
Pharmacological and Metabolic Significance of Bile Acids in Retinal Diseases

 ,
, 


Abstract
:1. Introduction
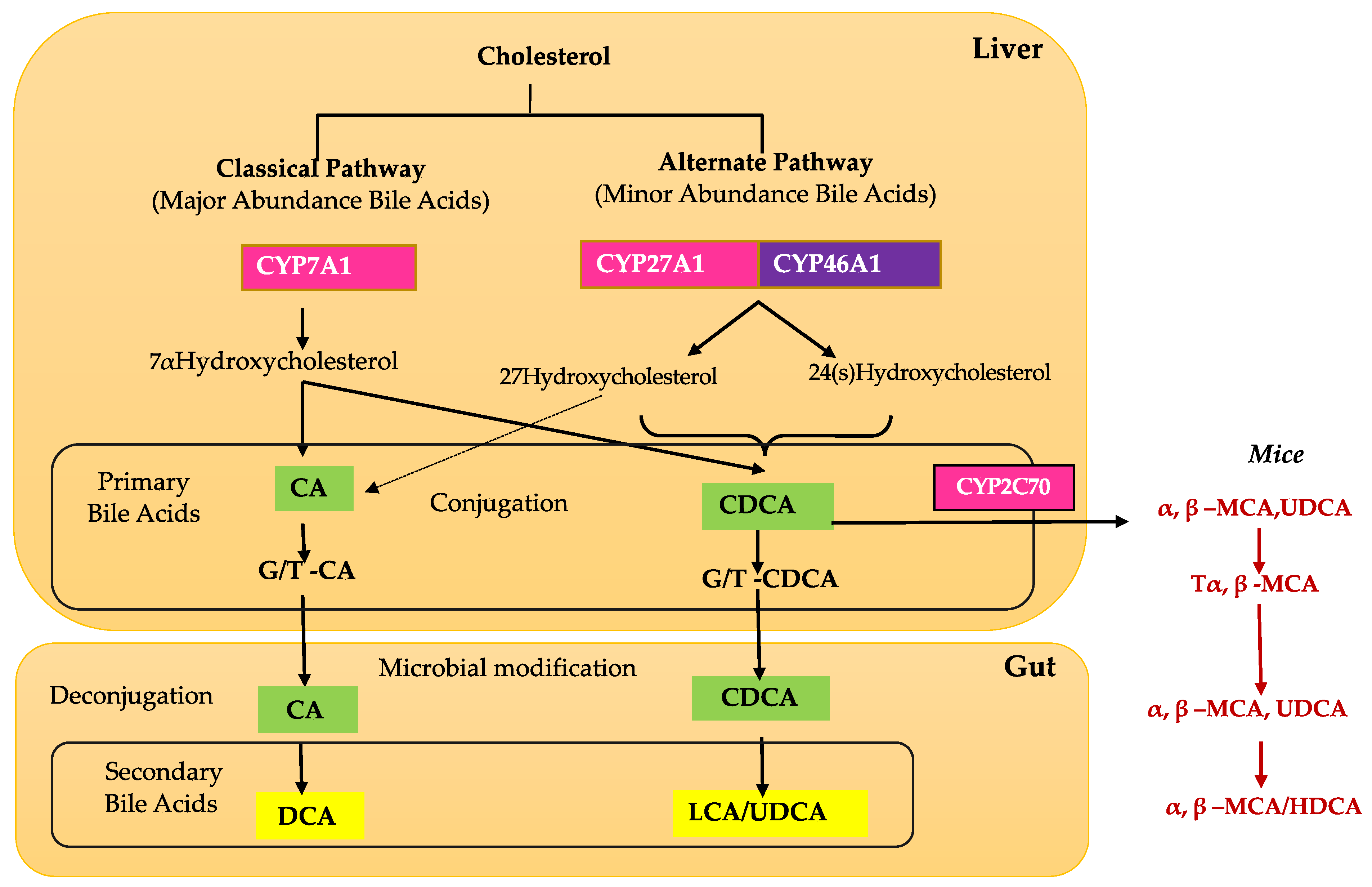
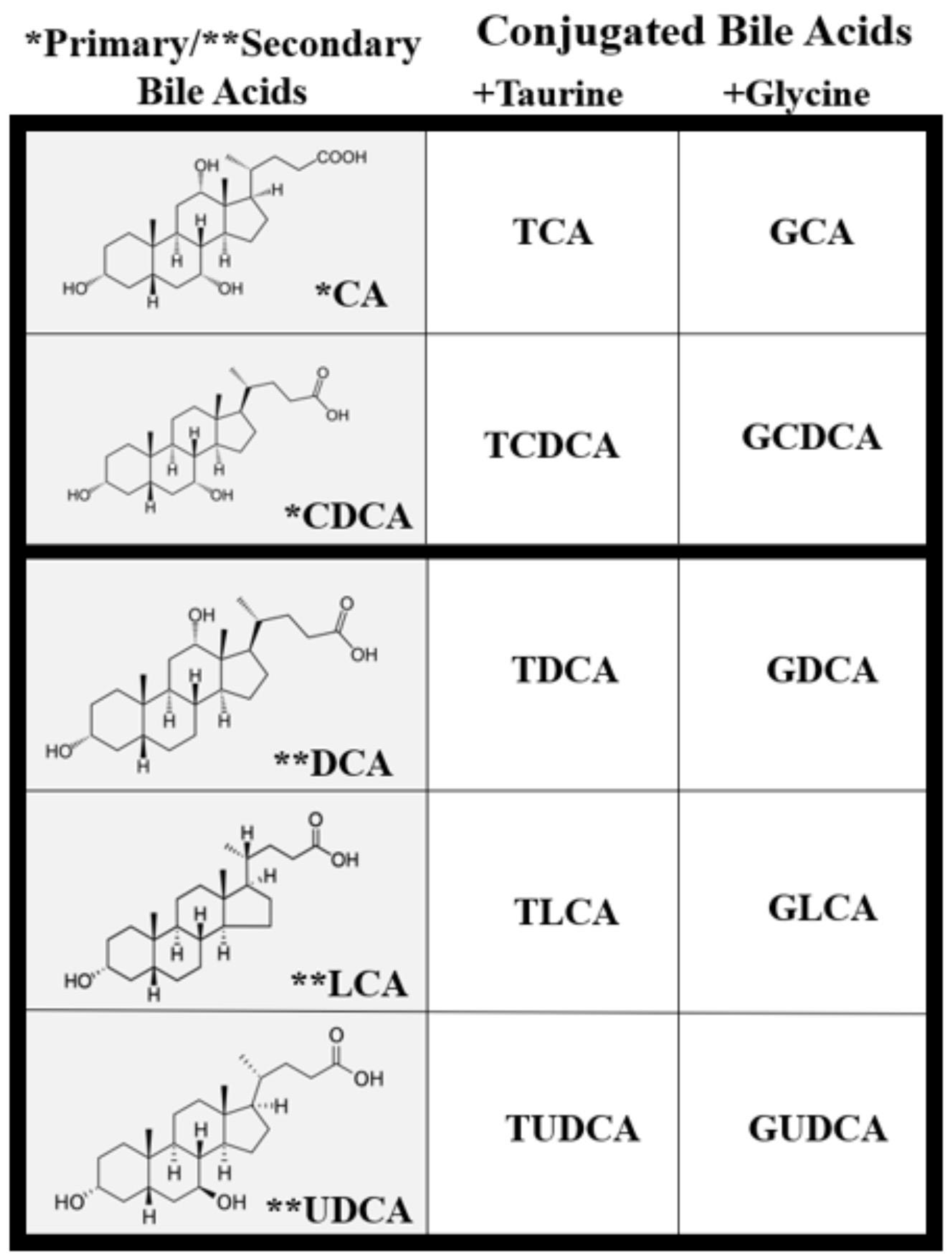
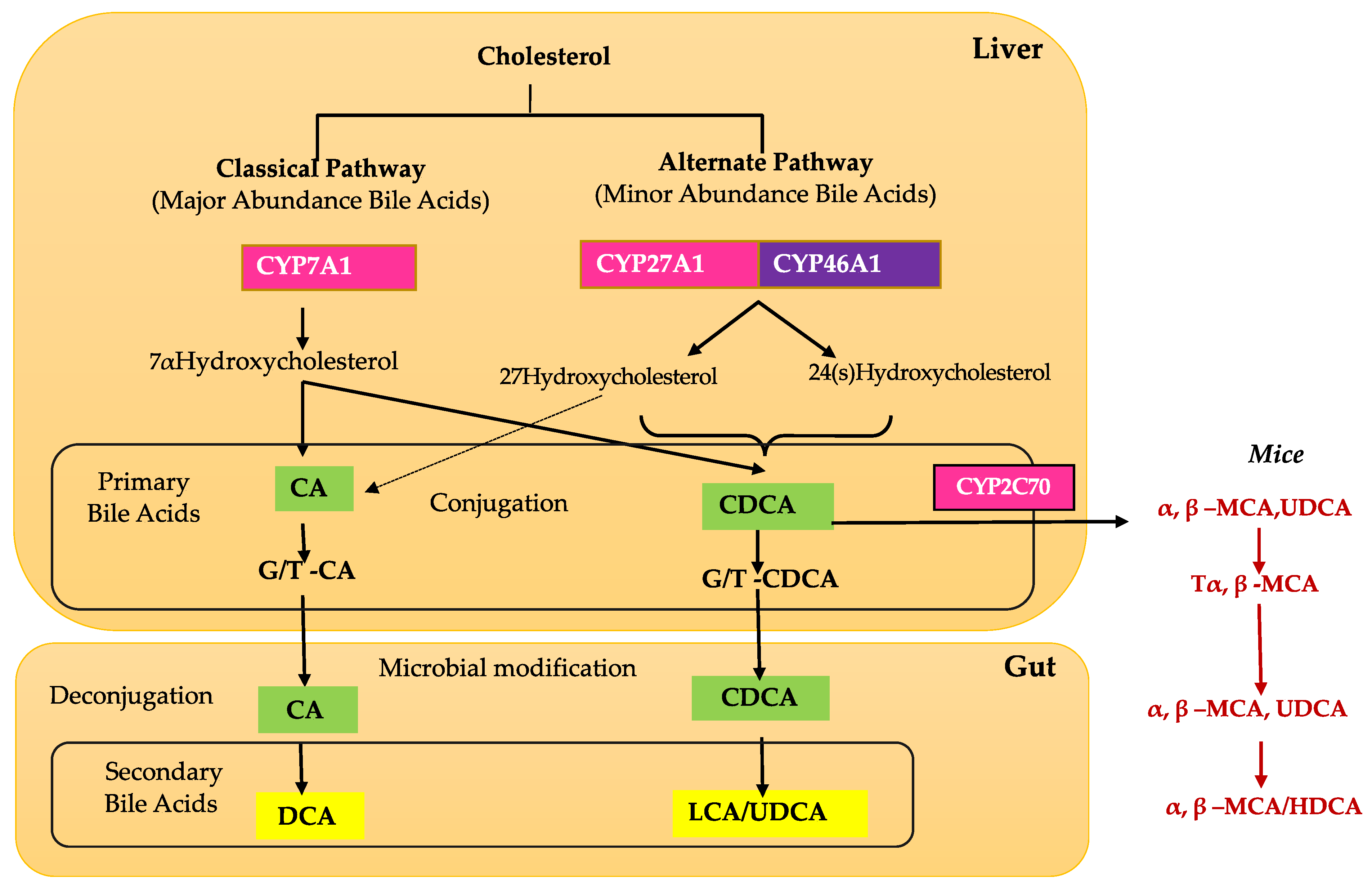
2. Bile Acid Physiology and Metabolism
3. Cytotoxic and Cytoprotective Effects of Bile Acids
4. Retinal Bile Acid Metabolism
5. Bile Acid Transporter and Receptor in the Retina
6. Pharmacological Effects of Bile Acids in Ocular Disease

{kind=link}
{kind=link}
| Ocular Disease | Bile Acid /Agonist | Dose | Findings | References |
|---|---|---|---|---|
| Leber congenital amaurosis | TUDCA | Systemic injection; 500 mg/kg b.w./3 days | TUDCA is a potential agent in reducing ER stress, to prevent apoptosis, and preserve cones in the LCA model | [61] |
| Retinal detachment | TUDCA | Intraperitoneal injection; 500 mg/kg b.w. | TUDCA preserves photoreceptors after retinal detachment, inhibits caspase activity, and reduces ER and oxidative stress | [67] |
| Cataracts | TUDCA | Subcutaneous injection; 500 mg/kg body weight (b.w.)/day | TUDCA treatment alleviates cataract formation via the UPR-dependent pathway | [68] |
| Oxidative stress-induced retinal degeneration | TUDCA | Intraperitoneal injection; 500 mg/kg every 3 days | TUDCA produces modest preservation of outer nuclear layer thickness and rod function at P30. And significant preservation of cone cell number and cone function at P50 | [74] |
| Retinal degeneration | TUDCA | Subcutaneous injection; 500 mg/kg b.w. | TUDCA greatly slowed retinal degeneration in LIRD, and rd10 mice protected photoreceptor and suppressed apoptosis | [75] |
| Rpgr-Associated Retinitis pigmentosa | TUDCA | Intraperitoneal injection; 500 mg/kg b.w. | TUDCA suppresses microglial activation, inhibits inflammation, and prevents photoreceptor degeneration | [79] |
| Retinal degeneration | TUDCA | Systemically injected; 500 mg/kg every 3 days from P6 to P30 | TUDCA-treated rd10 retinas had fivefold more photoreceptors than vehicle-treated retinas. TUDCA treatments did not alter the retinal function or morphology of wild-type mice when administered to age-matched mice. | [80] |
| Retinal degeneration | TUDCA | Subcutaneous injection; 500 mg/kg b.w./ day | TUDCA treatment: reduces caspase 3 activation and apoptosis, slows the loss of photoreceptors and retinal function, and delays retinal damage | [83] |
| Retinal degeneration | TUDCA | Subcutaneous injection; 500 mg/kg b.w. | TUDCA inhibits photoreceptor degeneration and decreases visual impairments. TUDCA rectifies abnormalities in visual signal transmission | [84] |
| Diabetic Retinopathy | UDCA | Oral delivery, intragastric administration; 30 mg/kg b.w. | UDCA reverses the breakdown of the blood-retinal barrier and reduces retinal inflammation | [87] |
| Diabetic Retinopathy | UDCA | Intraperitoneal injection; 100 mg/kg/d b.w. | vascular integrity was improved and pericyte loss reduced in the retina of STZ-induced diabetic mice | [92] |
| Diabetic Retinopathy | UDCA | Subcutaneous injection / daily for P7-9 neonates; 100 mg/kg. | UDCA reduced the increased expression of angiogenic factors and inflammatory mediators (vascular endothelial growth factor, intercellular adhesion molecule-1, and monocyte chemotactic protein-1 | [93] |
| Diabetic Retinopathy | INT-777 (semisynthetic bile acid) a TGR5 agonist | 50 ng/μL, 5 μL was injected into the vitreous cavity | Upregulation or activation of TGR5 may inhibit RhoA/ROCK-dependent actin remodeling and represent an important therapeutic intervention for DR. | [96] |
| Choroidal neovascularization (CNV) | UDCA TUDCA | Intraperitoneal injection; UDCA 500 mg/kg, TUDCA 100 mg/kg, | The systemic administration of UDCA and TUDCA suppressed laser-induced CNV formation | [100] |
| Choroidal neovascularization (CNV) | UDCA | Oral delivery; 125 or 250 mg/kg b.w./day | UDCA inhibits CNV and promotes functional recovery in mice retinas | [101] |
| Retinopathy of prematurity | UDCA TUDCA GUDCA | Intraperitoneal injection; 50 mg/kg | UDCA decreased the extension of neovascular and avascular areas, whereas treatments with TUDCA and GUDCA showed no changes. UDCA also prevented reactive gliosis, preserved ganglion cell survival, and ameliorated OIR-induced blood-retinal barrier dysfunction. | [104] |
7. Influence of Changes in Gut Microbiota on BA Circulation and Retinal Diseases
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.Q.; Carey, M.C. Therapeutic uses of animal biles in traditional Chinese medicine: An ethnopharmacological, biophysical chemical and medicinal review. World J. Gastroenterol. 2014, 20, 9952–9975. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Hong, M.; Li, L.; Cheung, F.; Feng, Y. Substitutes for Bear Bile for the Treatment of Liver Diseases: Research Progress and Future Perspective. Evid. Based. Complement. Alternat. Med. 2016, 2016, 4305074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Siu, K.; Wang, N.; Ng, K.M.; Tsao, S.W.; Nagamatsu, T.; Tong, Y. Bear bile: Dilemma of traditional medicinal use and animal protection. J. Ethnobiol. Ethnomed. 2009, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusaczuk, M. Tauroursodeoxycholate-Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, D.; Arab, J.P.; Arrese, M. UDCA, NorUDCA, and TUDCA in Liver Diseases: A Review of Their Mechanisms of Action and Clinical Applications. Handb. Exp. Pharmacol. 2019, 256, 237–264. [Google Scholar] [CrossRef]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Ethanic, M.; Stanimirov, B.; Pavlovic, N.; Golocorbin-Kon, S.; Al-Salami, H.; Stankov, K.; Mikov, M. Pharmacological Applications of Bile Acids and Their Derivatives in the Treatment of Metabolic Syndrome. Front. Pharmacol. 2018, 9, 1382. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Hagey, L.R. Key discoveries in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, J.D.; Guo, G.L. Pharmacologic Modulation of Bile Acid-FXR-FGF15/FGF19 Pathway for the Treatment of Nonalcoholic Steatohepatitis. Handb. Exp. Pharmacol. 2019, 256, 325–357. [Google Scholar] [CrossRef]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int J. Mol. Sci. 2018, 19, 69. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.H.; Carey, E.J.; Lindor, K.D. Recent advances in the development of farnesoid X receptor agonists. Ann. Transl. Med. 2015, 3, 5. [Google Scholar] [CrossRef]
- Staels, B.; Fonseca, V.A. Bile acids and metabolic regulation: Mechanisms and clinical responses to bile acid sequestration. Diabetes Care 2009, 32 (Suppl. 2), S237–S245. [Google Scholar] [CrossRef] [Green Version]
- Kiriyama, Y.; Nochi, H. The Biosynthesis, Signaling, and Neurological Functions of Bile Acids. Biomolecules 2019, 9, 232. [Google Scholar] [CrossRef] [Green Version]
- Mertens, K.L.; Kalsbeek, A.; Soeters, M.R.; Eggink, H.M. Bile Acid Signaling Pathways from the Enterohepatic Circulation to the Central Nervous System. Front. Neurosci. 2017, 11, 617. [Google Scholar] [CrossRef] [Green Version]
- Pols, T.W.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J. Hepatol. 2011, 54, 1263–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, H.; Kolodziejczyk, A.A.; Halstuch, D.; Elinav, E. Bile acids in glucose metabolism in health and disease. J. Exp. Med. 2018, 215, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Insull, W., Jr. Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: A scientific review. South. Med. J. 2006, 99, 257–273. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Borgstroem, B. The Intraluminal Phase of Fat Digestion in Man: The Lipid Content of the Micellar and Oil Phases of Intestinal Content Obtained during Fat Digestion and Absorption. J. Clin. Investig. 1964, 43, 247–257. [Google Scholar] [CrossRef]
- McMillin, M.; DeMorrow, S. Effects of bile acids on neurological function and disease. FASEB J. 2016, 30, 3658–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daruich, A.; Picard, E.; Boatright, J.H.; Behar-Cohen, F. Review: The bile acids urso- and tauroursodeoxycholic acid as neuroprotective therapies in retinal disease. Mol. Vis. 2019, 25, 610–624. [Google Scholar]
- Li, T.; Chiang, J.Y. Bile Acid signaling in liver metabolism and diseases. J. Lipids 2012, 2012, 754067. [Google Scholar] [CrossRef] [Green Version]
- Myant, N.B.; Mitropoulos, K.A. Cholesterol 7 alpha-hydroxylase. J. Lipid Res. 1977, 18, 135–153. [Google Scholar] [CrossRef]
- Schwarz, M.; Russell, D.W.; Dietschy, J.M.; Turley, S.D. Alternate pathways of bile acid synthesis in the cholesterol 7alpha-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. J. Lipid Res. 2001, 42, 1594–1603. [Google Scholar] [CrossRef]
- Priamukhina, N.S.; Semina, N.A. The differentiation of intestinal Escherichia coli infections. Zhurnal Mikrobiol. Epidemiol. Immunobiol. 1991, 2, 81–87. [Google Scholar]
- Hofmann, A.F. The enterohepatic circulation of bile acids in man. Clin. Gastroenterol. 1977, 6, 3–24. [Google Scholar]
- Urdaneta, V.; Casadesus, J. Interactions between Bacteria and Bile Salts in the Gastrointestinal and Hepatobiliary Tracts. Front. Med. 2017, 4, 163. [Google Scholar] [CrossRef]
- Long, S.L.; Gahan, C.G.M.; Joyce, S.A. Interactions between gut bacteria and bile in health and disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Hill, C.; Gahan, C.G. Bile salt hydrolase activity in probiotics. Appl. Environ. Microbiol 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016, 7, 22–39. [Google Scholar] [CrossRef] [Green Version]
- Dawson, P.A. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb. Exp. Pharmacol. 2011, 169–203. [Google Scholar] [CrossRef] [Green Version]
- Voronova, V.; Sokolov, V.; Al-Khaifi, A.; Straniero, S.; Kumar, C.; Peskov, K.; Helmlinger, G.; Rudling, M.; Angelin, B. A Physiology-Based Model of Bile Acid Distribution and Metabolism Under Healthy and Pathologic Conditions in Human Beings. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 149–170. [Google Scholar] [CrossRef] [Green Version]
- Reinicke, M.; Schroter, J.; Muller-Klieser, D.; Helmschrodt, C.; Ceglarek, U. Free oxysterols and bile acids including conjugates - Simultaneous quantification in human plasma and cerebrospinal fluid by liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta. 2018, 1037, 245–255. [Google Scholar] [CrossRef]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef]
- Araki, Y.; Andoh, A.; Bamba, H.; Yoshikawa, K.; Doi, H.; Komai, Y.; Higuchi, A.; Fujiyama, Y. The cytotoxicity of hydrophobic bile acids is ameliorated by more hydrophilic bile acids in intestinal cell lines IEC-6 and Caco-2. Oncol. Rep. 2003, 10, 1931–1936. [Google Scholar] [CrossRef]
- Benedetti, A.; Alvaro, D.; Bassotti, C.; Gigliozzi, A.; Ferretti, G.; La Rosa, T.; Di Sario, A.; Baiocchi, L.; Jezequel, A.M. Cytotoxicity of bile salts against biliary epithelium: A study in isolated bile ductule fragments and isolated perfused rat liver. Hepatology 1997, 26, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Godschalk, M. Activity of single neurons in monkey cortex preceding sensory cued limb movements. Electroencephalogr. Clin. Neurophysiol. Suppl. 1991, 42, 71–79. [Google Scholar]
- Monte, M.J.; Marin, J.J.; Antelo, A.; Vazquez-Tato, J. Bile acids: Chemistry, physiology, and pathophysiology. World J. Gastroenterol. 2009, 15, 804–816. [Google Scholar] [CrossRef]
- Khurana, S.; Raufman, J.P.; Pallone, T.L. Bile acids regulate cardiovascular function. Clin. Transl. Sci. 2011, 4, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhu, C.; Shao, L.; Ye, J.; Shen, Y.; Ren, Y. Role of Bile Acids in Dysbiosis and Treatment of Nonalcoholic Fatty Liver Disease. Mediat. Inflamm. 2019, 2019, 7659509. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Dorko, K.; Antoine, D.J.; Clarke, J.I.; Gholami, P.; Li, F.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Fan, F.; et al. Bile acid-induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol. Appl. Pharmacol. 2015, 283, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mello-Vieira, J.; Sousa, T.; Coutinho, A.; Fedorov, A.; Lucas, S.D.; Moreira, R.; Castro, R.E.; Rodrigues, C.M.; Prieto, M.; Fernandes, F. Cytotoxic bile acids, but not cytoprotective species, inhibit the ordering effect of cholesterol in model membranes at physiologically active concentrations. Biochim. Biophys. Acta 2013, 1828, 2152–2163. [Google Scholar] [CrossRef] [Green Version]
- Palmela, I.; Correia, L.; Silva, R.F.; Sasaki, H.; Kim, K.S.; Brites, D.; Brito, M.A. Hydrophilic bile acids protect human blood-brain barrier endothelial cells from disruption by unconjugated bilirubin: An in vitro study. Front. Neurosci. 2015, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, J.W.; Silbert, P.L. Zeta waves: A distinctive type of intermittent delta wave studied prospectively. Clin. Exp. Neurol. 1991, 28, 238–243. [Google Scholar]
- Hofmann, A.F. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef]
- Omarova, S.; Charvet, C.D.; Reem, R.E.; Mast, N.; Zheng, W.; Huang, S.; Peachey, N.S.; Pikuleva, I.A. Abnormal vascularization in mouse retina with dysregulated retinal cholesterol homeostasis. J. Clin. Investig. 2012, 122, 3012–3023. [Google Scholar] [CrossRef] [Green Version]
- Saadane, A.; Mast, N.; Charvet, C.D.; Omarova, S.; Zheng, W.; Huang, S.S.; Kern, T.S.; Peachey, N.S.; Pikuleva, I.A. Retinal and nonocular abnormalities in Cyp27a1(-/-)Cyp46a1(-/-) mice with dysfunctional metabolism of cholesterol. Am. J. Pathol. 2014, 184, 2403–2419. [Google Scholar] [CrossRef] [Green Version]
- Bretillon, L.; Diczfalusy, U.; Bjorkhem, I.; Maire, M.A.; Martine, L.; Joffre, C.; Acar, N.; Bron, A.; Creuzot-Garcher, C. Cholesterol-24S-hydroxylase (CYP46A1) is specifically expressed in neurons of the neural retina. Curr. Eye Res. 2007, 32, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.T.; Abbas, S.; Chandra, A.; Singh, L.; Rizvi, S.; Mahdi, F. Association of angiotensin-converting enzyme, CYP46A1 genes polymorphism with senile cataract. Oman J. Ophthalmol. 2017, 10, 21–25. [Google Scholar] [CrossRef]
- Geyer, J.; Fernandes, C.F.; Doring, B.; Burger, S.; Godoy, J.R.; Rafalzik, S.; Hubschle, T.; Gerstberger, R.; Petzinger, E. Cloning and molecular characterization of the orphan carrier protein Slc10a4: Expression in cholinergic neurons of the rat central nervous system. Neuroscience 2008, 152, 990–1005. [Google Scholar] [CrossRef]
- Geyer, J.; Wilke, T.; Petzinger, E. The solute carrier family SLC10: More than a family of bile acid transporters regarding function and phylogenetic relationships. Naunyn Schmied. Arch. Pharmacol. 2006, 372, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Beli, E.; Yan, Y.; Moldovan, L.; Vieira, C.P.; Gao, R.; Duan, Y.; Prasad, R.; Bhatwadekar, A.; White, F.A.; Townsend, S.D.; et al. Restructuring of the Gut Microbiome by Intermittent Fasting Prevents Retinopathy and Prolongs Survival in db/db Mice. Diabetes 2018, 67, 1867–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobysheva, E.; Taylor, C.M.; Marshall, G.R.; Kisselev, O.G. Tauroursodeoxycholic acid binds to the G-protein site on light activated rhodopsin. Exp. Eye Res. 2018, 170, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Murase, H.; Tsuruma, K.; Shimazawa, M.; Hara, H. TUDCA Promotes Phagocytosis by Retinal Pigment Epithelium via MerTK Activation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2511–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.J.; Wang, L. Bile Acid-Activated Receptors: A Review on FXR and Other Nuclear Receptors. Handb. Exp. Pharmacol. 2019, 256, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [Green Version]
- Duane, W.C.; Javitt, N.B. 27-hydroxycholesterol: Production rates in normal human subjects. J. Lipid Res. 1999, 40, 1194–1199. [Google Scholar] [CrossRef]
- Pardue, M.T.; Allen, R.S. Neuroprotective strategies for retinal disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef]
- Ackerman, H.D.; Gerhard, G.S. Bile Acids in Neurodegenerative Disorders. Front. Aging Neurosci. 2016, 8, 263. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Sanchez, L.; Lax, P.; Noailles, A.; Angulo, A.; Maneu, V.; Cuenca, N. Natural Compounds from Saffron and Bear Bile Prevent Vision Loss and Retinal Degeneration. Molecules 2015, 20, 13875–13893. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, J.M.; Martins, A.; Cruz, R.; Rodrigues, C.M.; Ambrosio, A.F.; Santiago, A.R. Tauroursodeoxycholic acid protects retinal neural cells from cell death induced by prolonged exposure to elevated glucose. Neuroscience 2013, 253, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Baehr, W.; Fu, Y. Chemical chaperone TUDCA preserves cone photoreceptors in a mouse model of Leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3349–3356. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Notomi, S.; Hisatomi, T.; Nakazawa, T.; Ishibashi, T.; Miller, J.W.; Vavvas, D.G. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog. Retin. Eye Res. 2013, 37, 114–140. [Google Scholar] [CrossRef] [Green Version]
- Park, K.H.; Hwang, J.M.; Choi, M.Y.; Yu, Y.S.; Chung, H. Retinal detachment of regressed retinopathy of prematurity in children aged 2 to 15 years. Retina 2004, 24, 368–375. [Google Scholar] [CrossRef]
- Dunaief, J.L.; Dentchev, T.; Ying, G.S.; Milam, A.H. The role of apoptosis in age-related macular degeneration. Arch. Ophthalmol. 2002, 120, 1435–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Cook, B.; Lewis, G.P.; Fisher, S.K.; Adler, R. Apoptotic photoreceptor degeneration in experimental retinal detachment. Investig. Ophthalmol. Vis. Sci. 1995, 36, 990–996. [Google Scholar]
- Mantopoulos, D.; Murakami, Y.; Comander, J.; Thanos, A.; Roh, M.; Miller, J.W.; Vavvas, D.G. Tauroursodeoxycholic acid (TUDCA) protects photoreceptors from cell death after experimental retinal detachment. PLoS ONE 2011, 6, e24245. [Google Scholar] [CrossRef]
- Mulhern, M.L.; Madson, C.J.; Kador, P.F.; Randazzo, J.; Shinohara, T. Cellular osmolytes reduce lens epithelial cell death and alleviate cataract formation in galactosemic rats. Mol. Vis. 2007, 13, 1397–1405. [Google Scholar]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell. Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012, 72, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Enemchukwu, N.; Zhong, X.; Zhang, O.; Fu, Y. Deletion of M-Opsin Prevents M Cone Degeneration in a Mouse Model of Leber Congenital Amaurosis. Am. J. Pathol. 2020, 190, 1059–1067. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, T. Pathophysilogical mechanism and treatment strategies for Leber congenital amaurosis. Adv. Exp. Med. Biol. 2014, 801, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, N.; Baehr, W.; Fu, Y. Cone opsin determines the time course of cone photoreceptor degeneration in Leber congenital amaurosis. Proc. Natl. Acad. Sci. USA 2011, 108, 8879–8884. [Google Scholar] [CrossRef] [Green Version]
- Oveson, B.C.; Iwase, T.; Hackett, S.F.; Lee, S.Y.; Usui, S.; Sedlak, T.W.; Snyder, S.H.; Campochiaro, P.A.; Sung, J.U. Constituents of bile, bilirubin and TUDCA, protect against oxidative stress-induced retinal degeneration. J. Neurochem. 2011, 116, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Boatright, J.H.; Moring, A.G.; McElroy, C.; Phillips, M.J.; Do, V.T.; Chang, B.; Hawes, N.L.; Boyd, A.P.; Sidney, S.S.; Stewart, R.E.; et al. Tool from ancient pharmacopoeia prevents vision loss. Mol. Vis. 2006, 12, 1706–1714. [Google Scholar]
- O’Neal, T.B.; Luther, E.E. Retinitis Pigmentosa. In StatPearls; Statpearls Publishing: San Francisco, CA, USA, 2020. [Google Scholar]
- Narayan, D.S.; Wood, J.P.; Chidlow, G.; Casson, R.J. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016, 94, 748–754. [Google Scholar] [CrossRef]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Zhang, X.; Shahani, U.; Reilly, J.; Shu, X. Disease mechanisms and neuroprotection by tauroursodeoxycholic acid in Rpgr knockout mice. J. Cell Physiol. 2019, 234, 18801–18812. [Google Scholar] [CrossRef]
- Phillips, M.J.; Walker, T.A.; Choi, H.Y.; Faulkner, A.E.; Kim, M.K.; Sidney, S.S.; Boyd, A.P.; Nickerson, J.M.; Boatright, J.H.; Pardue, M.T. Tauroursodeoxycholic acid preservation of photoreceptor structure and function in the rd10 mouse through postnatal day 30. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2148–2155. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.; Hawes, N.L.; Pardue, M.T.; German, A.M.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Rengarajan, K.; Boyd, A.P.; Sidney, S.S.; et al. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vis. Res. 2007, 47, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.; Fishman, G.A.; Anderson, R.J.; Tozatti, M.S.; Heckenlively, J.R.; Weleber, R.G.; Edwards, A.O.; Brown, J., Jr. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology 1999, 106, 1780–1785. [Google Scholar] [CrossRef]
- Boatright, J.H.; Nickerson, J.M.; Moring, A.G.; Pardue, M.T. Bile acids in treatment of ocular disease. J. Ocul. Biol. Dis. Inform. 2009, 2, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Dong, X.; Lu, X.; Qu, Y.; Wang, C.; Peng, G.; Zhang, J. Subcutaneous delivery of tauroursodeoxycholic acid rescues the cone photoreceptors in degenerative retina: A promising therapeutic molecule for retinopathy. Biomed. Pharmacother. 2019, 117, 109021. [Google Scholar] [CrossRef]
- Shukla, U.V.; Tripathy, K. Diabetic Retinopathy. In StatPearls; Statpearls Publishing: San Francisco, CA, USA, 2020. [Google Scholar]
- Kastelan, S.; Oreskovic, I.; Biscan, F.; Kastelan, H.; Gverovic Antunica, A. Inflammatory and angiogenic biomarkers in diabetic retinopathy. Biochem. Med. 2020, 30, 030502. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Mei, X.; Zhang, T.; Lu, B.; Ji, L. Ursodeoxycholic acid ameliorates diabetic retinopathy via reducing retinal inflammation and reversing the breakdown of blood-retinal barrier. Eur. J. Pharmacol. 2018, 840, 20–27. [Google Scholar] [CrossRef]
- Park, D.Y.; Lee, J.; Kim, J.; Kim, K.; Hong, S.; Han, S.; Kubota, Y.; Augustin, H.G.; Ding, L.; Kim, J.W.; et al. Plastic roles of pericytes in the blood-retinal barrier. Nat. Commun. 2017, 8, 15296. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Ejaz, S.; Chekarova, I.; Ejaz, A.; Sohail, A.; Lim, C.W. Importance of pericytes and mechanisms of pericyte loss during diabetes retinopathy. Diabetes Obes. Metab. 2008, 10, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P. Pericytes and the pathogenesis of diabetic retinopathy. Horm. Metab. Res. 2005, 37 (Suppl. 1), 39–43. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.R.; Choi, J.A.; Koh, J.Y.; Yoon, Y.H. Ursodeoxycholic Acid Attenuates Endoplasmic Reticulum Stress-Related Retinal Pericyte Loss in Streptozotocin-Induced Diabetic Mice. J. Diabetes Res. 2017, 2017, 1763292. [Google Scholar] [CrossRef]
- Shiraya, T.; Araki, F.; Ueta, T.; Fukunaga, H.; Totsuka, K.; Arai, T.; Uemura, A.; Moriya, K.; Kato, S. Ursodeoxycholic Acid Attenuates the Retinal Vascular Abnormalities in Anti-PDGFR-beta Antibody-Induced Pericyte Depletion Mouse Models. Sci. Rep. 2020, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Aung, M.H.; Kim, M.K.; Olson, D.E.; Thule, P.M.; Pardue, M.T. Early visual deficits in streptozotocin-induced diabetic long evans rats. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1370–1377. [Google Scholar] [CrossRef]
- Kohzaki, K.; Vingrys, A.J.; Bui, B.V. Early inner retinal dysfunction in streptozotocin-induced diabetic rats. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3595–3604. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, W.; Xie, T.H.; Zou, J.; Nie, X.; Wang, X.; Zhang, M.Y.; Wang, Z.Y.; Gu, S.; Zhuang, M.; et al. TGR5 receptor activation attenuates diabetic retinopathy through suppression of RhoA/ROCK signaling. FASEB J. 2020, 34, 4189–4203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Yonekawa, Y.; Kim, I.K. Clinical characteristics and current treatment of age-related macular degeneration. Cold Spring Harb. Perspect Med. 2014, 5, a017178. [Google Scholar] [CrossRef] [Green Version]
- Gehrs, K.M.; Anderson, D.H.; Johnson, L.V.; Hageman, G.S. Age-related macular degeneration--emerging pathogenetic and therapeutic concepts. Ann. Med. 2006, 38, 450–471. [Google Scholar] [CrossRef]
- Woo, S.J.; Kim, J.H.; Yu, H.G. Ursodeoxycholic acid and tauroursodeoxycholic acid suppress choroidal neovascularization in a laser-treated rat model. J. Ocul. Pharmacol. Ther. 2010, 26, 223–229. [Google Scholar] [CrossRef]
- Maharjan, P.; Kim, D.; Jin, M.; Ko, H.J.; Song, Y.H.; Lee, Y.; Ahn, B.N.; Kim, S.K.; Lee, Y.; Shin, M.C.; et al. Preclinical Evaluation of UDCA-Containing Oral Formulation in Mice for the Treatment of Wet Age-Related Macular Degeneration. Pharmaceutics 2019, 11, 561. [Google Scholar] [CrossRef] [Green Version]
- Graziosi, A.; Perrotta, M.; Russo, D.; Gasparroni, G.; D’Egidio, C.; Marinelli, B.; Di Marzio, G.; Falconio, G.; Mastropasqua, L.; Li Volti, G.; et al. Oxidative Stress Markers and the Retinopathy of Prematurity. J. Clin. Med. 2020, 9, 2711. [Google Scholar] [CrossRef] [PubMed]
- Vahatupa, M.; Jarvinen, T.A.H.; Uusitalo-Jarvinen, H. Exploration of Oxygen-Induced Retinopathy Model to Discover New Therapeutic Drug Targets in Retinopathies. Front. Pharmacol. 2020, 11, 873. [Google Scholar] [CrossRef] [PubMed]
- Thounaojam, M.C.; Jadeja, R.N.; Rajpurohit, S.; Gutsaeva, D.R.; Stansfield, B.K.; Martin, P.M.; Bartoli, M. Ursodeoxycholic Acid Halts Pathological Neovascularization in a Mouse Model of Oxygen-Induced Retinopathy. J. Clin. Med. 2020, 9, 1921. [Google Scholar] [CrossRef]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Nunez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Hills, R.D.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef] [Green Version]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef] [Green Version]
- Joyce, S.A.; MacSharry, J.; Casey, P.G.; Kinsella, M.; Murphy, E.F.; Shanahan, F.; Hill, C.; Gahan, C.G. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. USA 2014, 111, 7421–7426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, A.; Dumonceau, J.M.; Lescuyer, P. Proteomic analysis of human bile and potential applications for cancer diagnosis. Expert Rev. Proteom. 2009, 6, 285–301. [Google Scholar] [CrossRef]
- Kriaa, A.; Bourgin, M.; Potiron, A.; Mkaouar, H.; Jablaoui, A.; Gerard, P.; Maguin, E.; Rhimi, M. Microbial impact on cholesterol and bile acid metabolism: Current status and future prospects. J. Lipid Res. 2019, 60, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinero, N.; Ruiz, L.; Sanchez, B.; Margolles, A.; Delgado, S. Intestinal Bacteria Interplay With Bile and Cholesterol Metabolism: Implications on Host Physiology. Front. Physiol. 2019, 10, 185. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentella, M.C.; Scaldaferri, F.; Pizzoferrato, M.; Gasbarrini, A.; Miggiano, G.A.D. Nutrition, IBD and Gut Microbiota: A Review. Nutrients 2020, 12, 944. [Google Scholar] [CrossRef] [Green Version]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel. Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Ferreira, C.M.; Vieira, A.T.; Vinolo, M.A.; Oliveira, F.A.; Curi, R.; Martins Fdos, S. The central role of the gut microbiota in chronic inflammatory diseases. J. Immunol. Res. 2014, 2014, 689492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yi, G.; Peng, H.; Li, Z.; Chen, S.; Zhong, H.; Chen, Y.; Wang, Z.; Deng, Q.; Fu, M. How Ocular Surface Microbiota Debuts in Type 2 Diabetes Mellitus. Front. Cell Infect. Microbiol. 2019, 9, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
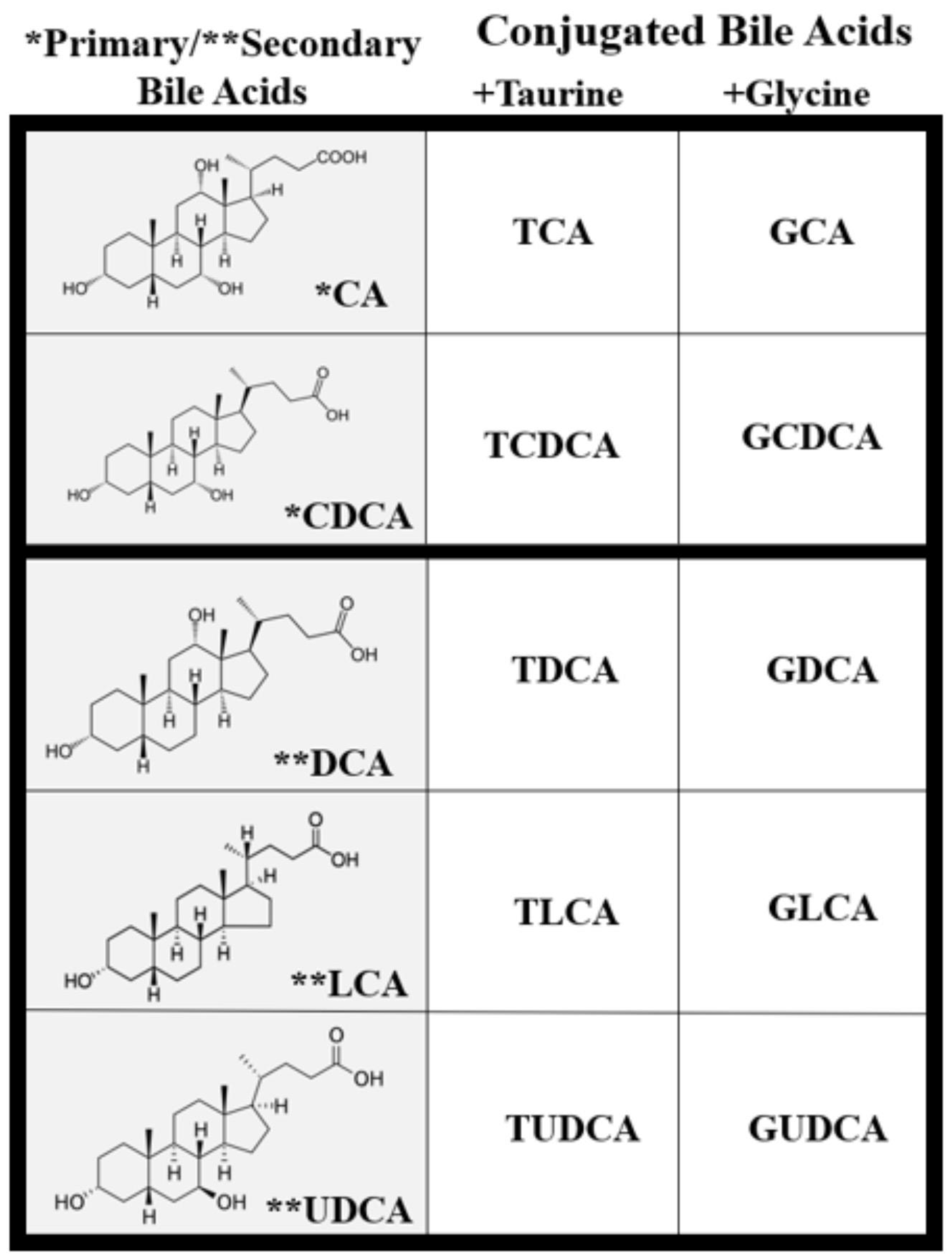
| Nuclear Receptors | Affinity | Location |
|---|---|---|
| FXR | CDCA, CA, LCA, DCA | Liver, intestine, brain |
| PXR | LCA, CDCA | Liver, intestine, brain, retina (RPE cells) |
| VDR | CDCA, CA, LCA | Intestine, brain retina, kidney, retina, bone |
| GR | UDCA TCA GDCA TUDCA | Liver, brain, retina |
| CAR | LCA | Liver, brain, kidney, adrenal |
| Membrane Receptor | Affinity | Location |
| TGR5 | LCA, DCA, CDCA, CA | Liver, intestine, brain, eye (primary retina ganglion cells) spleen, lung, monocytes |
| S1PR2 | TCA, DCA, TDCA, GDCA, TUDCA | Liver, brain, eye, lung, ear |
| α5β1 | TUDCA | Liver brain, retina |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Win, A.; Delgado, A.; Jadeja, R.N.; Martin, P.M.; Bartoli, M.; Thounaojam, M.C. Pharmacological and Metabolic Significance of Bile Acids in Retinal Diseases. Biomolecules 2021, 11, 292. https://doi.org/10.3390/biom11020292
Win A, Delgado A, Jadeja RN, Martin PM, Bartoli M, Thounaojam MC. Pharmacological and Metabolic Significance of Bile Acids in Retinal Diseases. Biomolecules. 2021; 11(2):292. https://doi.org/10.3390/biom11020292
Chicago/Turabian StyleWin, Alice, Amanda Delgado, Ravirajsinh N. Jadeja, Pamela M. Martin, Manuela Bartoli, and Menaka C. Thounaojam. 2021. "Pharmacological and Metabolic Significance of Bile Acids in Retinal Diseases" Biomolecules 11, no. 2: 292. https://doi.org/10.3390/biom11020292
APA StyleWin, A., Delgado, A., Jadeja, R. N., Martin, P. M., Bartoli, M., & Thounaojam, M. C. (2021). Pharmacological and Metabolic Significance of Bile Acids in Retinal Diseases. Biomolecules, 11(2), 292. https://doi.org/10.3390/biom11020292