger, G.; et al. Lipidomics profiling and risk of cardiovascular disease in the prospective population-based Bruneck study. Circulation 2014, 129, 1821–1831. [Google Scholar] [CrossRef] [Green Version]
- Laaksonen, R.; Ekroos, K.; Sysi-Aho, M.; Hilvo, M.; Vihervaara, T.; Kauhanen, D.; Suoniemi, M.; Hurme, R.; März, W.; Scharnagl, H.; et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur. Heart J. 2016, 37, 1967–1976. [Google Scholar] [CrossRef]
- Hoogeveen, R.M.; Pereira, J.P.B.; Nurmohamed, N.S.; Zampoleri, V.; Bom, M.J.; Baragetti, A.; Boekholdt, S.M.; Knaapen, P.; Khaw, K.-T.; Wareham, N.J.; et al. Improved cardiovascular risk prediction using targeted plasma proteomics in primary prevention. Eur. Heart J. 2020, 41, 3998–4007. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Opstal, T.S.; Hoogeveen, R.M.; Fiolet, A.T.; Silvis, M.J.; The, S.H.; Bax, W.A.; De Kleijn, D.P.; Mosterd, A.; Stroes, E.S.; Cornel, J.H. Colchicine Attenuates Inflammation Beyond the Inflammasome in Chronic Coronary Artery Disease: A LoDoCo2 Proteomic Substudy. Circulation 2020, 142, 1996–1998. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.H.; Newgard, C.B. Integrated metabolomics and genomics: Systems approaches to biomarkers and mechanisms of cardiovascular disease. Circ. Cardiovasc. Genet. 2015, 8, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Tahir, U.A.; Gerszten, R.E. Omics and Cardiometabolic Disease Risk Prediction. Annu. Rev. Med. 2020, 71, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Hicks, A.A.; Pramstaller, P.P.; Johansson, Å.; Vitart, V.; Rudan, I.; Ugocsai, P.; Aulchenko, Y.; Franklin, C.S.; Liebisch, G.; Erdmann, J.; et al. Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genet. 2009, 5, e1000672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Zhou, F.; Ren, J.; Li, X.; Jiang, Y.; Ma, S. A Selective Review of Multi-Level Omics Data Integration Using Variable Selection. High.-Throughput 2019, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Ma, S. A selective review of robust variable selection with applications in bioinformatics. Brief. Bioinform. 2015, 16, 873–883. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usova, E.I.; Alieva, A.S.; Yakovlev, A.N.; Alieva, M.S.; Prokhorikhin, A.A.; Konradi, A.O.; Shlyakhto, E.V.; Magni, P.; Catapano, A.L.; Baragetti, A. Integrative Analysis of Multi-Omics and Genetic Approaches—A New Level in Atherosclerotic Cardiovascular Risk Prediction. Biomolecules 2021, 11, 1597. https://doi.org/10.3390/biom11111597
Usova EI, Alieva AS, Yakovlev AN, Alieva MS, Prokhorikhin AA, Konradi AO, Shlyakhto EV, Magni P, Catapano AL, Baragetti A. Integrative Analysis of Multi-Omics and Genetic Approaches—A New Level in Atherosclerotic Cardiovascular Risk Prediction. Biomolecules. 2021; 11(11):1597. https://doi.org/10.3390/biom11111597
Chicago/Turabian StyleUsova, EIena I., Asiiat S. Alieva, Alexey N. Yakovlev, Madina S. Alieva, Alexey A. Prokhorikhin, Alexandra O. Konradi, Evgeny V. Shlyakhto, Paolo Magni, Alberico L. Catapano, and Andrea Baragetti. 2021. "Integrative Analysis of Multi-Omics and Genetic Approaches—A New Level in Atherosclerotic Cardiovascular Risk Prediction" Biomolecules 11, no. 11: 1597. https://doi.org/10.3390/biom11111597
APA StyleUsova, E. I., Alieva, A. S., Yakovlev, A. N., Alieva, M. S., Prokhorikhin, A. A., Konradi, A. O., Shlyakhto, E. V., Magni, P., Catapano, A. L., & Baragetti, A. (2021). Integrative Analysis of Multi-Omics and Genetic Approaches—A New Level in Atherosclerotic Cardiovascular Risk Prediction. Biomolecules, 11(11), 1597. https://doi.org/10.3390/biom11111597