
Lipoprotein(a)—The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation


Abstract
:1. Introduction
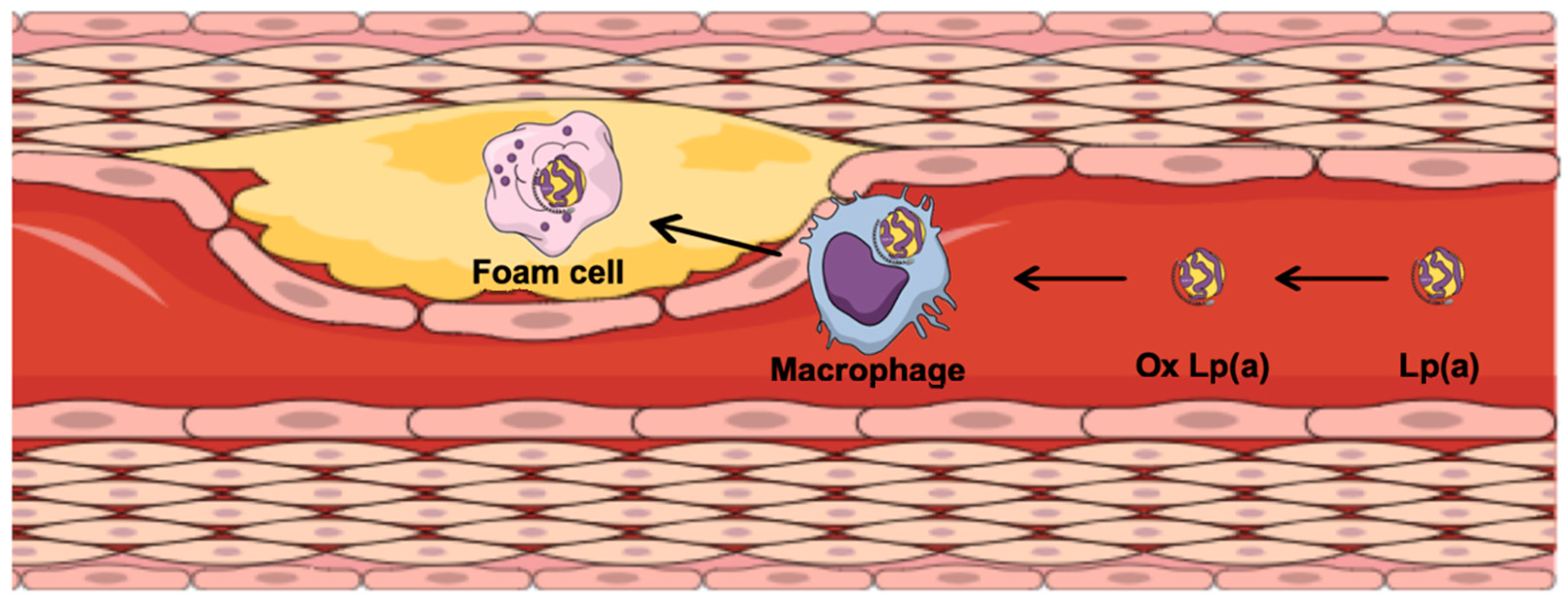
2. Lipoprotein(a) and Atherosclerosis
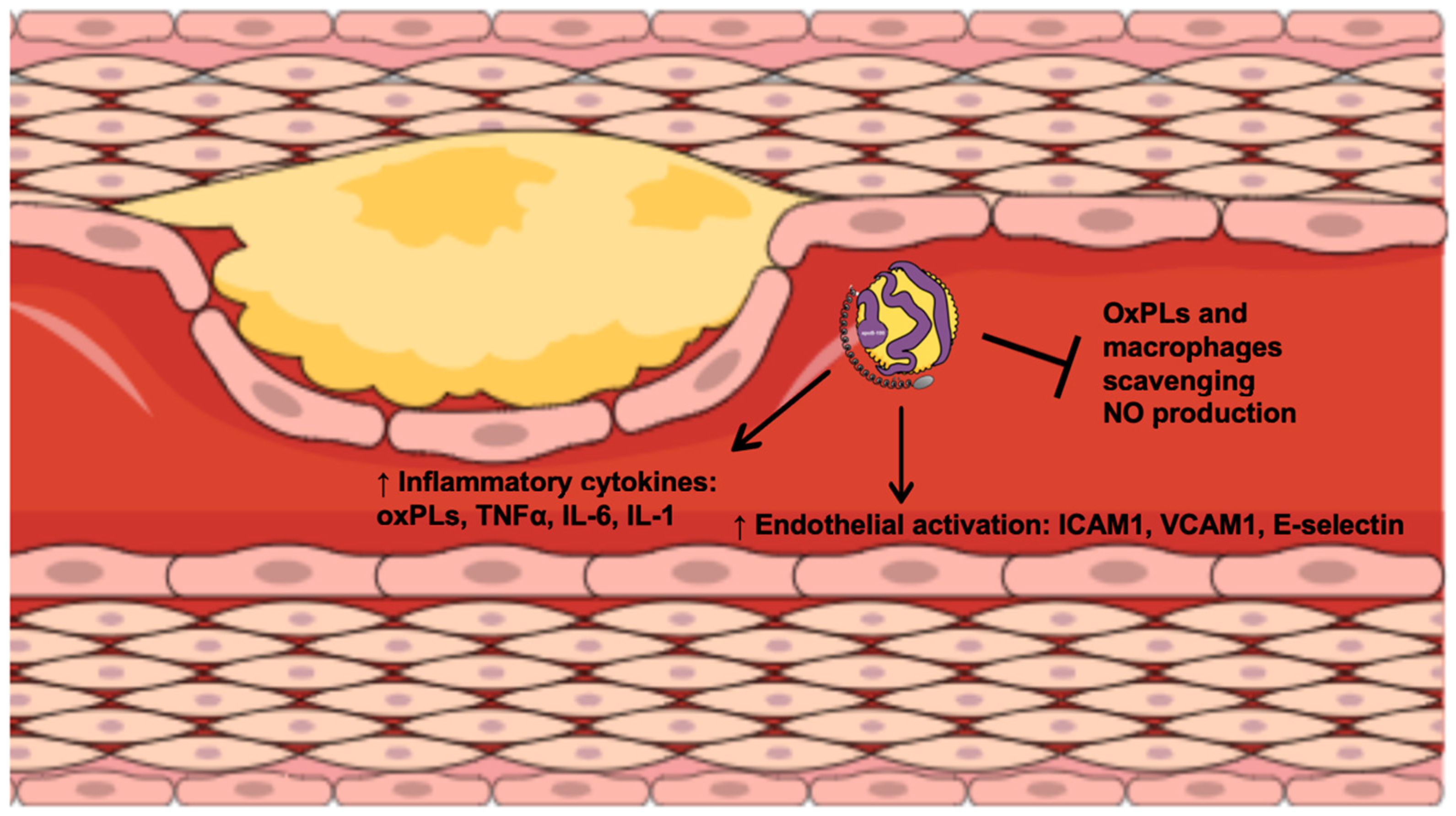
3. Lipoprotein(a) and Inflammation
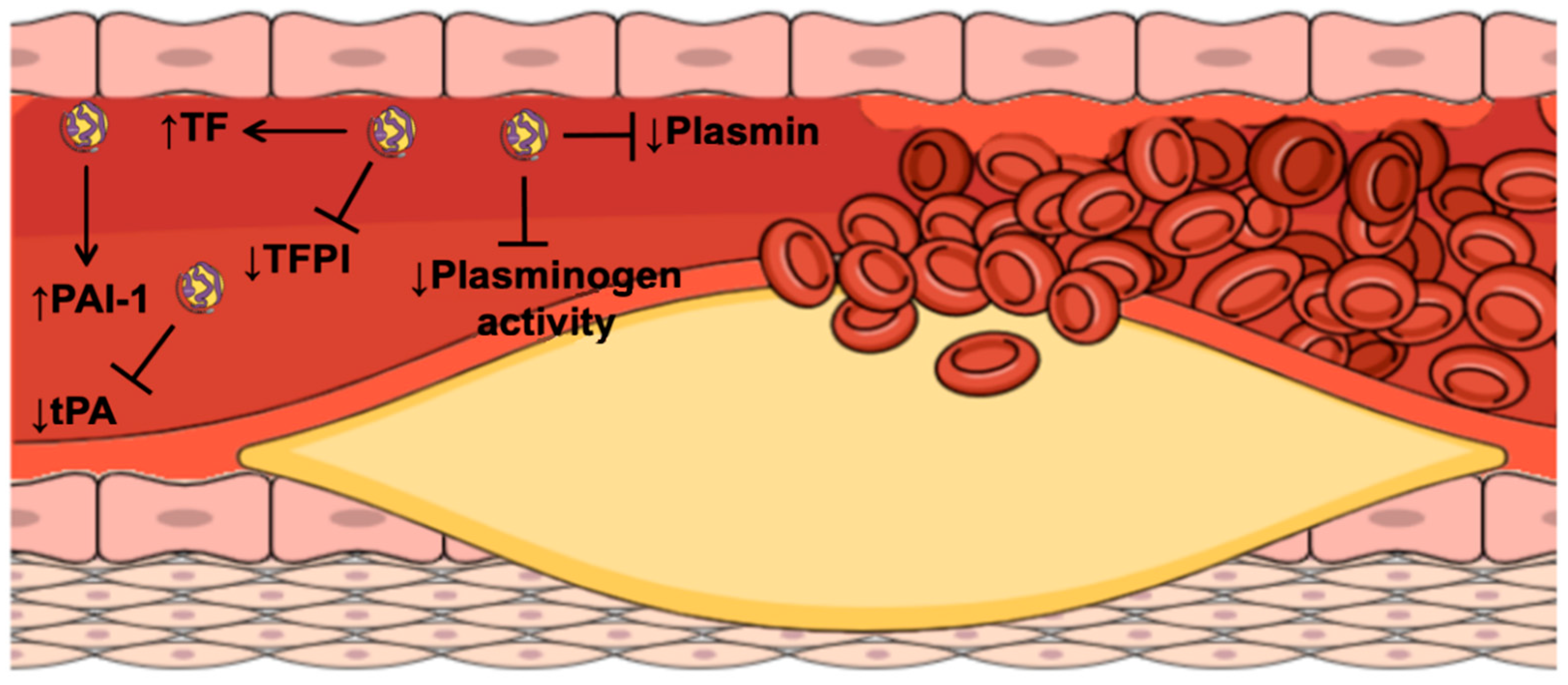
4. Lipoprotein(a) and Atherothrombosis
5. Prognostic Value of Lipoprotein(a) in Combination with Thrombotic and Inflammation Parameters
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cybulska, B.; Kłosiewicz-Latoszek, L.; Penson, P.E.; Banach, M. What do we know about the role of lipoprotein(a) in atherogenesis 57 years after its discovery? Prog. Cardiovasc. Dis. 2020, 63, 219–227. [Google Scholar] [CrossRef]
- Loretto, P.; Brian, K. Dietary and genetic interactions in the regulation of plasma lipoprotein(a). Curr. Opin. Lipidol. 1999, 10, 35–40. [Google Scholar]
- Orsó, E.; Schmitz, G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin. Res. Cardiol. Suppl. 2017, 12, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Enas, E.A.; Varkey, B.; Dharmarajan, T.S.; Pare, G.; Bahl, V.K. Lipoprotein(a): An independent, genetic, and causal factor for cardiovascular disease and acute myocardial infarction. Indian Heart J. 2019, 71, 99–112. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Ballantyne, C.M. Residual cardiovascular risk at low LDL: Remnants, lipoprotein(a), and inflammation. Clin. Chem. 2021, 67, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Saleheen, D.; Haycock, P.C.; Rasheed, A.; Taleb, A.; Imran, A.; Abbas, S.; Majeed, F.; Akhtar, S.; Qamar, N.; Shah Zaman, K.; et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: A mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017, 5, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Rehberger Likozar, A.; Zavrtanik, M.; Šebeštjen, M. Lipoprotein(a) in atherosclerosis: From pathophysiology to clinical relevance and treatment options. Ann. Med. 2020, 52, 162–177. [Google Scholar] [CrossRef]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Steffen, B.T.; Budoff, M.; Post, W.S.; Thanassoulis, G.; Kestenbaum, B.; McConnell, J.P.; Warnick, R.; Guan, W.; Tsai, M.Y. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in Caucasian and Black individuals: The multi-ethnic study of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1003–1009. [Google Scholar] [CrossRef] [Green Version]
- Rajamannan, N.M.; Evans, F.J.; Aikawa, E.; Grande-Allen, J.; Demer, L.L.; Heistad, D.D.; Simmons, C.A.; Masters, K.S.; Matheiu, P.; O’Brien, K.D.; et al. Calcific aortic valve disease: Not simply a degenerative process. Circulation 2011, 124, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Boren, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Bergmark, C.; Dewan, A.; Orsoni, A.; Merki, E.; Miller, E.R.; Shin, M.J.; Binder, C.J.; Hörkko, S.; Krauss, R.M.; Chapman, M.J.; et al. A novel function of lipoprotein(a) as a preferential carrier of oxidized phospholipids in human plasma. J. Lipid Res. 2008, 49, 2230–2239. [Google Scholar] [CrossRef] [Green Version]
- Kiechl, S.; Willeit, J. The mysteries of lipoprotein(a) and cardiovascular disease revisited. J. Am. Coll. Cardiol. 2010, 55, 2168–2170. [Google Scholar] [CrossRef] [Green Version]
- Nordestgaard, B.G.; Nielsen, L.B. Atherosclerosis and arterial influx of lipoproteins. Curr. Opin. Lipidol. 1994, 5, 252–257. [Google Scholar] [CrossRef]
- Tsimikas, S. A test in context: Lipoprotein(a): Diagnosis, prognosis, controversies, and emerging therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Tsimikas, S. Unmet needs in understanding lipoprotein(a) pathophysiology: NHLBI working group recommendations to reduce risk of cardiovascular disease and aortic stenosis. Physiol. Behav. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Deb, A.; Caplice, N.M. Lipoprotein(a): New insights into mechanisms of atherogenesis and thrombosis. Clin. Cardiol. 2004, 27, 258–264. [Google Scholar] [CrossRef]
- Labudovic, D.; Kostovska, I.; Tosheska Trajkovska, K.; Cekovska, S.; Brezovska Kavrakova, J.; Topuzovska, S. Lipoprotein(a)—Link between atherogenesis and thrombosis. Prague Med. Rep. 2019, 120, 39–51. [Google Scholar] [CrossRef]
- Syrovets, T.; Thllet, J.; Chapman, M.J.; Simmet, T. Lipoprotein(a) is a potent chemoattractant for human peripheral monocytes. Blood 1997, 90, 2027–2036. [Google Scholar] [CrossRef]
- Tsimikas, S.; Witztum, J.L. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr. Opin. Lipidol. 2008, 19, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; Danesh, J. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. The emerging risk factors collaboration. JAMA Cardiol. 2009, 302, 412–423. [Google Scholar] [CrossRef]
- Kotani, K.; Banach, M. Lipoprotein(a) and inhibitors of proprotein convertase subtilisin/kexin type 9. J. Thorac. Dis. 2017, 9, E78–E82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: The Copenhagen city heart study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Nave, A.H.; Lange, K.S.; Leonards, C.O.; Siegerink, B.; Doehner, W.; Landmesser, U.; Steinhagen-Thiessen, E.; Endres, M.; Ebinger, M. Lipoprotein(a) as a risk factor for ischemic stroke: A meta-analysis. Atherosclerosis 2015, 242, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Kiechl, S.; Kronenberg, F.; Witztum, J.L.; Santer, P.; Mayr, M.; Xu, Q.; Mayr, A.; Willeit, J.; Tsimikas, S. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): Prospective 15-year outcomes in the bruneck study. J. Am. Coll. Cardiol. 2014, 64, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J. Am. Coll. Cardiol. 2013, 61, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbeek, R.; Sandhu, M.S. Lipoprotein(a) improves cardiovascular risk prediction based on established risk algorithms. J. Am. Coll. Cardiol. 2017, 69, 1513–1515. [Google Scholar] [CrossRef]
- Cook, N.R.; Mora, S.; Ridker, P.M. Lipoprotein(a) and cardiovascular risk prediction among women. J. Am. Coll. Cardiol. 2018, 72, 287–296. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Pineda, A.L.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk insights from the FOURIER trial. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.Y.; Liu, J.; Jiang, H.X.; Hu, B.L.; Zhou, Y.; Olkkonen, V.M. Association between baseline lipoprotein(a) levels and restenosis after coronary stenting: Meta-analysis of nine cohort studies. Atherosclerosis 2013, 227, 360–366. [Google Scholar] [CrossRef]
- Khantalin, I.; Blanchard, V.; Viallet, N.; Lambert, G. Recurrent coronary syndromes in a patient with isolated very-high lipoprotein(a) and the prothrombin genetic variant rs1799963 (G20210A): A case report. Eur. Hear. J.-Case Rep. 2019, 3, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Daida, H.; Lee, Y.J.; Yokoi, H.; Kanoh, T.; Ishiwata, S.; Kato, K.; Nishikawa, H.; Takatsu, F.; Kato, H.; Kutsumi, Y.; et al. Prevention of restenosis after percutaneous transluminal coronary angioplasty by reducing lipoprotein(a) levels with low-density lipoprotein apheresis. Am. J. Cardiol. 1994, 73, 1037–1040. [Google Scholar] [CrossRef]
- Ozkan, U.; Ozcelik, F.; Yildiz, M.; Budak, M. Lipoprotein(a) gene polymorphism increases a risk factor for aortic valve calcification. J. Cardiovasc. Dev. Dis. 2019, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsenault, B.J.; Boekholdt, M.; Dubé, M.P.; Rhéaume, É.; Wareham, N.J.; Khaw, K.T.; Sandhu, M.S.; Tardif, J.C. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis a prospective mendelian randomization study and replication in a case-control cohort. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Rashedi, N.; Otto, C.M. Aortic stenosis: Changing disease concepts. J. Cardiovasc. Ultrasound. 2015, 23, 59–69. [Google Scholar] [CrossRef]
- Nanda, N.C. Genetic associations with valvular calcification and aortic stenosis. Cardiol. Rev. 2013, 29, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Langsted, A.; Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: A prospective cohort study. Lancet Diabetes Endocrinol. 2016, 4, 577–587. [Google Scholar] [CrossRef]
- Alonso, R.; Andres, E.; Mata, N.; Fuentes Jiménez, F.; Badimón, L.; López Miranda, J.; Padró, T.; Muñiz, O.; Díaz Díaz, J.L.; Mauri, M.; et al. Lipoprotein(a) levels in familial hypercholesterolemia: An important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J. Am. Coll. Cardiol. 2014, 63, 1982–1989. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Pirro, M.; Bianconi, V.; Paciullo, F.; Mannarino, M.R.; Bagaglia, F.; Sahebkar, A. Lipoprotein(a) and inflammation: A dangerous duet leading to endothelial loss of integrity. Pharmacol. Res. 2017, 119, 178–187. [Google Scholar] [CrossRef]
- Leibundgut, G.; Scipione, C.; Yin, H.; Schneider, M.; Boffa, M.B.; Green, S.; Yang, X.; Dennis, E.; Witztum, J.L.; Koschinsky, M.L.; et al. Determinants of binding of oxidized phospholipids on apolipoprotein(a) and lipoprotein(a). J. Lipid Res. 2013, 54, 2815–2830. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Tsironis, L.D.; Tselepis, A.D. New insights into the role of lipoprotein(a)-associated lipoprotein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2094–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, P.; Tafelmeier, M.; Chittka, D.; Choi, S.H.; Zhang, L.; Byun, Y.S.; Almazan, F.; Yang, X.; Iqbal, N.; Chowdhury, P.; et al. MCP-1 binds to oxidized LDL and is carried by lipoprotein(a) in human plasma. J. Lipid Res. 2013, 54, 1877–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbah, N.; Jaisson, S.; Garnotel, R.; Anglés-Cano, E.; Gillery, P. Small size apolipoprotein(a) isoforms enhance inflammatory and proteolytic potential of collagen-primed monocytes. Lipids Health Dis. 2019, 18, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, K.; Luke, M.M.; Koschinsky, M.L.; Miller, E.R.; Pullinger, C.R.; Witztum, J.L.; Kane, J.P.; Tsimikas, S. The I4399M variant of apolipoprotein(a) is associated with increased oxidized phospholipids on apolipoprotein B-100 particles. Atherosclerosis 2010, 209, 498–503. [Google Scholar] [CrossRef]
- Puri, R.; Nissen, S.E.; Arsenault, B.J.; St John, J.; Riesmeyer, J.S.; Ruotolo, G.; McErlean, E.; Menon, V.; Cho, L.; Wolski, K.; et al. Effect of C-reactive protein on lipoprotein(a)-associated cardiovascular risk in optimally treated patients with high-risk vascular disease: A prespecified secondary analysis of the ACCELERATE trial. JAMA Cardiol. 2020, 5, 1136–1143. [Google Scholar] [CrossRef]
- Topçiu-Shufta, V.; Haxhibeqiri, V.; Begolli, L.; Baruti Gafurri, Z.; Veseli, S.; Haxhibeqiri, S.; Miftari, R.; Kurti, L.; Avdiu, D. Correlation of inflammation and lipoprotein(a) with hypercoagulability in hemodialysis patients. Med. Arch. 2015, 69, 232–235. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Thompson, L.; Giles, J.T.; Bathon, J.M.; Salmon, J.E.; Beaulieu, A.D.; Codding, C.E.; Carlson, T.H.; Delles, C.; Lee, J.S.; et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann. Rheum. Dis. 2015, 74, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Dursunoǧlu, D.; Evrengül, H.; Polat, B.; Tanriverdi, H.; Çobankara, V.; Kaftan, A.; Kiliç, M. Lp(a) lipoprotein and lipids in patients with rheumatoid arthritis: Serum levels and relationship to inflammation. Rheumatol. Int. 2005, 25, 241–245. [Google Scholar] [CrossRef]
- Wang, J.; Hu, B.; Kong, L.; Cai, H.; Zhang, C. Native, oxidized lipoprotein(a) and lipoprotein(a) immune complex in patients with active and inactive rheumatoid arthritis: Plasma concentrations and relationship to inflammation. Clin. Chim. Acta. 2008, 390, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, J.; Herrlinger, S.; Pruy, A.; Metzger, T.; Wanner, C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int. 1999, 55, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langsted, A.; Varbo, A.; Kamstrup, P.R.; Nordestgaard, B.G. Elevated lipoprotein(a) does not cause low-grade inflammation despite causal association with aortic valve stenosis and myocardial infarction: A study of 100 578 individuals from the general population. J. Clin. Endocrinol. Metab. 2015, 100, 2690–2699. [Google Scholar] [CrossRef] [Green Version]
- Sueishi, K.; Ichikawa, K.; Kato, K.; Nakagawa, K.; Chen, Y.X. Atherosclerosis: Coagulation and fibrinolysis. Semin. Thromb. Hemost. 1998, 24, 255–260. [Google Scholar] [CrossRef]
- Barre, D. The molecular nature and consequences of lipoprotein(a)’s association with platelets. Protein Pept. Lett. 2007, 14, 839–842. [Google Scholar] [CrossRef]
- Tsironis, L.D.; Mitsios, J.V.; Milionis, H.J.; Elisaf, M.; Tselepis, A.D. Effect of lipoprotein(a) on platelet activation induced by platelet-activating factor: Role of apolipoprotein(a) and endogenous PAF-acetylhydrolase. Cardiovasc. Res. 2004, 63, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, G.; Bacchetti, T.; Johnston, T.P.; Banach, M.; Pirro, M.; Sahebkar, A. Lipoprotein(a): A missing culprit in the management of athero-thrombosis? J. Cell. Physiol. 2018, 233, 2966–2981. [Google Scholar] [CrossRef]
- Boffa, M.B.; Koschinsky, M.L. Thematic review series: Lipoprotein(a): Coming of age at last: Lipoprotein(a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Di Nisio, M.; Ten Wolde, M.; Meijers, J.C.M.; Buller, H.R. Effects of high plasma lipoprotein(a) levels on tissue factor pathway inhibitor and the protein C pathway. J. Thromb. Haemost. 2005, 3, 2123–2125. [Google Scholar] [CrossRef] [PubMed]
- Bilgen, D.; Sönmez, H.; Ekmekçi, H.; Ulutin, T.; Oztürk, Z.; Kökoglu, E.; Bayram, C.; Soner, A.; Domaniç, N. The relationship of TFPI, Lp(a), and oxidized LDL antibody levels in patients with coronary artery disease. Clin. Biochem. 2005, 38, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Stulnig, T.M.; Morozzi, C.; Reindl-Schwaighofer, R.; Stefanutti, C. Looking at Lp(a) and related cardiovascular risk: From scientific evidence and clinical practice. Curr. Atheroscler. Rep. 2019, 21, 37. [Google Scholar] [CrossRef]
- Romagnuolo, R.; Marcovina, S.M.; Boffa, M.B.; Koschinsky, M.L. Inhibition of plasminogen activation by apo(a): Role of carboxyl-terminal lysines and identification of inhibitory domains in apo(a). J. Lipid Res. 2014, 55, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Plow, E.F.; Hoover-Plow, J. The functions of plasminogen in cardiovascular disease. Trends Cardiovasc. Med. 2004, 14, 180–186. [Google Scholar] [CrossRef]
- Ma, Q.; Ozel, A.B.; Ramdas, S.; McGee, B.; Khoriaty, R.; Siemieniak, D.; Li, H.D.; Guan, Y.; Brody, L.C.; Mills, J.L.; et al. Genetic variants in PLG, LPA, and SIGLEC 14 as well as smoking contribute to plasma plasminogen levels. Blood 2014, 124, 3155–3164. [Google Scholar] [CrossRef]
- Wang, H.; Hong, C.E.; Lewis, J.P.; Zhu, Y.; Wang, X.; Chu, X.; Backman, J.; Hu, Z.; Yang, P.; Still, C.D.; et al. Effect of two lipoprotein(a)-associated genetic variants on plasminogen levels and fibrinolysis. G3 Genes Genomes Genet. 2016, 6, 3525–3532. [Google Scholar] [CrossRef] [Green Version]
- Rowland, C.M.; Pullinger, C.R.; Luke, M.M.; Shiffman, D.; Green, L.; Movsesyan, I.; Devlin, J.J.; Malloy, M.J.; Kane, J.P.; Undas, A. Lipoprotein(a), LPA Ile4399Met, and fibrin clot properties. Thromb. Res. 2014, 133, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Scipione, C.A.; McAiney, J.T.; Simard, D.J.; Bazzi, Z.A.; Gemin, M.; Romagnuolo, R.; Macrae, F.L.; Ariëns, R.A.; Hegele, R.A.; Auld, J.; et al. Characterization of the I4399M variant of apolipoprotein(a): Implications for altered prothrombotic properties of lipoprotein(a). J. Thromb. Haemostasis. 2017, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tziomalos, K.; Athyros, V.G.; Wierzbicki, A.S.; Mikhailidis, D.P. Lipoprotein a: Where are we now? Curr. Opin. Cardiol. 2009, 24, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Šebeštjen, M.; Žegura, B.; Gužič-Salobir, B.; Keber, I. Fibrinolytic parameters and insulin resistance in young survivors of myocardial infarction with heterozygous familial hypercholesterolemia. Wien. Klin. Wochenschr. 2001, 113, 113–118. [Google Scholar] [PubMed]
- Etingin, O.R.; Hajjar, D.P.; Hajjar, K.A.; Harpel, P.C.; Nachman, R.L. Lipoprotein(a) regulates plasminogen activator inhibitor-1 expression in endothelial cells: A potential mechanism in thrombogenesis. J. Biol. Chem. 1991, 266, 2459–2465. [Google Scholar] [CrossRef]
- Shindo, J.; Ishibashi, T.; Kijima, M.; Nakazato, K.; Nagata, K.; Yokoyama, K.; Hirosaka, A.; Sato, E.; Kunii, H.; Yamaguchi, H.; et al. Increased plasminogen activator inhibitor-1 and apolipoprotein(a) in coronary atherectomy specimens in acute coronary syndromes. Coron. Artery Dis. 2001, 12, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between plasminogen activator inhibitor-1 and cardiovascular events: A systematic review and meta-analysis. Thromb. J. 2018, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Moss, A.J.; Goldstein, R.E.; Marder, V.J.; Sparks, C.E.; Oakes, D.; Greenberg, H.; Weiss, H.J.; Zareba, W.; Brown, M.W.; Liang, C.S.; et al. Thrombogenic factors and recurrent coronary events. Circulation 1999, 99, 2517–2522. [Google Scholar] [CrossRef] [Green Version]
- Bogaty, P.; Poirier, P.; Simard, S.; Boyer, L.; Solymoss, S.; Dagenais, G.R. Biological profiles in subjects with recurrent acute coronary events compared with subjects with long-standing stable angina. Circulation 2001, 103, 3062–3068. [Google Scholar] [CrossRef]
- Pineda, J.; Marín, F.; Marco, P.; Roldán, V.; Valencia, J.; Ruiz Nodar, J.M.; Sogorb, F.; Lip, G.Y.H. Premature coronary artery disease in young (age < 45) subjects: Interactions of lipid profile, thrombophilic and haemostatic markers. Int. J. Cardiol. 2009, 136, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Park, D.; Lesty, C.; Soria, J.; Soria, C.; Montalescot, G.; Weisel, J.W. Influence of fibrin network conformation and fibrin diameter on fibrinolysis speed: Dynamic and structural approaches by confocal microscopy. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1354–1361. [Google Scholar] [CrossRef] [Green Version]
- Hervio, L.; Chapman, M.J.; Thillet, J.; Loyau, S.; Angles-Cano, E. Does apolipoprotein(a) heterogeneity influence lipoprotein(a) effects on fibrinolysis? Blood 1993, 82, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Galvano, F.; Malaguarnera, M.; Vacante, M.; Motta, M.; Russo, C.; Malaguarnera, G.; D’Orazio, N.; Malaguarnera, L. The physiopathology of lipoprotein(a). Front. Biosci. 2010, S2, 866–875. [Google Scholar] [CrossRef]
- Hancock, M.A.; Boffa, M.B.; Marcovina, S.M.; Nesheim, M.E.; Koschinsky, M.L. Inhibition of plasminogen activation by lipoprotein(a). Critical domains in apolipoprotein(a) and mechanism of inhibition on fibrin and degraded fibrin surfaces. J. Biol. Chem. 2003, 278, 23260–23269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangrar, W.; Gabel, B.R.; Boffa, M.B.; Walker, J.B.; Hancock, M.A.; Marcovina, S.M.; Horrevoets, A.J.G.; Nesheim, M.E.; Koschinsky, M.L. The solution phase interaction between apolipoprotein(a) and plasminogen inhibits the binding of plasminogen to a plasmin-modified fibrinogen surface. Biochemistry 1997, 36, 10353–10363. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. Lipoprotein(a): Fasting and nonfasting levels, inflammation, and cardiovascular risk. Atherosclerosis 2014, 234, 95–101. [Google Scholar] [CrossRef]
- Cremer, P.; Nagel, D.; Mann, H.; Labrot, B.; Müller Berninger, R.; Elster, H.; Seidel, D. Ten-year follow-up results from the Goettingen Risk, Incidence and Prevalence Study (GRIPS). I. Risk factors for myocardial infarction in a cohort of 5790 men. Atherosclerosis 1997, 129, 221–230. [Google Scholar] [CrossRef]
- Seed, M.; Ayres, K.L.; Humphries, S.E.; Miller, G.J. Lipoprotein(a) as a predictor of myocardial infarction in middle-aged men. Am. J. Med. 2001, 110, 22–27. [Google Scholar] [CrossRef]
- Sagastagoitia, J.D.; Sáez, Y.; Vacas, M.; Narváez, I.; Sáez de Lafuente, J.P.; Molinero, E.; Magro, A.; Lafita, M.; Santos, M.; Escobar, A.; et al. Association between inflammation, lipid and hemostatic factors in patients with stable angina. Thromb. Res. 2007, 120, 53–59. [Google Scholar] [CrossRef]
- Rohde, L.E.P.; Hennekens, C.H.; Ridker, P.M. Survey of C-reactive protein and cardiovascular risk factors in apparently healty men. Am. J. Cardiol. 2004, 84, 1018–1022. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, J.L.; Cao, Y.X.; Liu, H.H.; Zhang, H.W.; Guo, Y.L.; Wu, N.Q.; Gao, Y.; Hua, Q.; Li, Y.F.; et al. Prognostic utility of lipoprotein(a) combined with fibrinogen in patients with stable coronary artery disease: A prospective, large cohort study. J. Transl. Med. 2020, 18, 1–8. [Google Scholar] [CrossRef]
- Niessner, A.; Graf, S.; Nikfardjam, M.; Speidl, W.S.; Huber Beckmann, R.; Zorn, G.; Wojta, J.; Huber, K. Circulating t-PA antigen predicts major adverse coronary events in patients with stable coronary artery disease-13-year follow-up. Thromb. Haemost. 2003, 90, 344–350. [Google Scholar] [CrossRef]
- Alaigh, P.; Hoffman, C.J.; Korlipara, G.; Neuroth, A.; Dervan, J.P.; Lawson, W.E.; Hultin, M.B. Lipoprotein(a) level does not predict restenosis after percutaneous transluminal coronary angioplasty. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1281–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardys, I.; Oemrawsingh, R.M.; Kay, I.P.; Jones, G.T.; McCormick, S.P.A.; Daemen, J.; Van Geuns, J.R.; Boersma, E.; Van Domburg, R.T.; Serruys, P.W. Lipoprotein(a), interleukin-10, c-reactive protein, and 8-year outcome after percutaneous coronary intervention. Clin. Cardiol. 2012, 35, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Zairis, M.N.; Ambrose, J.A.; Manousakis, S.J.; Stefanidis, A.S.; Papadaki, O.A.; Bilianou, H.I.; DeVoe, M.C.; Fakiolas, C.N.; Pissimissis, E.G.; Olympios, C.D.; et al. The impact of plasma levels of C-reactive protein, lipoprotein(a) and homocysteine on the long-term prognosis after successful coronary stenting: The global evaluation of new events and restenosis after stent implantation study. J. Am. Coll. Cardiol. 2002, 40, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Speidl, W.S.; Nikfardjam, M.; Niessner, A.; Zeiner, A.; Jordanova, N.; Zorn, G.; Maurer, G.; Schreiber, W.; Wojta, J.; Huber, K. Mild hyperhomocysteinemia is associated with a decreased fibrinolytic activity in patients after ST-elevation myocardial infarction. Thromb. Res. 2007, 119, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, R.; Brogi, D.; Sofi, F.; Giglioli, C.; Valente, S.; Alessandrello Liotta, A.; Lenti, M.; Gori, A.M.; Prisco, D.; Abbate, R.; et al. PAI-1 and homocysteine, but not lipoprotein(a) and thrombophilic polymorphisms, are independently associated with the occurrence of major adverse cardiac events after successful coronary stenting. Heart 2006, 92, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Stefanini, G.G.; Holmes, D.R. Drug-eluting coronary-artery stents. N. Engl. J. Med. 2013, 368, 254–265. [Google Scholar] [CrossRef] [Green Version]
- Bønaa, K.H.; Mannsverk, J.; Wiseth, R.; Aaberge, L.; Myreng, Y.; Nygard, O.; Nilsen, D.W.; Kløw, N.E.; Uchto, M.; Trovik, T.; et al. Drug-eluting or bare-metal stents for coronary artery disease. N. Engl. J. Med. 2016, 375, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Thogersen, A.M.; Söderberg, S.; Jansson, J.H.; Dahlén, G.; Boman, K.; Nilsson, T.K.; Lindahl, B.; Weinehall, L.; Stenlund, H.; Lundberg, V. Interactions between fibrinolysis, lipoproteins and leptin related to a first myocardial infarction. Eur. J. Prev. Cardiol. 2003, 11, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Pineda, J.; Marín, F.; Marco, P.; Roldán, V.; Valencia, J.; Ruiz Nodar, J.M.; Hernández Romero, D.; Sogorb, F.; Lip, G.Y.H. The prognostic value of biomarkers after a premature myocardial infarction. Int. J. Cardiol. 2010, 143, 249–254. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.; Kastelein, J.J.P. Effect of an siRNA therapeutic targeting PCSK9 on atherogenic lipoproteins: Prespecified secondary end points in ORION 1. Circulation 2018, 138, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Nishikido, T.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.L.J.; Kastelein, J.J.P. Effect of 1 or 2 doses of inclisiran on low-density lipoprotein cholesterol levels: One-year follow-up of the ORION-1 randomized clinical trial. JAMA Cardiol. 2019, 4, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Koren, M.J.; Moriarty, P.M.; Neutel, J.; Baum, S.J.; Hernandez-Illas, M.; Weintraub, H.S.; Hellawell, J.; Varrieur, T.; Sohn, W.; Wang, H.; et al. Abstract 13951: Safety, tolerability and efficacy of single-dose Amg 890, a novel siRNA targeting Lp(a), in healthy subjects and subjects with elevated Lp(a). Circulation 2020, 142, A13951. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Valanti, E.K.; Dalakoura-Karagkouni, K.; Siasos, G.; Kardassis, D.; Eliopoulos, A.G.; Sanoudou, D. Advances in biological therapies for dyslipidemias and atherosclerosis. Metabolism 2021, 116, 154461. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Study | Parameter Included | Study Population (n) | Primary Endpoint | Independent Predictor | Ref. | ||
|---|---|---|---|---|---|---|---|
| (n) | Age (Years) | Characteristics | |||||
| Langsted et al. (2014) | Lp(a), CRP | 34,829 | - | General population | Ischemic heart disease | Lp(a), independent of CRP levels | [84] |
| Cremer et al. (1997) | Lp(a), fibrinogen | 5790 | 40–60 | Male, without previous CAD | MI after 10 years | Lp(a) | [85] |
| Seed et al. (2001) | Lp(a), fibrinogen | 2616 | 51–61 | Male, without previous CAD | CAD after 6 years | Lp(a) | [86] |
| Zhang et al. (2020) | Lp(a), fibrinogen | 8417 | - | Stable CAD, no stent implantation | MACE after 37 months | Lp(a), fibrinogen; combination of both superior | [89] |
| Niessner et al. (2003) | Lp(a), tPA | 141 | - | CAD after PTCA, no stent implantation | MACE after 13 years | t-PA | [90] |
| Alaigh et al. (1998) | Lp(a), PAI-1 | 163 | - | CAD after PTCA, no stent implantation | Restenosis after 6 months | None | [91] |
| Kardys et al. (2012) | Lp(a), CRP, IL-10 | 161 | - | CAD after sirolimus-eluting stent implantation | MACE after 1, 6 years | 1 year: Lp(a); 6 years: CRP | [92] |
| Zairis et al. (2002) | Lp(a), CRP | 483 | - | ACS after PCI, various stent implantations | MACE after 3 years | Lp(a), CRP | [93] |
| Marcucci et al. (2006) | Lp(a) PAI-1, homocysteine | 520 | - | ACS, bar metal stent implantation | MACE after 24 months | PAI-1, homocysteine | [95] |
| Thogersen et al. (2003) | Lp(a), PAI-1, t-PA, leptin | 62, plus 124 controls | Sex and age matched | - | First myocardial infarction | Lp(a), t-PA | [98] |
| Pineda et al. (2010) | Lp(a), CRP, PAI-1, t-PA, fibrinogen, D-dimer, homocysteine | 142; 56; 10 | - | MI before 45 years; treated with fibrinolysis; PCI | MACE after 36 months | Homocysteine | [99] |
| Moss et al. (1999) | Lp(a), PAI-1, fibrinogen, D-dimer | 1045 | - | Post-MI (2 months), treated with thrombolysis or PCI | Coronary death or nonfatal MI after 26 months | D-dimer | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugovšek, S.; Šebeštjen, M. Lipoprotein(a)—The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules 2022, 12, 26. https://doi.org/10.3390/biom12010026
Ugovšek S, Šebeštjen M. Lipoprotein(a)—The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules. 2022; 12(1):26. https://doi.org/10.3390/biom12010026
Chicago/Turabian StyleUgovšek, Sabina, and Miran Šebeštjen. 2022. "Lipoprotein(a)—The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation" Biomolecules 12, no. 1: 26. https://doi.org/10.3390/biom12010026
APA StyleUgovšek, S., & Šebeštjen, M. (2022). Lipoprotein(a)—The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules, 12(1), 26. https://doi.org/10.3390/biom12010026