
Long-Term Consequences of Placental Vascular Pathology on the Maternal and Offspring Cardiovascular Systems


Abstract
:1. Introduction
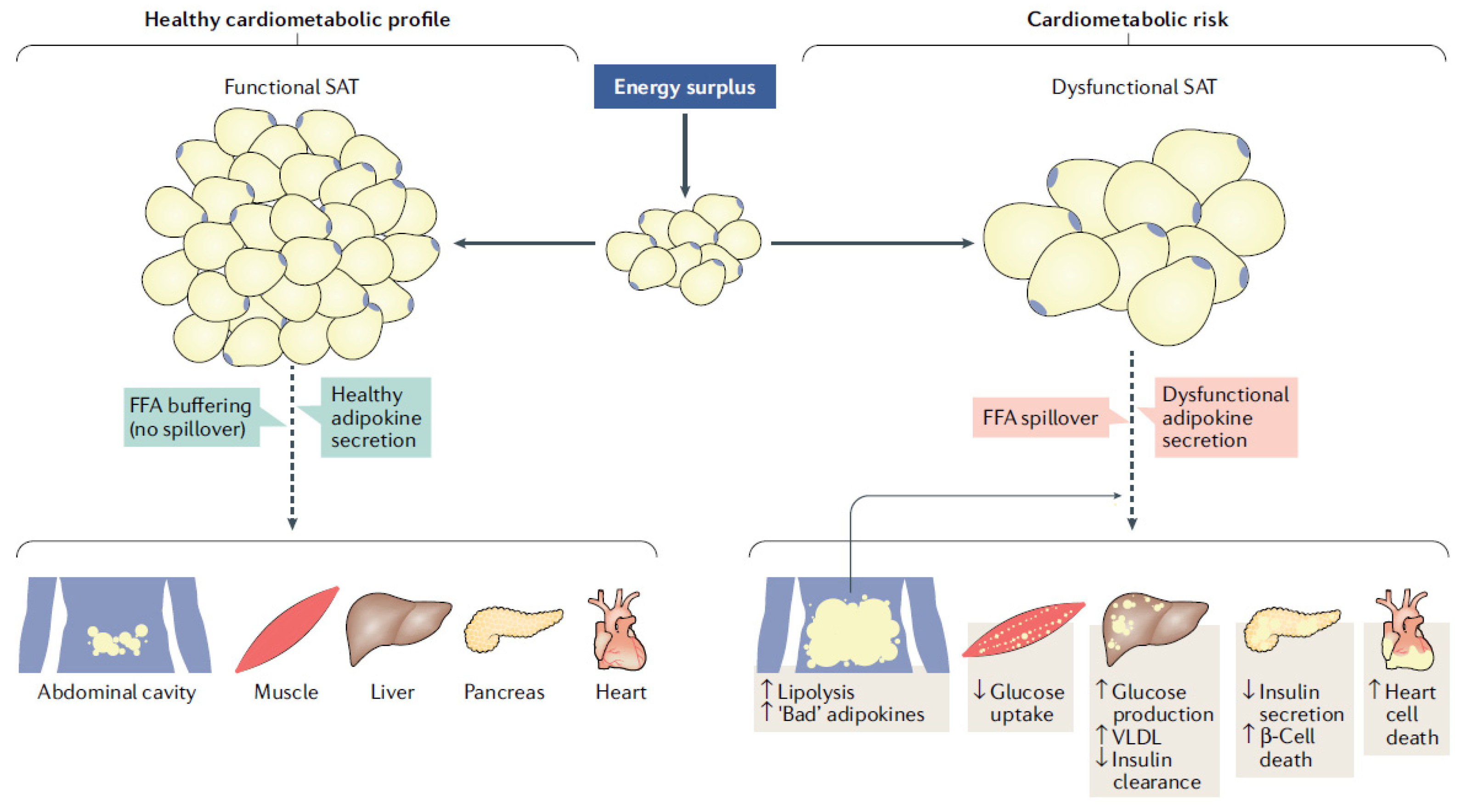
2. Cardiovascular and Metabolic Consequences of the Great Obstetrical Syndromes
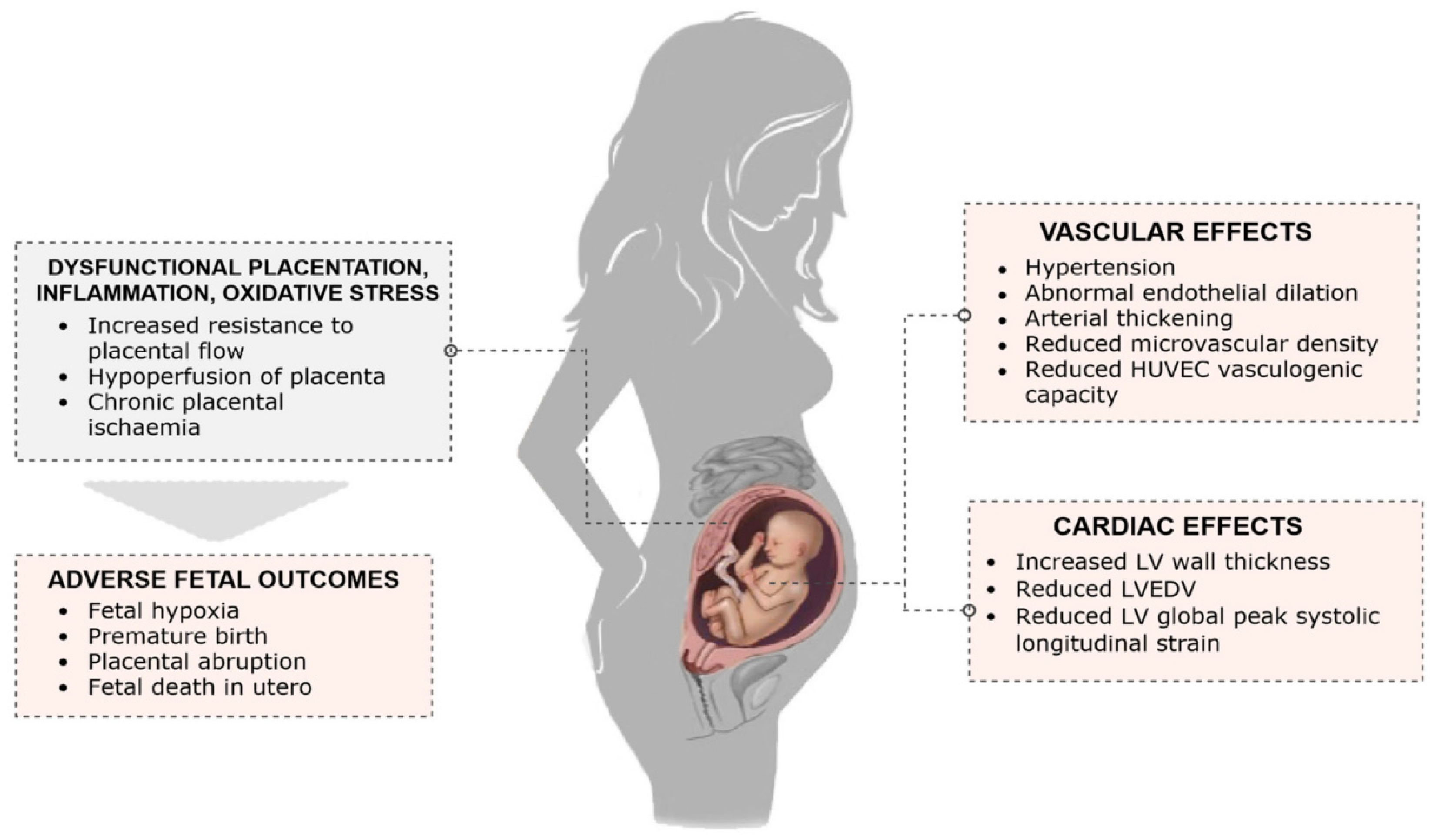
3. Consequences for the Pregnant Woman
- (a)
- Hypertension: Relative risk (RR) = 3.7, 95% CI: 2.70–5.05, after 14.1 years weighted mean follow-up.
- (b)
- IHD: RR = 0.16, 95% CI: 1.86–2.52, after 11.7 years.
- (c)
- Stroke: RR = 1.81, 95% CI: 1.45–2.27, after 10.4 years.
- (d)
- Venous thrombo-embolism: RR = 1.79, 95% CI: 1.37–2.33, after 4.7 years.
- (a)
- Fatal or diagnosed CVD: Odds Ratio (OR) = 2.28, 95% CI: 1.87–2.78.
- (b)
- Cerebrovascular accident: OR = 1.76, 95% CI: 1.43–2.21;
- (c)
- Hypertension: RR = 3.13, 95% CI: 2.51–3.89.
- (a)
- Any CVD: RR = 1.80, 95%CI: 1.67–1.94.
- (b)
- Coronary artery disease: RR = 1.66, 95% CI: 1.49–1.84
- (c)
- Heart failure: RR = 2.87, 95% CI: 2.14–3.85.
- (d)
- Peripheral vascular disease: RR = 1.60, 95% CI: 1.29–2.00.
- (e)
- Stroke: RR = 1.72, 95% CI: 1.50–1.97.
- (f)
- CVD-related mortality: RR = 1.78, 95% CI 1.58–2.00.
- (g)
- Hypertension: RR = 3.16, 95% CI: 2.74–3.64)
3.1. Pathogenetic Mechanisms of Delayed Effects of Preeclampsia
3.2. Biomolecules Involved in the Pathogenesis of Long-Term Effects
3.2.1. Inflammation and the Role of Inflammatory Biomolecules
3.2.2. In Vivo Acute-Phase Response Markers
3.2.3. Metabolic Syndrome Biomolecules
3.2.4. Oxidative-Stress Biomolecules
3.2.5. Proangiogenic and Antiangiogenic Biomolecules
3.2.6. Altered Placental Biomolecules
3.3. Biophysical Markers Involved in the Pathogenesis of Long-Term Effects
- (a)
- Carotid Intima-Media Thickness (CIMT): Conflicting evidence raises the possibility that vascular structural changes may manifest as a result of hypertension, possibly as an adaptive response to increased arterial stress. Discrepant findings have been reported in the post-partum with respect to the persistence of increased CIMT. The evidence of long-term changes comes from a meta-analysis of women with a history of PE up to 10 years postpartum, showing greater CIMT in the PE group: 0.18 mm (95% CI, 0.05–0.30 mm) [105].
- (b)
- Cardiac Computed Tomography and Calcium Score: There is evidence in the post-partum period of a strong association between PE and vascular structural changes. HDP is significantly associated with coronary artery calcification even after adjusting fo serum creatinine levels, urinary albumin/creatinine ratio, menopause and diabetes status and antihypertensive medication use.
- (c)
- Retinal Microvasculature: An investigation found that during PE there is a significant decrease in central retinal artery and vein equivalent diameters: 1 year postpartum, the decrease persisted [106].
- (d)
- Flow Mediated Dilatation (FMD): Evaluation through FMD, indicates that the ED seen in PE is likely to persist after delivery, at least over the short period (up to 6 months).
- (e)
- Pulse Wave Velocity (PWV): A consistent finding is that preceding onset of PE, there is an increase in PWV, lasting at least up to 2–3 years postpartum.
4. Consequences for the Offspring
4.1. Hypertension
4.2. Cardiovascular Diseases
4.3. Possible Pathogenic Mechanisms of the Effects on the Newborn
4.3.1. Endothelial Dysfunction
4.3.2. Genetic Alterations
4.3.3. Comparison of Biochemical and Biophysical Markers in Offspring
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Okada, H.; Tsuzuki, T.; Shindoh, H.; Nishigaki, A.; Yasuda, K.; Kanzaki, H. Regulation of decidualization and angiogenesis in the human endometrium: Mini review. J. Obstet. Gynaecol. Res. 2014, 40, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Plaisier, M. Decidualisation and angiogenesis. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Pijnenborg, R.; Bland, J.M.; Robertson, W.; Brosens, I. Uteroplacental arterial changes related to interstitial trophoblast migration in early human pregnancy. Placenta 1983, 4, 397–413. [Google Scholar] [CrossRef]
- Craven, C.M.; Morgan, T.; Ward, K. Decidual spiral artery remodelling begins before cellular interaction with cytotrophoblasts. Placenta 1998, 19, 241–252. [Google Scholar] [CrossRef]
- Gaynor, L.M.; Colucci, F. Uterine natural killer cells: Functional distinctions and influence on pregnancy in humans and mice. Front. Immunol. 2017, 8, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sojka, D.K.; Yang, L.; Plougastel-Douglas, B.; Higuchi, D.A.; Croy, B.A.; Yokoyama, W.M. Cutting Edge: Local proliferation of uterine tissue resident NK cells during decidualization in mice. J. Immunol. 2018, 201, 2551–2556. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosens, I.; Puttemans, P.; Benagiano, G. Placental bed research: I. The placental bed: From spiral arteries remodeling to the great obstetrical syndromes. Am. J. Obstet. Gynecol. 2019, 221, 437–456. [Google Scholar] [CrossRef]
- Khong, Y.; Brosens, I. Defective deep placentation. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Labarrere, C.A.; Di Carlo, H.L.; Bammerlin, E.; Hardin, J.W.; Kim, Y.M.; Chaemsaithong, P.; Haas, D.M.; Kassab, G.S.; Romero, R. Failure of physiologic transformation of spiral arteries, endothelial and trophoblast cell activation, and acute atherosis in the basal plate of the placenta. Am. J. Obstet. Gynecol. 2017, 216, 287.e1–287.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosens, I.; Brosens, J.J.; Muter, J.; Benagiano, G. Acute atherosis and diffuse lipid infiltration of the placental bed: A review of historical lipid studies. Placenta 2020, 97, 36–41. [Google Scholar] [CrossRef]
- Brosens, I.A. The Placental Bed. Ph.D. Thesis, University of London, London, UK, 1965. [Google Scholar]
- Hertig, A.T. Vascular pathology in hypertensive pregnancy in albuminuric toxemias of pregnancy. Clinics 1945, 4, 602–614. [Google Scholar]
- Sexton, L.I.; Hertig, A.T.; Reid, D.E.; Kellogs, F.S.; Patterson, W.S. Premature separation of the normally implanted placenta; a clinicopathological study of 476 cases. Am. J. Obstet. Gynecol. 1950, 59, 13–24. [Google Scholar] [CrossRef]
- Zeek, P.M.; Assali, N.S. Vascular changes in the decidua associated with eclamptogenic toxemia of pregnancy. Am. J. Clin. Pathol. 1950, 20, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, K.; Nakashima, Y. Pathologic intimal thickening in human atherosclerosis is formed by extracellular accumulation of plasma-derived lipids and dispersion of intimal smooth muscle cells. Atherosclerosis 2018, 274, 235–242. [Google Scholar] [CrossRef]
- Alnaes-Katjavivi, P.; Lyall, F.; Roald, B.; Redman, C.W.; Staff, A.C. Acute atherosis invacuum suction biopsies of decidua basalis: An evidence based research definition. Placenta 2016, 37, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Lendrum, A.C.; Dobsin, J.; Fawkes, R.S.; Morrison, S.M. Plasmatic vasculosis in the terminal vasculature. Biorheology 1978, 15, 49–50. [Google Scholar] [PubMed]
- Robertson, W.B.; Brosens, I.; Dixon, G. Uteroplacental vascular pathology. Eur. J. Obstet. Gynecol. Reprod. Biol. 1975, 5, 47–65. [Google Scholar] [CrossRef]
- Kim, Y.M.; Chaemsaithong, P.; Romero, R.; Shaman, M.; Kim, C.J.; Kim, J.S.; Qureshi, F.; Jacques, S.M.; Ahmed, A.I.; Chaiworapongsa, T.; et al. The frequency of acute atherosis in normal pregnancy and preterm labor, preeclampsia, small-for-gestational age, fetal death and midtrimester spontaneous abortion. J. Matern. Fetal Neonatal Med. 2015, 28, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiologyand clinical implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef] [Green Version]
- Fosheim, I.K.; Alnaes-Katjavivi, P.; Redman, C.; Roald, B.; Staff, A.C.; Storvold, G.L. Acute atherosis of decidua basalis; characterization of spiral arteries, endothelialstatus and activation. Placenta 2019, 82, 10–16. [Google Scholar] [CrossRef]
- World Health Organization. Cardiovascular Diseases. Definition. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 18 October 2021).
- Napoli, C.; D’Armiento, F.P.; Mancini, F.P.; Postiglione, A.; Witztum, J.L.; Palumbo, G.; Palinski, W. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J. Clin. Investig. 1997, 100, 2680–2690. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef]
- Kaperonis, A.; Liapis, C.D.; Kakisis, J.D.; Dimitroulis, D.; Papavassilliou, V.G. Inflammation and Atherosclerosis. Eur. J. Vasc. Endovasc. Surg. 2006, 31, 386–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benagiano, M.; Azzurri, A.; Ciervo, A.; Amedei, A.; Tamburini, C.; Ferrari, M.; Telford, J.L.; Baldari, C.T.; Romagnani, S.; Cassone, A.; et al. T helper type-1 lymphocyte-driven inflammation in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 6658–6663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemme, S.; Faber, B.; Holm, J.; Wiklund, O.; Witztum, J.L.; Hansson, G.K. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1995, 92, 3893–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenson, G.S.; Srinivasan, S.R.; Bao, W.; Newman, W.P., 3rd; Tracy, R.E.; Wattigney, W.A. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N. Engl. J. Med. 1998, 338, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Berenson, G.S.; Wattigney, W.A.; Tracy, R.E.; Newman, W.P., 3rd; Srinivasan, S.R.; Webber, L.S.; Dalferes, E.R., Jr.; Strong, J.P. Atherosclerosis of the aorta and coronary arteries and cardiovascular risk factors in persons aged 6 to 30 years and studied at necropsy (The Bogalusa Heart Study). Am. J. Cardiol. 1992, 70, 851–858. [Google Scholar] [CrossRef]
- Strong, J.P.; Malcom, G.T.; McMahan, C.A.; Tracy, R.E.; Newman, W.P., III; Herderick, E.E.; Cornhill, J.F. Prevalence and extent of atherosclerosis in adolescents and young adults: Implications for prevention from the pathobiological determinants of atherosclerosis in youth study. JAMA 1999, 281, 727–735. [Google Scholar] [CrossRef]
- Ross, R.; Neeland, I.J.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; Cuevas, A.; Hu, F.B.; et al. Waist circumference as a vital sign in clinical practice: A Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat. Rev. Endocrinol. 2020, 16, 177–189. [Google Scholar] [CrossRef]
- Krantz, S.; Mack, W.J.; Hodis, H.N.; Liu, C.-R.; Liu, C.-H.; Kaufman, F.R. Early onset of subclinical atherosclerosis in young persons with type 1 diabetes. J. Pediat. 2004, 145, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Kasiske, B.L. Risk factors for accelerated atherosclerosis in renal transplant recipients. Am. J. Med. 1988, 84, 985–992. [Google Scholar] [CrossRef]
- Marenberg, M.E.; Risch, N.; Berkman, L.F.; Floderus, B.; De Faire, U. Genetic susceptibility to death from coronary heart disease in a study of twins. N. Engl. J. Med. 1994, 330, 1041–1046. [Google Scholar] [CrossRef]
- Shah, S.H.; Freedman, N.J.; Zhang, L.; Crosslin, D.R.; Stone, D.H.; Haynes, C.; Johnson, J.; Nelson, S.; Wang, L.; Connelly, J.J.; et al. Neuropeptide Y Gene Polymorphisms Confer Risk of Early-Onset Atherosclerosis. PLoS Genet. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Robertson, W.B.; Brosens, I.; Dixon, H.G. The pathological response of the vessels of the placental bed to hypertensive pregnancy. J. Pathol. Bacteriol. 1967, 93, 581–592. [Google Scholar] [CrossRef] [PubMed]
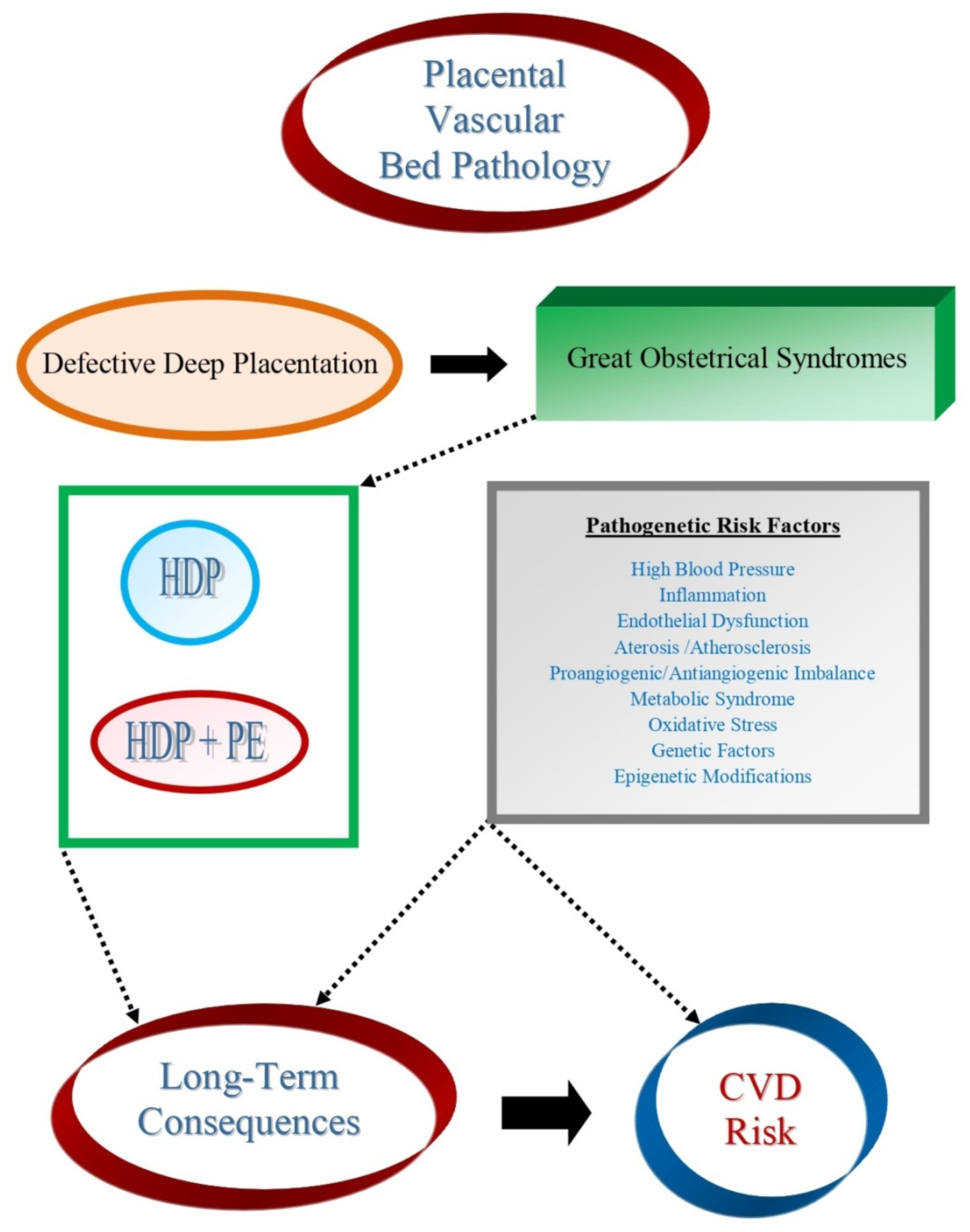
- Di Renzo, G.C. The great obstetrical syndromes. J. Matern. Fetal Neonatal Med. 2009, 22, 633–635. [Google Scholar] [CrossRef]
- Brosens, J.J.; Parker, M.G.; McIndoe, A.; Pijnenborg, R.; Brosens, I.A. A role for menstruation in-preconditioning the uterus for successful pregnancy. Am. J. Obstet. Gynecol. 2009, 200, 615.e1–615.e6. [Google Scholar] [CrossRef] [PubMed]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of Deep Placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryans, C.I. The remote prognosis in toxaemia of pregnancy. Clin. Obstet. Gynecol. 1966, 9, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Chesley, L.C.; Annitto, J.E.; Cosgrove, R.A. The remote prognosis of eclamptic women: Sixth periodic report. Am. J. Obstet. Gynecol. 1976, 124, 446–459. [Google Scholar] [CrossRef]
- Fisher, K.A.; Luger, A.; Spargo, B.H.; Lindheimer, M.D. Hypertension in pregnancy: Clinical pathological correlations and remote prognosis. Medicine 1981, 60, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Sibai, B.; el Nazer, A.; Gonzalez Ruiz, A. Severe preeclampsia eclampsia in young primigravid women: Subsequent pregnancy outcome and remoteprognosis. Am. J. Obstet. Gynecol. 1986, 155, 1011–1016. [Google Scholar] [CrossRef]
- Langford, H.G.; Watson, R. Prepregnant Blood Pressure, Hypertension during Pregnancy, and later Blood Pressure of Mothers and Offspring. Hypertension 1980, 2, I130–I133. [Google Scholar] [CrossRef] [Green Version]
- Bokslag, A.; van Weissenbruch, M.; Mol, B.W.; de Groot, C.J. Preeclampsia; short and long-term consequences for mother and neonate. Early Hum. Dev. 2016, 102, 47–50. [Google Scholar] [CrossRef]
- Groenhof, T.K.J.; van Rijn, B.B.; Franx, A.; Roeters van Lennep, J.E.; Bots, M.L.; Lely, A.T. Preventing cardiovascular disease after hypertensive disorders of pregnancy: Searching for the how and when. Eur. J. Prev. Cardiol. 2017, 24, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Muijsers, H.E.C.; Roeleveld, N.; van der Heijden, O.W.H.; Maas, A.H.E.M. Consider Preeclampsia as a First Cardiovascular Event. Curr. Cardiovasc. Risk Rep. 2019, 13, 21. [Google Scholar] [CrossRef] [Green Version]
- Sattar, N.; Greer, I.A. Pregnancy complications and maternal cardiovascular risk: Opportunities for intervention and screening? BMJ 2002, 32, 157–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haug, E.B.; Horn, J.; Markovitz, A.R.; Fraser, A.; Klykken, B.; Dalen, H.; Vatten, L.J.; Romundstad, P.R.; Rich-Edwards, J.W.; Åsvold, B.O. Association of Conventional Cardiovascular Risk Factors With Cardiovascular Disease After Hypertensive Disorders of Pregnancy: Analysis of the Nord-Trøndelag Health Study. J. AMA Cardiol. 2019, 4, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Bellamy, L.; Casas, J.-P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ 2007, 335, 974. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.C.; Best, K.E.; Pearce, M.S.; Waugh, J.; Robson, S.C.; Bell, R. Cardiovascular disease risk in women with pre-eclampsia: Systematic review and meta-analysis. Eur. J. Epidemiol. 2013, 28, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, T.; Gu, R.; Xing, D.; Ye, C.; Chen, Y.; Liu, X.; Chen, L. Hypertensive Disorders of Pregnancy and Risk of Cardiovascular Disease-Related Morbidity and Mortality: A Systematic Review and Meta-Analysis. Cardiology 2020, 145, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Hermes, W.; Ket, J.C.; van Pampus, M.G.; Franx, A.; Veenendaal, M.V.; Kolster, C.; Tamsma, J.T.; Bloemenkamp, K.W.; Ponjee, G.; van der Hout, E.; et al. Biochemical cardiovascular risk factors after hypertensive pregnancy disorders: A systematic review and meta-analysis. Obstet. Gynecol. Surv. 2012, 67, 793–809. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, L.; van der Meiden-van Roest, A.J.; Savelkoul, C.; Vogelvang, T.E.; Lely, A.T.; Franx, A.; van Rijn, B.B. Recurrence of pre-eclampsia and the risk of future hypertension and cardiovascular disease: A systematic review and meta-analysis. BJOG 2018, 125, 1642–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.C.S.; Pell, J.P.; Walsh, D. Pregnancy complications and maternal risk of ischaemic heart disease: A retrospective cohort study of 129′290 births. Lancet 2001, 357, 2002–2006. [Google Scholar] [CrossRef]
- Brosens, I.; Benagiano, M.; Puttemans, P.; D’Elios, M.M.; Benagiano, G. The placental bed vascular pathology revisited: A risk indicator for cardiovascular disease. J. Matern. Fetal Neonatal Med. 2019, 32, 1556–1564. [Google Scholar] [CrossRef]
- Staff, A.C.; Dechend, R.; Pijnenborg, R. Learning from the placenta: Acute atherosis and vascular remodeling in preeclampsia—Novel aspects for atherosclerosis and future cardiovascular health. Hypertension 2010, 56, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, D.U.; Al-Nasiry, S.; Fajta, M.M.; Bulten, J.; Van Dijk, A.P.; Van Der Vlugt, M.J.; Oyen, W.J.; van Vugt, J.M.; Spaanderman, M.E. Cardiovascular and thrombogenic risk of decidual vasculopathy in preeclampsia. Am. J. Obstet. Gynecol. 2014, 210, 545.e1–545.e6. [Google Scholar] [CrossRef] [PubMed]
- Milic, N.M.; Milin-Lazovic, J.; Weissgerber, T.L.; Trajkovic, G.; White, W.M.; Garovic, V.D. Preclinical atherosclerosis at the time of pre-eclamptic pregnancy and up to 10 years postpartum: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2017, 49, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosens, I.; Muter, J.; Ewington, L.; Puttemans, P.; Petraglia, F.; Brosens, J.J.; Benagiano, G. Adolescent Preeclampsia: Pathological Drivers and Clinical Prevention. Reprod. Sci. 2019, 26, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Catov, J.M.; Ouyang, P. Hypertensive Disorders of Pregnancy and Future Maternal Cardiovascular Risk. J. Am. Heart Assoc. 2018, 7, e009382. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.; Nelson, S.M.; Macdonald-Wallis, C.; Cherry, L.; Butler, E.; Sattar, N.; Lawlor, D.A. Associations of pregnancy complications with calculated cardiovascular disease risk and cardiovascular risk factors in middle age: The Avon Longitudinal Study of Parents and Children. Circulation 2012, 125, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Aykas, F.; Solak, Y.; Erden, A.; Bulut, K.; Dogan, S.; Sarli, B.; Acmaz, G.; Afsar, B.; Siriopol, D.; Covic, A.; et al. Persistence of cardiovascular risk factors in women with previous preeclampsia: A long-term follow-up study. J. Investig. Med. 2015, 63, 641–645. [Google Scholar] [CrossRef]
- Stuart, J.J.; Tanz, L.J.; Missmer, S.A.; Rimm, E.B.; Spiegelman, D.; James-Todd, T.M.; Rich-Edwards, J.W. Hypertensive disorders of pregnancy and maternal cardiovascular disease risk factor development: An observational cohort study. Ann. Intern. Med. 2018, 169, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Agatisa, P.K.; Ness, R.B.; Roberts, J.M.; Costantino, J.P.; Kuller, L.H.; McLaughlin, M.K. Impairment of endothelial function in women with a history of preeclampsia: An indicator of cardiovascular risk. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1389–H1393. [Google Scholar] [CrossRef] [PubMed]
- Hubel, C.A.; Wallukat, G.; Wolf, M.; Herse, F.; Rajakumar, A.; Roberts, J.M.; Markovic, N.; Thadhani, R.; Luft, F.C.; Dechend, R. Agonistic angiotensin II type 1 receptor autoantibodies in postpartum women with a history of preeclampsia. Hypertension 2007, 49, 612–617. [Google Scholar] [CrossRef] [Green Version]
- Kvehaugen, A.S.; Dechend, R.; Ramstad, H.B.; Troisi, R.; Fugelseth, D.; Staff, A.C. Endothelial function and circulating biomarkers are disturbed in women and children after preeclampsia. Hypertension 2011, 58, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Mangos, G.J.; Spaan, J.J.; Pirabhahar, S.; Brown, M.A. Markers of cardiovascular disease risk after hypertension in pregnancy. J. Hypertens. 2012, 30, 351–358. [Google Scholar] [CrossRef]
- van Rijn, B.B.; Bruinse, H.W.; Veerbeek, J.H.; Post Uiterweer, E.D.; Koenen, S.V.; van der Bom, J.G.; Rijkers, G.T.; Roest, M.; Franx, A. Postpartum circulating markers of inflammation and the systemic acute-phase response after early-onset preeclampsia. Hypertension 2016, 67, 404–414. [Google Scholar] [CrossRef]
- Sandvik, M.K.; Leirgul, E.; Nygard, O.; Ueland, P.M.; Berg, A.; Svarstad, E.; Vikse, B.E. Preeclampsia in healthy women and ED10 years later. Am. J. Obstet. Gynecol. 2013, 209, 569.e561–569.e510. [Google Scholar] [CrossRef] [PubMed]
- Osol, G.; Bernstein, I. Preeclampsia and maternal cardiovascular disease: Consequence or predisposition? J. Vasc. Res. 2014, 51, 290–304. [Google Scholar] [CrossRef]
- Lane-Cordova, A.D.; Khan, S.S.; Grobman, W.A.; Greenland, P.; Shah, S.J. Long-Term Cardiovascular Risks Associated With Adverse Pregnancy Outcomes: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.; Friedman, J.E. Epigenetics and microRNAs in preeclampsia. Clin. Exp. Hypertens. 2012, 34, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.M.; Eggink, A.J.; Willemsen, S.P.; Slieker, R.C.; Wijnands, K.P.J.; Felix, J.F.; Chen, J.; Stubbs, A.; van der Spek, P.J.; van Meurs, J.B.; et al. Early- and late-onset preeclampsia and the tissue-specific epigenome of the placenta and newborn. Placenta 2017, 58, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Turbeville, H.R.; Sasser, J.M. Preeclampsia beyond pregnancy: Long-term consequences for mother and child. Am. J. Physiol. Renal Physiol. 2020, 318, F1315–F1326. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis: An inflammatory disease. NEJM 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Poston, L.; Burton, G.J. Placental-related diseases of pregnancy: Involvement of oxidative stress and implications in human evolution. Hum. Reprod. Update 2006, 12, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.F.; Newton, L.; Lewandowski, A.J.; Lazdam, M.; Kelly, B.A.; Kyriakou, T.; Leeson, P. Pre-eclampsia and offspring cardiovascular health: Mechanistic insights from experimental studies. Clin. Sci. 2012, 123, 53–72. [Google Scholar] [CrossRef] [Green Version]
- Hermes, W.; Tamsma, J.T.; Grootendorst, D.C.; Franx, A.; van der Post, J.; van Pampus, M.G.; Bloemenkamp, K.W.; Porath, M.; Mol, B.W.; de Groot, C.J. Cardiovascular risk estimation in women with a history of hypertensive pregnancy disorders at term: A longitudinal follow-up study. BMC Pregnancy Childbirth 2013, 13, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauspurg, A.; Countouris, M.E.; Jeyabalan, A.; Hubel, C.A.; Roberts, J.M.; Schwarz, E.B.; Catov, J.M. Risk of hypertension and abnormal biomarkers in the first year postpartum associated with hypertensive disorders of pregnancy among overweight and obese women. Pregnancy Hypertens. 2019, 15, 1–6. [Google Scholar] [CrossRef]
- Bang, O.Y.; Lee, M.A.; Lee, J.H.; Kim, J.W.; Lee, P.H.; Joo, I.S.; Huh, K.J. Association of Metabolic Syndrome and C reactive Protein Levels with Intracranial Atherosclerotic Stroke. J. Clin. Neurol. 2005, 1, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Crowe, T.; Sasiela, W.J.; Tsai, J.; Orazem, J.; Magorien, R.D.; O’Shaughnessy, C.; Ganz, P. Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) Investigators. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N. Engl. J. Med. 2005, 352, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Anti-inflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Ross, R.; Després, J.P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: A position statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725. [Google Scholar] [CrossRef]
- Syme, C.; Pelletier, S.; Shin, J.; Abrahamowicz, M.; Leonard, G.; Perron, M.; Richer, L.; Veillette, S.; Gaudet, D.; Pike, B.; et al. Visceral fat-related systemic inflammation and the adolescent brain: A mediating role of circulating glycerophosphocholines. Int. J. Obes. 2019, 43, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Hui, X.; Hoo, R.L.C.; Ye, D.; Chan, C.Y.C.; Feng, T.; Wang, Y.; Lam, K.S.L.; Xu, A.J. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J. Clin. Investig. 2019, 129, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Bastien, M.; Poirier, P.; Lemieux, I.; Després, J.P. Overview of epidemiology and contribution of obesity to cardiovascular disease. Prog. Cardiovasc. Dis. 2014, 56, 369–381. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.P.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, G.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Malý, M.; Hajšl, M.; Bechynská, K.; Kučerka, O.; Šrámek, M.; Suttnar, J.; Hlavácková, A.; Hajšlová, J.; Kosek, V. Lipidomic Analysis to Assess Oxidative Stress in Acute Coronary Syndrome and Acute Stroke Patients. Metabolites 2021, 11, 412. [Google Scholar] [CrossRef]
- Myatt, L.; Webster, R.P. Vascular biology of preeclampsia. J. Thromb. Haemost. 2009, 7, 375–384. [Google Scholar] [CrossRef]
- Trundley, A.; Moffett, A. Human uterine leucocytes and pregnancy. Tissue Antigens 2004, 63, 1–12. [Google Scholar] [CrossRef]
- Stark, J.M. Pre-eclampsia and cytokine induced oxidative stress. Br. J. Obstet. Gynaecol. 1993, 100, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Dai, A.; Alexenko, A.P.; Liu, Y.; Stephens, A.J.; Schulz, L.C.; Schust, D.J.; Roberts, R.M.; Ezashi, T. Abnormal Oxidative Stress Responses in Fibroblasts from Preeclampsia Infants. PLoS ONE 2014, 9, e103110. [Google Scholar] [CrossRef]
- Furuya, M.; Kurasawa, K.; Nagahama, K.; Kawachi, K.; Nozawa, A.; Takahashi, T.; Aoki, I. Disrupted balance of angiogenic and antiangiogenic signaling in preeclampsia. J. Pregnancy 2011, 2011, 123717. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, R.; Steinthorsdottir, V.; Williams, N.O.; Thorleifsson, G.; Shooter, S.; Hjartardottir, S.; Bumpstead, S.; Stefansdottir, L.; Hildyard, L.; Sigurdsson, J.K.; et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat. Genet. 2017, 49, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.J.; Saxena, R.; Karumanchi, S.A. Genetic predisposition to preeclampsia is conferred by fetal DNA variants near FLT1, a gene involved in the regulation of angiogenesis. Am. J. Obstet. Gynecol. 2018, 218, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- Benschop, L.; Schalekamp-Timmermans, S.; Broere-Brown, Z.A.; Roeters van Lennep, J.E.; Jaddoe, V.W.V.; Roos-Hesselink, J.W.; Kamran Ikram, M.; Steegers, E.A.P.; Roberts, J.M.; Gandley, R.E. Placental Growth Factor as an Indicator of Maternal Cardiovascular Risk After Pregnancy. Circulation 2019, 139, 1698–1709. [Google Scholar] [CrossRef]
- Neuman, R.I.; Figaroa, A.M.J.; Nieboer, D.; Saleh, L.; Verdonk, K.; Danser, A.H.J.; Duvekot, H.J.J.; van den Meiracker, A.H.; van Lennep, J.; Visser, W. Angiogenic markers during preeclampsia: Are they associated with hypertension 1 year postpartum? Pregnancy Hypertens. 2021, 23, 116–122. [Google Scholar] [CrossRef]
- Wilson, S.L.; Blair, J.D.; Hogg, K.; Langlois, S.; von Dadelszen, P.; Robinson, W.P. Placental DNA methylation at term reflects maternal serum levels of INHA and FN1, but not PAPPA, early in pregnancy. BMC Med. Genet. 2015, 16, 111. [Google Scholar] [CrossRef] [Green Version]
- Manokhina, I.; Del Gobbo, G.F.; Konwar, C.; Wilson, S.L.; Robinson, W.P. Placental biomarkers for assessing fetal health. Hum. Mol. Gen. 2017, 26, R237–R245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirollos, S.; Skilton, M.; Patel, S.; Clare Arnott, C. A Systematic Review of Vascular Structure and Function in Pre-eclampsia: Non-invasive Assessment and Mechanistic Links. Front. Cardiovasc. Med. 2019, 15, 166. [Google Scholar] [CrossRef] [PubMed]
- Garovic, V.D.; Milic, N.M.; Weissgerber, T.L.; Mielke, M.M.; Bailey, K.R.; Lahr, B.; Jayachandran, M.; White, W.M.; Hodis, H.N.; Miller, V.M. Carotid artery intima-media thickness and subclinical atherosclerosis in women with remote histories of preeclampsia: Results from a Rochester epidemiology project-based study and meta-analysis. Mayo Clin. Proc. 2017, 92, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Soma-Pillay, P.; Pillay, R.; Wong, T.Y.; Makin, J.D.; Pattinson, R.C. The effect of preeclampsia on retinal microvascular caliber at delivery and post-partum. Obstet. Med. 2018, 11, 116–120. [Google Scholar] [CrossRef]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsdahl, A. Are poor living conditions in childhood and adolescence an important risk factor forarteriosclerotic heart disease? Br. J. Prev. Soc. Med. 1977, 31, 91–95. [Google Scholar] [PubMed] [Green Version]
- Romero, R. Prenatal medicine: The child is the father of the man. 1996. J. Matern. Fetal Neonatal Med. 2009, 22, 636–639. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E.J. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. Br. Med. J. 1989, 298, 576–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.P.; Bull, A.R.; Osmond, C.; Simmones, S.J. Fetal and placental size and risk of hypertension in adult life. Br. Med. J. 1990, 301, 259–262. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Godfrey, K.M.; Osmond, C.; Bull, A. The relation of fetal length, ponderal index and head circumference to blood pressure and the risk hypertension in adult life. Paediatr. Perinat. Epidemiol. 1992, 6, 35–44. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Gluckman, P.D.; Godfrey, K.M.; Harding, J.E.; Owens, J.A.; Robinson, J.S. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993, 341, 938–941. [Google Scholar] [CrossRef]
- Law, C.M.; deSwiet, M.; Osmond, C.; Fayers, P.M.; Barker, D.J.P.; Crucdas, A.M.; Fall, C.H.D. Initiation of hypertension in utero and its amplification throughout life. Br. Med. J. 1993, 306, 24–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, B.M.; Garcia, D.L.; Anderson, S. Glomeruli and blood pressure. Less of one, more the other? Am. J. Hypertens. 1988, 1, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Perico, N.; Somaschini, M.; Manfellotto, D.; Valensise, H.; Cetin, I.; Simeoni, U.; Allegaert, K.; Vikse, B.E.; Steegers, E.A.; et al. A Developmental Approach to the Prevention of Hypertension and Kidney Disease—A report from the Birth Weight and Nephron Number Working Group. Lancet 2017, 390, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, T.V.; Brunetto, S.; Ramos, J.G.; Bernardi, J.R.; Goldani, M.Z. Hypertensive disorders during pregnancy and health outcomes in the offspring: A systematic review. J. Dev. Orig. Health Dis. 2016, 7, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Palti, H.; Rothschild, E. Blood pressure and growth at 6 years of age among offsprings of mothers with hypertension of pregnancy. Early Hum. Dev. 1989, 19, 263–269. [Google Scholar] [CrossRef]
- Seidman, D.S.; Laor, A.; Gale, R.; Stevenson, D.K.; Mashiach, S.; Danon, Y.L. Pre-eclampsia and offspring’s blood pressure, cognitive ability and physical development at 17-years-of-age. Br. J. Obstet. Gynaecol. 1991, 98, 1009–1014. [Google Scholar] [CrossRef]
- Vatten, L.J.; Romundstad, P.R.; Holmen, T.L.; Hsieh, C.C.; Trichopoulos, D.; Stuver, S.O. Intrauterine exposure to preeclampsia and adolescent blood pressure, body size, and age at menarche in female offspring. Obstet. Gynecol. 2003, 101, 529–533. [Google Scholar]
- Mamun, A.A.; Kinarivala, M.K.; O’Callaghan, M.; Williams, G.; Najman, J.; Callaway, L. Does hypertensive disorder of pregnancy predict offspring blood pressure at 21 years? Evidence from a birth cohort study. J. Hum. Hypertens. 2012, 26, 288–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, I.; Peeters, L.L.; Stehouwer, C.D. Preeclampsia and increased blood pressure in the offspring: Meta-analysis and critical review of the evidence. J. Hypertens. 2009, 27, 1955–1959. [Google Scholar] [CrossRef]
- Yu, H.; He, Y.; Mao, Z.; Dong, W.; Fu, X.; Lei, X. Hypertensive disorders during pregnancy and elevated blood pressure in the offspring: A systematical review and meta-analysis protocol. Medicine 2019, 98, e15677. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.P. Fetal origins of coronary heart disease. BMJ 1995, 311, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Tenhola, S.; Rahiala, E.; Martikainen, A.; Halonen, P.; Voutilainen, R. Blood Pressure, Serum Lipids, Fasting Insulin, and Adrenal Hormones in 12-Year-old Children Born with Maternal Preeclampsia. J. Clin. Endocrinol. Metab. 2003, 88, 1217–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timpka, S.; Macdonald-Wallis, C.; Hughes, A.D.; Chaturvedi, N.; Franks, P.W.; Debbie, A.; Lawlor, D.A.; Fraser, A. Hypertensive Disorders of Pregnancy and Offspring Cardiac Structure and Function in Adolescence. J. Am. Heart Assoc. 2016, 5, e003906. [Google Scholar] [CrossRef] [Green Version]
- Golding, J.; ALSPAC Study Team. The Avon Longitudinal Study of Parents and Children. Eur. J. Endocrinol. 2004, 151, U119–U123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmsten, K.; Buka, S.L.; Karin, B.; Michels, K.B. Maternal Pregnancy-Related Hypertension and Risk for Hypertension in Offspring Later in Life. Obstet. Gynecol. 2010, 116, 858–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammad, I.A.; Meeks, H.; Fraser, A.; Theilen, L.H.; Esplin, M.S.; Smith, K.R.; Varner, M.W. Risks of cause-specific mortality in offspring of pregnancies complicated by hypertensive disease of pregnancy. Am. J. Obstet. Gynecol. 2020, 222, e1–e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karatza, A.A.; Dimitriou, G. Preeclampsia Emerging as a Novel Risk Factor for Cardiovascular Disease in the Offspring. Curr. Pediatr. Rev. 2020, 16, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.; Kitt, J.; Leeson, P.; Aye, C.Y.L.; Lewandowski, A.J. Preeclampsia: Risk Factors, Diagnosis, Management, and the Cardiovascular Impact on the Offspring. J. Clin. Med. 2019, 8, 1625. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Garcia, G.; Contag, S. Maternal Preeclampsia and Risk for Cardiovascular Disease in Offspring. Curr. Hypertens. Rep. 2014, 16, 475. [Google Scholar] [CrossRef] [PubMed]
- Lazdam, M.; de la Horra, A.; Pitcher, P.; Mannie, Z.; Diesch, J.; Trevitt, C.; Kylintireas, I.; Contractor, H.; Singhal, A.; Lucas, A.; et al. Elevated Blood Pressure in Offspring Born Premature to Hypertensive Pregnancy. Is Endothelial Dysfunction the Underlying Vascular Mechanism? Hypertension 2010, 56, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giachini, F.R.; Galaviz-Hernandez, C.; Damiano, A.E.; Viana, M.; Cadavid, A.; Asturizaga, P.; Teran, E.; Clapes, S.; Alcala, M.; Bueno, J.; et al. Vascular Dysfunction in Mother and Offspring During Preeclampsia: Contributions from Latin-American Countries. Curr. Hypertens. Rep. 2017, 19, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayet, P.-Y.; Rimoldi, S.F.; Stuber, T.; Salinas Salmòn, C.; Hutter, D.; Rexhaj, E.; Thalmann, S.; Schwab, M.; Turini, P.; Sartori-Cucchia, C.; et al. Pulmonary and Systemic Vascular Dysfunction in Young Offspring of Mothers With Preeclampsia. Circulation 2010, 122, 488–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.-C.; Ying Jiang, Y.; Yang, M.-M.; He, S.-N.; Xi, X.; Xu, Y.T.; Hu, W.-S.; Luo, Q. Hypermethylation of delta-like homolog 1/maternally expressed gene 3 loci in human umbilical veins: Insights into offspring vascular dysfunction born after preeclampsia. J. Hypertens. 2019, 37, 581–589. [Google Scholar] [CrossRef]
- Hromadnikova, I.; Kotlabova, K.; Hympanova, L.; Krofta, L. Cardiovascular and Cerebrovascular Disease Associated microRNAs Are Dysregulated in Placental Tissues Affected with Gestational Hypertension, Preeclampsia and Intrauterine Growth Restriction. PLoS ONE 2015, 10, e0138383. [Google Scholar] [CrossRef] [Green Version]
- Hakim, J.; Senterman, M.K.; Hakim, A.M. Preeclampsia is a biomarker for vascular disease in both mother and child: The need for a medical alert system. Int. J. Pediatr. 2013, 2013, 953150. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, N.; Sharp, G.C.; Reese, S.E.; Vehmeijer, F.O.; Lahti, J.; Page, C.M.; Zhang, W.; Rifas-Shiman, S.L.; Rezwan, F.I.; Simpkin, A.J.; et al. Hypertensive Disorders of Pregnancy and DNA Methylation in Newborns. Findings from the Pregnancy and Childhood Epigenetics Consortium. Hypertension 2019, 74, 375–383. [Google Scholar] [CrossRef]
- Herzog, E.M.; Eggink, A.J.; Reijnierse, A.; Kerkhof, M.A.M.; de Krijger, R.R.; Roks, A.J.M.; Reiss, I.K.M.; Nigg, A.L.; Eilers, P.H.C.; Steegers, E.A.P.; et al. Impact of early- and late-onset preeclampsia on features of placental and newborn vascular health. Placenta 2017, 49, 72–79. [Google Scholar] [CrossRef]
- Henry, A.; Canoy, D. Editorial: Hypertension During Pregnancy and Future Risk of Cardiovascular and Other Long-Term Health Outcomes. Front. Cardiovasc. Med. 2020, 7, 569735. [Google Scholar] [CrossRef]
- Rowley, D.A.; Köhler, H.; Cowan, J.D. An immunologic network. Contemp. Top. Immunobiol. 1980, 9, 205–230. [Google Scholar]
- Harris, L.K.; Benagiano, M.; D’Elios, M.M.; Brosens, I.; Benagiano, G. Placental bed research: 2. Functional and immunological investigations of the placental bed. Am. J. Obstet. Gynecol. 2019, 221, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Lazdam, M.; Davis, E.F.; Lewandowski, A.J.; Worton, S.A.; Kenworthy, Y.; Kelly, B.; Leeson, P. Prevention of Vascular Dysfunction after Preeclampsia: A Potential Long-Term Outcome Measure and an Emerging Goal for Treatment. J. Pregnancy 2012, 2012, 704146. [Google Scholar] [CrossRef]
- Bianco-Miotto, T.; Mayne, B.T.; Buckberry, S.; Breen, J.; Rodriguez Lopez, C.M.; Roberts, C.T. Recent progress towards understanding the role of DNA methylation in human placental development. Reproduction 2016, 152, R23–R30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamble, D.T.; Brikinns, B.; Myint, P.K.; Bhattacharya, S. Hypertensive Disorders of Pregnancy and Subsequent Cardiovascular Disease: Current National and International Guidelines and the Need for Future Research. Front. Cardiovasc. Med. 2019, 6, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pregnancy Outcome | Incidence in Pregnancy % | Risk Factors Shown to be Perturbed after Pregnancy | Association or Risk Ratio (95% Cl) |
|---|---|---|---|
| Gestational diabetes | 1.9–5.0 | Lipids | Increased risk for type 2 diabetes, especially if recurrence of gestational diabetes in a subsequent Pregnancy. No data on Coronary heart disease |
| Blood pressure | |||
| Large vessel function | |||
| Small vessel function | |||
| Preeclampsia (PE) | 2–4 | Lipids | 1.9 (1.0–3.5) vs. pregnancy |
| Clotting | induced hypertension alone | ||
| Fasting insulin | 1.7 (1.3–2.2) vs. no-PE | ||
| Large vessels function | 2.0 (1.5–2.5) vs. no-PE | ||
| Low birth weight (<2500 g) | 5 | Not studied | 11.3 (2.5–36.1) vs. ≥3500 g |
| 7.1 (2.6–18.7) vs. ≥3500 g | |||
| Preterm delivery (<37 weeks) | 5–6 | Not studied | 1.8 (1.3–2.5) vs. term deliv |
| 2.1.(1.2–3.5) vs. term deliv |
| The Following Modifications in Values for the Parameters Investigated Were Observed: | |
|---|---|
| Glucose: | +0.17 mmol/L (95% CI: 0.08–0.25 mmol/L) |
| Insulin: | +3.46 mU/mL (95% CI: 2.34–4.58 mU/mL) |
| Triglycerides: | +0.13 mmol/L (95% CI: 0.05–0.21) |
| Total cholesterol: | +0.22 mmol/L (95% CI: 0.11–0.33 mmol/L) |
| HDL-cholesterol: | −0.11 mmol/L (95% CI: −0.18 to −0.04 mmol/L) |
| LDH-cholesterol: | +0.21 mmol/L (95% CI: 0.10–0.32) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benagiano, M.; Mancuso, S.; Brosens, J.J.; Benagiano, G. Long-Term Consequences of Placental Vascular Pathology on the Maternal and Offspring Cardiovascular Systems. Biomolecules 2021, 11, 1625. https://doi.org/10.3390/biom11111625
Benagiano M, Mancuso S, Brosens JJ, Benagiano G. Long-Term Consequences of Placental Vascular Pathology on the Maternal and Offspring Cardiovascular Systems. Biomolecules. 2021; 11(11):1625. https://doi.org/10.3390/biom11111625
Chicago/Turabian StyleBenagiano, Marisa, Salvatore Mancuso, Jan J. Brosens, and Giuseppe Benagiano. 2021. "Long-Term Consequences of Placental Vascular Pathology on the Maternal and Offspring Cardiovascular Systems" Biomolecules 11, no. 11: 1625. https://doi.org/10.3390/biom11111625