
RAS Nanoclusters: Dynamic Signaling Platforms Amenable to Therapeutic Intervention

 ,
,
Abstract
:1. Introduction
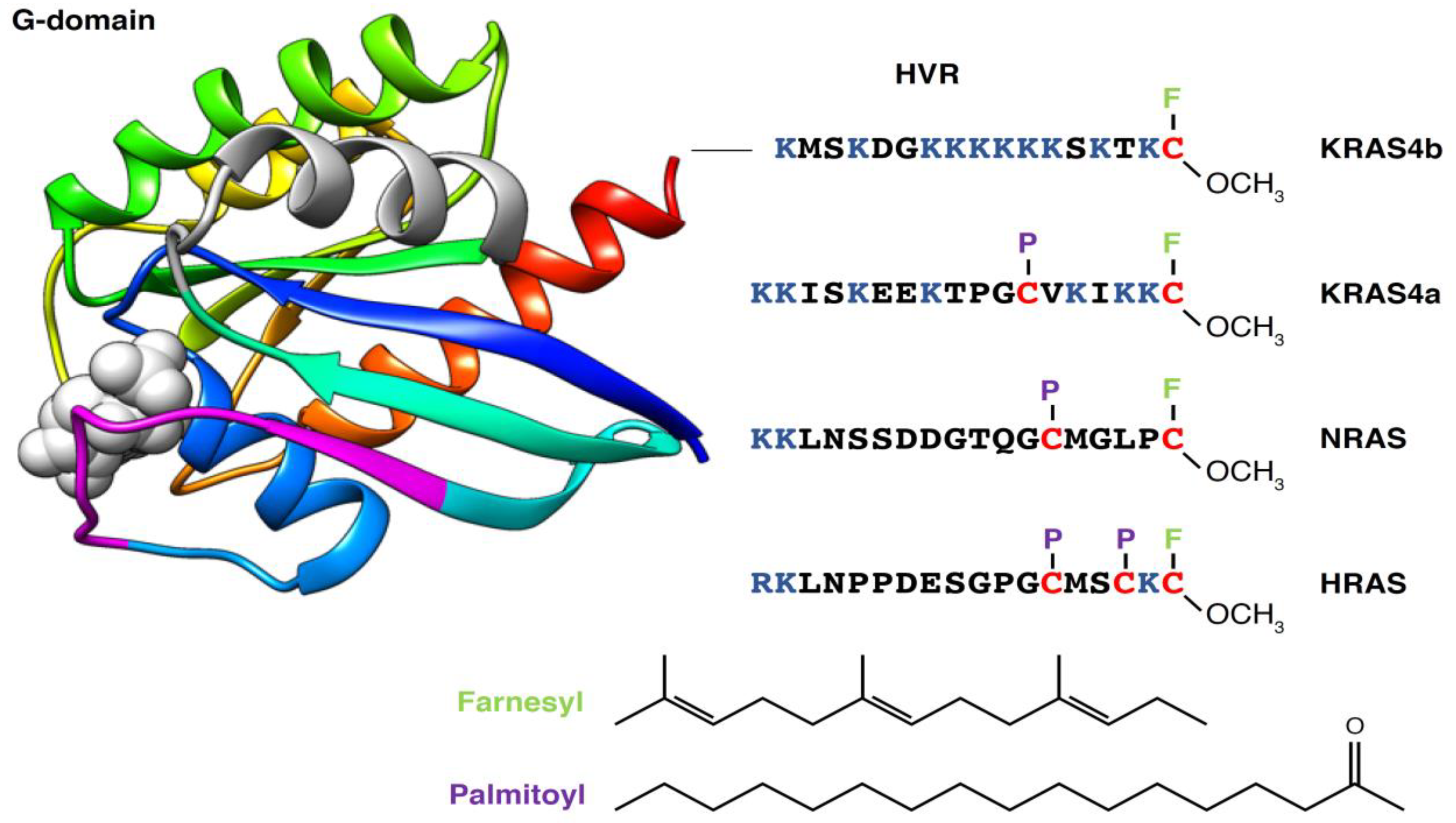
2. RAS Proteins as Membrane-Bound Molecular Switches
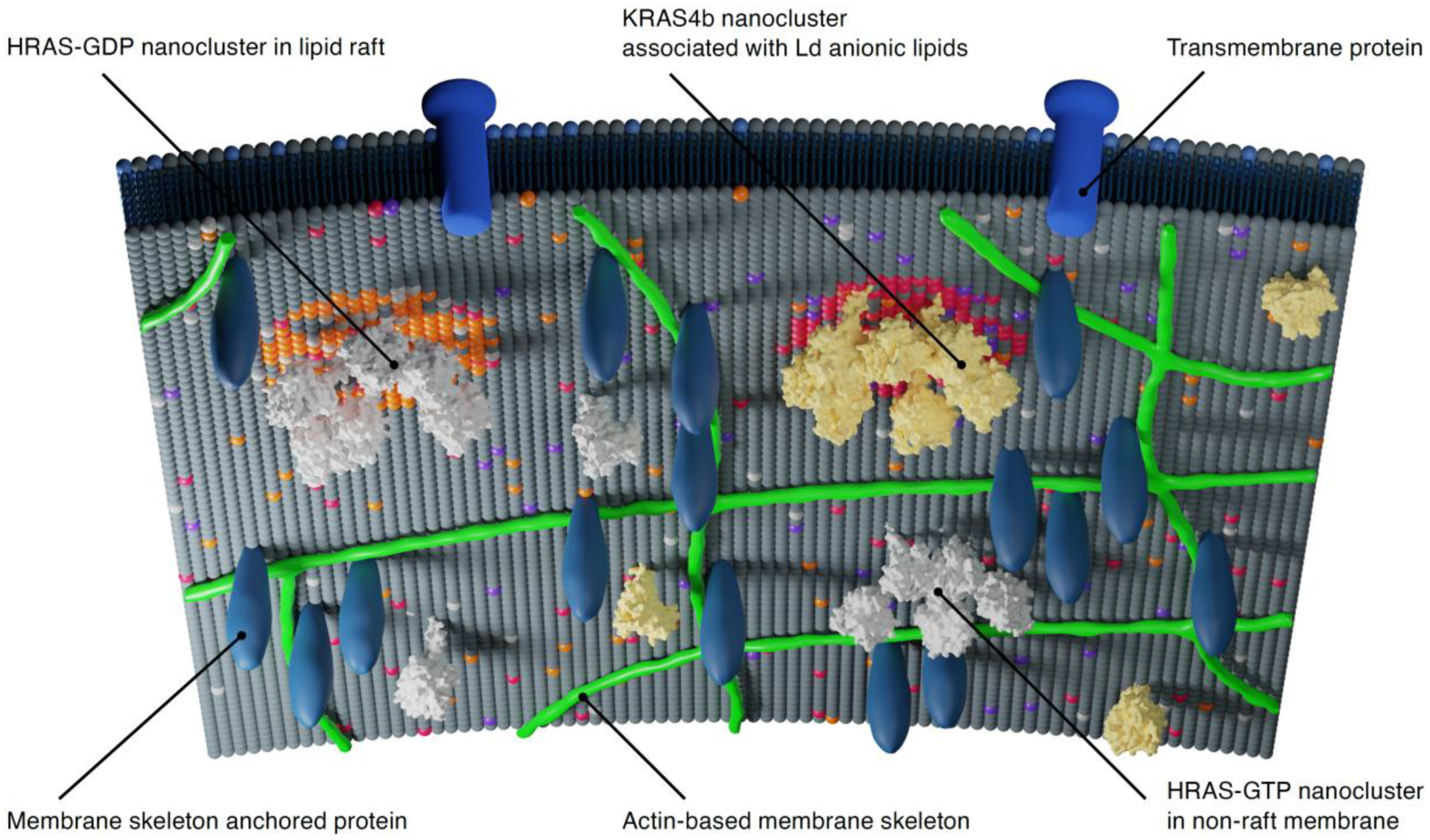
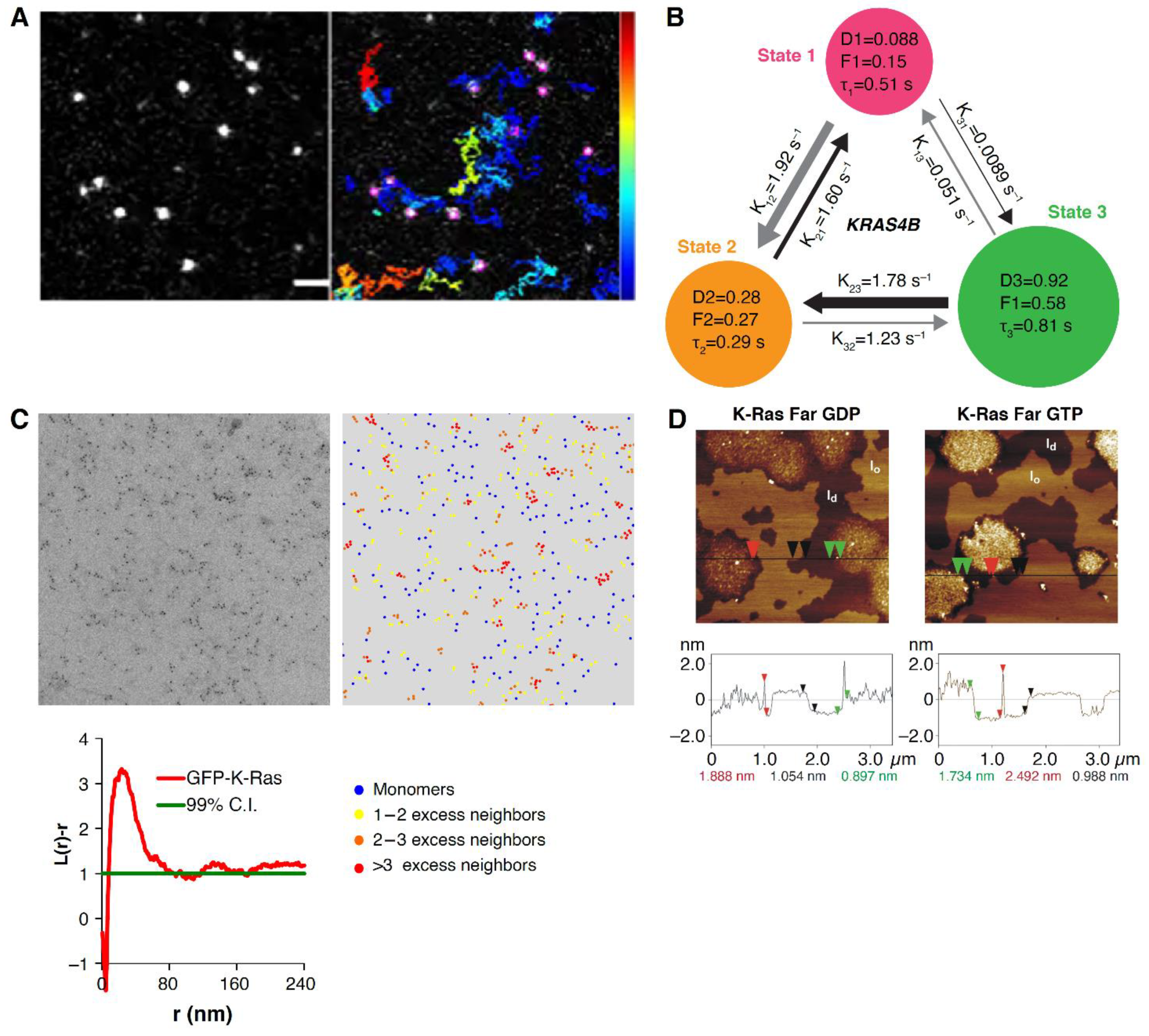
3. Membrane Organization, RAS Dynamics and RAS Clustering
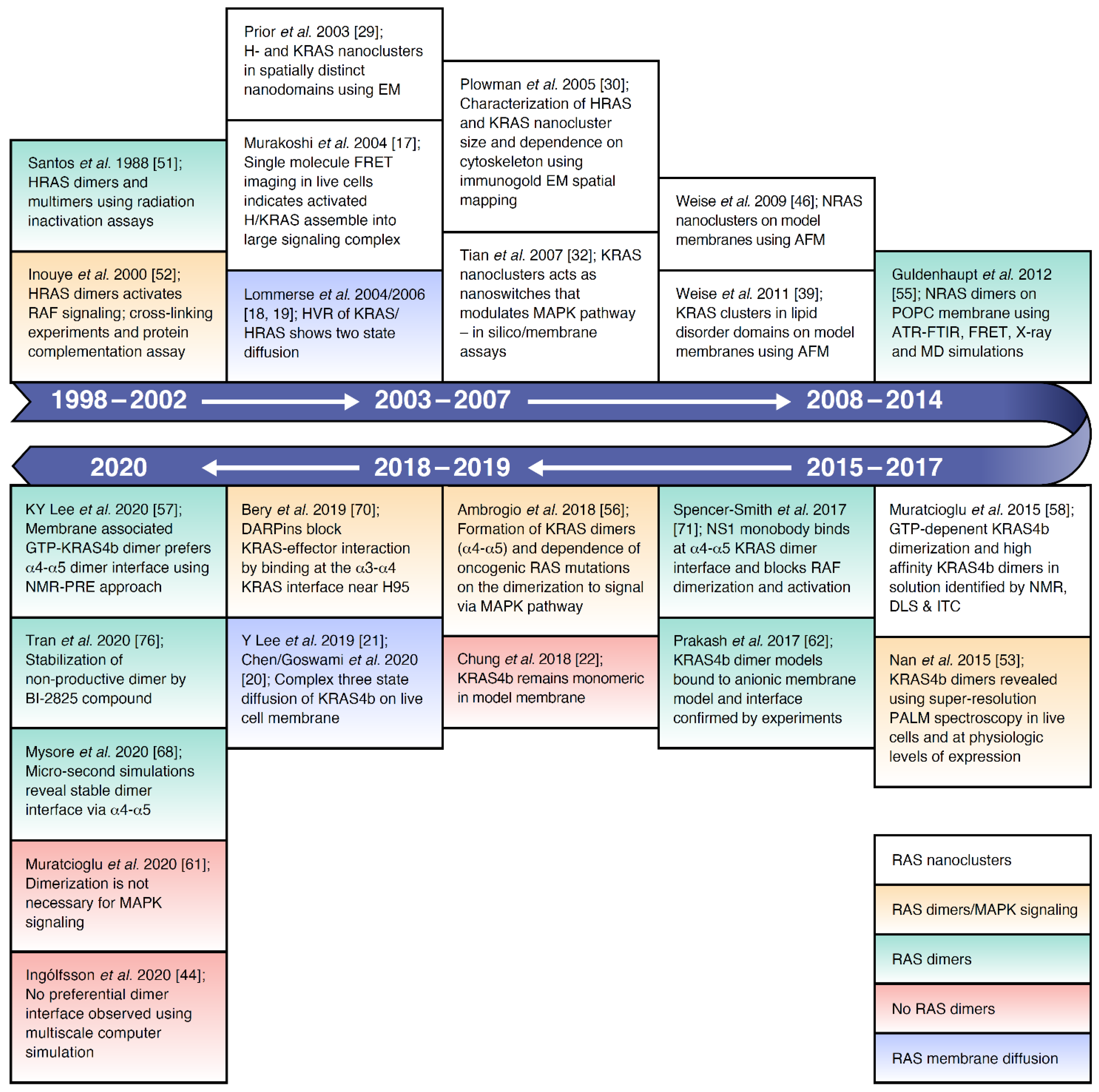
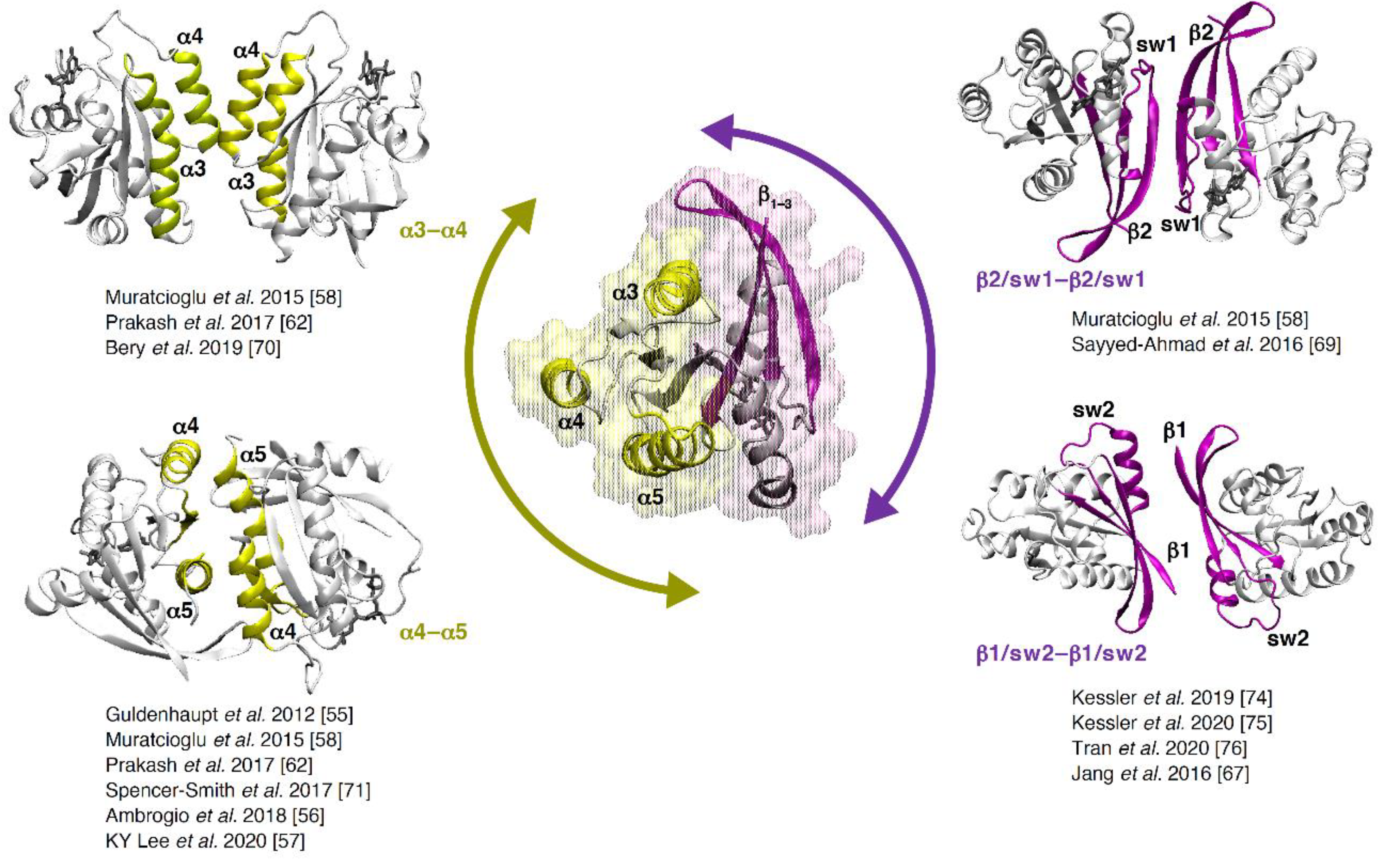
4. Formation of RAS Dimers and Higher Multimers.
5. Modulation of RAS Dimers and Nanoclusters as a Therapeutic Strategy
6. Outlook and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Trahey, M.; McCormick, F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 1987, 238, 542–545. [Google Scholar] [CrossRef]
- Willumsen, B.M.; Christensen, A.; Hubbert, N.L.; Papageorge, A.G.; Lowy, D.R. The p21 ras C-terminus is required for transformation and membrane association. Nat. Cell Biol. 1984, 310, 583–586. [Google Scholar] [CrossRef]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Stokoe, D.; Macdonald, S.G.; Cadwallader, K.; Symons, M.; Hancock, J.F. Activation of Raf as a result of recruitment to the plasma membrane. Scince 1994, 264, 1463–1467. [Google Scholar] [CrossRef]
- Ahearn, I.M.; Haigis, K.M.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.J.; Snitkin, H.; Gumper, I.; Philips, M.R.; Sabatini, D.; Pellicer, A. The Differential Palmitoylation States of N-Ras and H-Ras Determine Their Distinct Golgi Subcompartment Localizations. J. Cell. Physiol. 2014, 230, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmick, M.; Vartak, N.; Papke, B.; Kovacevic, M.; Truxius, D.C.; Rossmannek, L.; Bastiaens, P.I. KRas Localizes to the Plasma Membrane by Spatial Cycles of Solubilization, Trapping and Vesicular Transport. Cell 2014, 157, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Kholodenko, B.N.; Hoek, J.B.; Westerhoff, H.V. Why cytoplasmic signaling proteins should be recruited to the plasma membrane. Trends Cell Biol. 2000, 10, 173–178. [Google Scholar] [CrossRef]
- Mugler, A.; Tostevin, F.; Wolde, P.R.T. Spatial partitioning improves the reliability of biochemical signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 5927–5932. [Google Scholar] [CrossRef] [Green Version]
- Kusumi, A.; Nakada, C.; Ritchie, K.; Murase, K.; Suzuki, K.; Murakoshi, H.; Kasai, R.S.; Kondo, J.; Fujiwara, T. Paradigm Shift of the Plasma Membrane Concept from the Two-Dimensional Continuum Fluid to the Partitioned Fluid: High-Speed Single-Molecule Tracking of Membrane Molecules. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 351–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusumi, A.; Suzuki, K.G.; Kasai, R.S.; Ritchie, K.; Fujiwara, T.K. Hierarchical mesoscale domain organization of the plasma membrane. Trends Biochem. Sci. 2011, 36, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, H.; Iino, R.; Kobayashi, T.; Fujiwara, T.; Ohshima, C.; Yoshimura, A.; Kusumi, A. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7317–7322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lommerse, P.H.M.; Blab, G.A.; Cognet, L.; Harms, G.S.; Snaar-Jagalska, B.E.; Spaink, H.P.; Schmidt, T. Single-Molecule Imaging of the H-Ras Membrane-Anchor Reveals Domains in the Cytoplasmic Leaflet of the Cell Membrane. Biophys. J. 2004, 86, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Lommerse, P.H.M.; Vastenhoud, K.; Pirinen, N.J.; Magee, A.I.; Spaink, H.P.; Schmidt, T. Single-Molecule Diffusion Reveals Similar Mobility for the Lck, H-Ras, and K-Ras Membrane Anchors. Biophys. J. 2006, 91, 1090–1097. [Google Scholar] [CrossRef] [Green Version]
- Goswami, D.; Chen, D.; Yang, Y.; Gudla, P.R.; Columbus, J.; Worthy, K.; Rigby, M.; Wheeler, M.; Mukhopadhyay, S.; Powell, K.; et al. Membrane interactions of the globular domain and the hypervariable region of KRAS4b define its unique diffusion behavior. eLife 2020, 9. [Google Scholar] [CrossRef]
- Lee, Y.; Phelps, C.; Huang, T.; Mostofian, B.; Wu, L.; Zhang, Y.; Tao, K.; Chang, Y.H.; Stork, P.J.; Gray, J.W.; et al. High-throughput, single-particle tracking reveals nested membrane domains that dictate KRasG12D diffusion and trafficking. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.K.; Lee, Y.K.; Denson, J.-P.; Gillette, W.K.; Alvarez, S.; Stephen, A.G.; Groves, J.T. K-Ras4B Remains Monomeric on Membranes over a Wide Range of Surface Densities and Lipid Compositions. Biophys. J. 2018, 114, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, J.F.; Parton, R.G. Ras plasma membrane signalling platforms. Biochem. J. 2005, 389, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abankwa, D.; Gorfe, A.A.; Hancock, J.F. Ras nanoclusters: Molecular structure and assembly. Semin. Cell Dev. Biol. 2007, 18, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hancock, J.F. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol. 2012, 23, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Hancock, J.F. Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Jang, H.; Tsai, C.-J. Oligomerization and nanocluster organization render specificity. Biol. Rev. 2015, 90, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Prakash, P.; Gorfe, A.A.; Hancock, J.F. Ras and the Plasma Membrane: A Complicated Relationship. Cold Spring Harb. Perspect. Med. 2018, 8, a031831. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plowman, S.J.; Muncke, C.; Parton, R.G.; Hancock, J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 102, 15500–15505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abankwa, D.; Gorfe, A.A.; Inder, K.; Hancock, J.F. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 1130–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Harding, A.; Inder, K.; Plowman, S.; Parton, R.G.; Hancock, J.F. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007, 9, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Mageean, C.J.; Griffiths, J.R.; Smith, D.L.; Clague, M.J.; Prior, I.A. Absolute Quantification of Endogenous Ras Isoform Abundance. PLoS ONE 2015, 10, e0142674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, A.S.; Hancock, J.F. Using plasma membrane nanoclusters to build better signaling circuits. Trends Cell Biol. 2008, 18, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Kholodenko, B.N.; Hancock, J.F.; Kolch, W. Signalling ballet in space and time. Nat. Rev. Mol. Cell Biol. 2010, 11, 414–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hancock, J.F. Compartmentalization of Ras proteins. J. Cell Sci. 2001, 114, 1603–1608. [Google Scholar] [PubMed]
- Prior, I.A.; Harding, A.; Yan, J.; Sluimer, J.; Parton, R.G.; Hancock, J.F. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 2001, 3, 368–375. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, H.; Rodkey, T.; Ariotti, N.; Parton, R.G.; Hancock, J.F. Signal Integration by Lipid-Mediated Spatial Cross Talk between Ras Nanoclusters. Mol. Cell. Biol. 2013, 34, 862–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, K.; Kapoor, S.; Denter, C.; Nikolaus, J.; Opitz, N.; Koch, S.; Triola, G.; Herrmann, A.; Waldmann, H.; Winter, R. Membrane-Mediated Induction and Sorting of K-Ras Microdomain Signaling Platforms. J. Am. Chem. Soc. 2011, 133, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Erwin, N.; Sperlich, B.; Garivet, G.; Waldmann, H.; Weise, K.; Winter, R. Lipoprotein insertion into membranes of various complexity: Lipid sorting, interfacial adsorption and protein clustering. Phys. Chem. Chem. Phys. 2016, 18, 8954–8962. [Google Scholar] [CrossRef]
- Li, L.; Dwivedi, M.; Patra, S.; Erwin, N.; Möbitz, S.; Winter, R.H.A.; Dwiwedi, M.; Möbitz, S. Probing Colocalization of N-Ras and K-Ras4B Lipoproteins in Model Biomembranes. ChemBioChem 2019, 20, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.C.; McLean, M.A.; Sligar, S.G. Interaction of KRas4b with anionic membranes: A special role for PIP 2. Biochem. Biophys. Res. Commun. 2017, 487, 351–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.; Chung, S.; Kim, S.; Li, Z.; Manor, D.; Buck, M. K-Ras G-domain binding with signaling lipid phosphatidylinositol (4,5)-phosphate (PIP2): Membrane association, protein orientation, and function. J. Biol. Chem. 2019, 294, 7068–7084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingólfsson, H.I.; Neale, C.; Carpenter, T.S.; Shrestha, R.; López, C.A.; Tran, T.H.; Oppelstrup, T.; Bhatia, H.; Stanton, L.G.; Zhang, X.; et al. Lipid-Dependent Dynamics of the RAS Signaling Protein. Nat. Commun. under review.
- Persson, F.; Lindén, M.; Unoson, C.; Elf, J. Extracting intracellular diffusive states and transition rates from single-molecule tracking data. Nat. Methods 2013, 10, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Weise, K.; Triola, G.; Brunsveld, L.; Waldmann, H.; Winter, R. Influence of the Lipidation Motif on the Partitioning and Association of N-Ras in Model Membrane Subdomains. J. Am. Chem. Soc. 2009, 131, 1557–1564. [Google Scholar] [CrossRef]
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Mattos, C. The Ras–Membrane Interface: Isoform-Specific Differences in the Catalytic Domain. Mol. Cancer Res. 2015, 13, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-C.; Lytle, N.K.; Gammon, S.T.; Wang, L.; Hayes, T.K.; Sutton, M.N.; Barst, R., Jr.; Der, C.J.; Piwnica-Worms, D.; McCormick, F.; et al. Analysis of RAS protein interactions in living cells reveals a mechanism for pan-RAS depletion by membrane-targeted RAS binders. Proc. Natl. Acad. Sci. USA 2020, 117, 12121–12130. [Google Scholar] [CrossRef]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. The importance of Raf dimerization in cell signaling. Small GTPases 2013, 4, 180–185. [Google Scholar] [CrossRef]
- Santos, E.; Nebreda, A.R.; Bryan, T.; Kempner, E.S. Oligomeric structure of p21 ras proteins as determined by radiation inactivation. J. Biol. Chem. 1988, 263, 9853–9858. [Google Scholar] [CrossRef]
- Inouye, K.; Mizutani, S.; Koide, H.; Kaziro, Y. Formation of the Ras Dimer Is Essential for Raf-1 Activation. J. Biol. Chem. 2000, 275, 3737–3740. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Tamgüney, T.M.; Collisson, E.A.; Lin, L.-J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Collisson, E.A.; Lewis, S.; Huang, J.; Tamgüney, T.M.; Liphardt, J.T.; McCormick, F.; Gray, J.W.; Chu, S. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 18519–18524. [Google Scholar] [CrossRef] [Green Version]
- Güldenhaupt, J.; Rudack, T.; Bachler, P.; Mann, D.; Triola, G.; Waldmann, H.; Kötting, C.; Gerwert, K. N-Ras Forms Dimers at POPC Membranes. Biophys. J. 2012, 103, 1585–1593. [Google Scholar] [CrossRef]
- Ambrogio, C.; Köhler, J.; Zhou, Z.-W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Fang, Z.; Enomoto, M.; Gasmi-Seabrook, G.; Zheng, L.; Koide, S.; Ikura, M.; Marshall, C.B. Two Distinct Structures of Membrane-Associated Homodimers of GTP- and GDP-Bound KRAS4B Revealed by Paramagnetic Relaxation Enhancement. Angew. Chem. Int. Ed. 2020, 59, 11037–11045. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Chavan, T.S.; Freed, B.C.; Jang, H.; Khavrutskii, L.; Freed, R.N.; Dyba, M.A.; Stefanisko, K.; Tarasov, S.G.; Gursoy, A.; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogmen, U.; Keskin, O.; Aytuna, A.S.; Nussinov, R.; Gursoy, A. PRISM: Protein interactions by structural matching. Nucleic Acids Res. 2005, 33, W331–W336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuncbag, N.; Gursoy, A.; Nussinov, R.; Keskin, O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat. Protoc. 2011, 6, 1341–1354. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Aydin, C.; Odabasi, E.; Ozdemir, E.S.; Firat-Karalar, E.N.; Jang, H.; Tsai, C.-J.; Nussinov, R.; Kavakli, I.H.; Gursoy, A.; et al. Oncogenic K-Ras4B Dimerization Enhances Downstream Mitogen-activated Protein Kinase Signaling. J. Mol. Biol. 2020, 432, 1199–1215. [Google Scholar] [CrossRef]
- Prakash, P.; Sayyed-Ahmad, A.; Cho, K.-J.; Dolino, D.M.; Chen, W.; Li, H.; Grant, B.J.; Hancock, J.F.; Gorfe, A.A. Computational and biochemical characterization of two partially overlapping interfaces and multiple weak-affinity K-Ras dimers. Sci. Rep. 2017, 7, 40109. [Google Scholar] [CrossRef] [Green Version]
- Sarkar-Banerjee, S.; Sayyed-Ahmad, A.; Prakash, P.; Cho, K.-J.; Waxham, M.N.; Hancock, J.F.; Gorfe, A.A. Spatiotemporal Analysis of K-Ras Plasma Membrane Interactions Reveals Multiple High Order Homo-oligomeric Complexes. J. Am. Chem. Soc. 2017, 139, 13466–13475. [Google Scholar] [CrossRef]
- Barklis, E.; Stephen, A.G.; Staubus, A.O.; Barklis, R.L.; Alfadhli, A. Organization of Farnesylated, Carboxymethylated KRAS4B on Membranes. J. Mol. Biol. 2019, 431, 3706–3717. [Google Scholar] [CrossRef] [PubMed]
- Kosoglu, K.; Omur, M.E.; Jang, H.; Nussinov, R.; Keskin, O.; Gursoy, A. The structural basis of the oncogenic mutant K-Ras4B homodimers. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; Huang, S.-Y. HSYMDOCK: A docking web server for predicting the structure of protein homo-oligomers with Cn or Dn symmetry. Nucleic Acids Res. 2018, 46, W423–W431. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R. Membrane-associated Ras dimers are isoform-specific: K-Ras dimers differ from H-Ras dimers. Biochem. J. 2016, 473, 1719–1732. [Google Scholar] [CrossRef]
- Mysore, V.P.; Zhou, Z.-W.; Ambrogio, C.; Li, L.; Kapp, J.N.; Lu, C.; Wang, Q.; Tucker, M.R.; Okoro, J.J.; Nagy-Davidescu, G.; et al. A structural model of a Ras-Raf signalosome. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sayyed-Ahmad, A.; Cho, K.-J.; Hancock, J.F.; Gorfe, A.A. Computational Equilibrium Thermodynamic and Kinetic Analysis of K-Ras Dimerization through an Effector Binding Surface Suggests Limited Functional Role. J. Phys. Chem. B 2016, 120, 8547–8556. [Google Scholar] [CrossRef] [Green Version]
- Bery, N.; Legg, S.; Debreczeni, J.; Breed, J.; Embrey, K.; Stubbs, C.; Kolasinska-Zwierz, P.; Barrett, N.; Marwood, R.; Watson, J.; et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Spencer-Smith, R.; Koide, A.; Zhou, Y.; Eguchi, R.R.; Sha, F.; Gajwani, P.; Santana, D.; Gupta, A.; O’Bryan, J.P.; Herrero-Garcia, E.; et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. [Google Scholar] [CrossRef]
- Packer, M.; Parker, J.A.; Chung, J.K.; Li, Z.; Lee, Y.K.; Cookis, T.; Guterres, H.; Alvarez, S.; Hossain, M.A.; Donnelly, D.P.; et al. Raf promotes dimerization of the Ras G-domain with increased allosteric connections. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cookis, T.; Mattos, C. Crystal Structure Reveals the Full Ras:Raf Interface and Advances Mechanistic Understanding of Raf Activation. Biophys. J. 2021, 120, 7a. [Google Scholar] [CrossRef]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, D.; Gollner, A.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Reply to Tran et al.: Dimeric KRAS protein–protein interaction stabilizers. Proc. Natl. Acad. Sci. USA 2020, 117, 3365–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.H.; Alexander, P.; Dharmaiah, S.; Agamasu, C.; Nissley, D.V.; McCormick, F.; Esposito, D.; Simanshu, D.K.; Stephen, A.G.; Balius, T.E. The small molecule BI-2852 induces a nonfunctional dimer of KRAS. Proc. Natl. Acad. Sci. USA 2020, 117, 3363–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.-C.; Iversen, L.; Tu, H.-L.; Rhodes, C.; Christensen, S.M.; Iwig, J.S.; Hansen, S.D.; Huang, W.Y.C.; Groves, J.T. H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc. Natl. Acad. Sci. USA 2014, 111, 2996–3001. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.K.; Lee, Y.K.; Lam, H.Y.M.; Groves, J.T. Covalent Ras Dimerization on Membrane Surfaces through Photosensitized Oxidation. J. Am. Chem. Soc. 2016, 138, 1800–1803. [Google Scholar] [CrossRef] [Green Version]
- Kovrigina, E.A.; Galiakhmetov, A.R.; Kovrigin, E.L. The Ras G Domain Lacks the Intrinsic Propensity to Form Dimers. Biophys. J. 2015, 109, 1000–1008. [Google Scholar] [CrossRef] [Green Version]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Förster resonance energy transfer. Science 2018, 359, eaan1133. [Google Scholar] [CrossRef] [Green Version]
- Algar, W.R.; Hildebrandt, N.; Vogel, S.S.; Medintz, I.L. FRET as a biomolecular research tool—Understanding its potential while avoiding pitfalls. Nat. Methods 2019, 16, 815–829. [Google Scholar] [CrossRef]
- Piston, D.W.; Kremers, G.-J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef]
- Cranfill, P.J.; Sell, B.R.; Baird, M.A.; Allen, J.R.; Lavagnino, Z.; de Gruiter, H.M.; Kremers, G.-J.; Davidson, M.W.; Ustione, A.; Piston, D.W. Quantitative assessment of fluorescent proteins. Nat. Methods 2016, 13, 557–562. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, J.; Cromm, P.M.; Zimmermann, G.; Grossmann, T.N.; Waldmann, H. Small-molecule modulation of Ras signaling. Nat. Chem. Biol. 2014, 10, 613–622. [Google Scholar] [CrossRef]
- Orgován, Z.; Keserű, G.M. Small molecule inhibitors of RAS proteins with oncogenic mutations. Cancer Metastasis Rev. 2020, 39, 1107–1126. [Google Scholar] [CrossRef]
- Zuberi, M.; Khan, I.; O’Bryan, J.P. Inhibition of RAS: Proven and potential vulnerabilities. Biochem. Soc. Trans. 2020, 48, 1831–1841. [Google Scholar] [CrossRef]
- Lee, H.W.; Sa, J.K.; Gualberto, A.; Scholz, C.; Sung, H.H.; Jeong, B.C.; Choi, H.Y.; Kwon, G.Y.; Park, S.H. A Phase II Trial of Tipifarnib for Patients with Previously Treated, Metastatic Urothelial Carcinoma Harboring HRAS Mutations. Clin. Cancer Res. 2020, 26, 5113–5119. [Google Scholar] [CrossRef]
- Kim, D.; Xue, J.Y.; Lito, P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell 2020, 183, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoeven, D.; Cho, K.-J.; Ma, X.; Chigurupati, S.; Parton, R.G.; Hancock, J.F. Fendiline Inhibits K-Ras Plasma Membrane Localization and Blocks K-Ras Signal Transmission. Mol. Cell. Biol. 2012, 33, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.-J.; van der Hoeven, D.; Zhou, Y.; Maekawa, M.; Ma, X.; Chen, W.; Fairn, G.D.; Hancock, J.F. Inhibition of acid sphingomyelinase depletes cellular phosphatidylserine and mislocalizes K-Ras from the plasma membrane. Mol. Cell. Biol. 2015, 36, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Kattan, W.E.; Hancock, J.F. RAS Function in cancer cells: Translating membrane biology and biochemistry into new therapeutics. Biochem. J. 2020, 477, 2893–2919. [Google Scholar] [CrossRef]
- Spencer-Smith, R.; Li, L.; Prasad, S.; Koide, A.; Koide, S.; O’Bryan, J.P. Targeting the α4-α5 interface of RAS results in multiple levels of inhibition. Small GTPases 2017, 10, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Spencer-Smith, R.; O’Bryan, J.P. Targeting the α4–α5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene 2019, 38, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.N.; Lu, Z.; Li, Y.-C.; Zhou, Y.; Huang, T.; Reger, A.S.; Hurwitz, A.M.; Palzkill, T.; Logsdon, C.; Liang, X.; et al. DIRAS3 (ARHI) Blocks RAS/MAPK Signaling by Binding Directly to RAS and Disrupting RAS Clusters. Cell Rep. 2019, 29, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [Green Version]
- Kessler, D.; Bergner, A.; Böttcher, J.; Fischer, G.; Döbel, S.; Hinkel, M.; Müllauer, B.; Weiss-Puxbaum, A.; McConnell, D.B. Drugging all RAS isoforms with one pocket. Future Med. Chem. 2020, 12, 1911–1923. [Google Scholar] [CrossRef]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2011, 11, 25–36. [Google Scholar] [CrossRef]
- Andrei, S.A.; Sijbesma, E.E.; Hann, M.; Davis, J.; O’Mahony, G.; Perry, M.W.D.; Karawajczyk, A.; Eickhoff, J.; Brunsveld, L.L.; Doveston, R.G.; et al. Stabilization of protein-protein interactions in drug discovery. Expert Opin. Drug Discov. 2017, 12, 925–940. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RAS Interfaces | Interface Residues | References | KRAS PDBs ‖,▲ | |
|---|---|---|---|---|
| Dimer | β2 switch 1 | I21, I24, Q25, H27, V29, E31, D33, I36, E37, D38, S39, Y40, R41, K42, Q43, L52 | [58,69] | 6MNX, 6QUX, 7ACQ (BI-5747) |
| α3–α4 | E62, E91, H94, R97, E98, K101, R102, D105, S106, E107, K128, Q129, D132, L133, R135, S136, Y137 | [58,62,70] | 4LUC, 4LV6 | |
| α4–α5 (α4–β6–α5) | E49, D108, K128, D132, R135, R164, E168, K172, K175 | [55,56,57,62,68,71,72,73] | 5US4, 5VP7, 5VPI, 5VPY, 5VPZ, 5VQ0, 5VQ1, 5VQ2, 5VQ6, 5VQ8, 5W22 | |
| β1 switch 2 (ligand induced) | M1, E3, K5, E37, R41, K42, Q43, L52, L53, D54, LIG(F0K201) † | [67,74,75,76] | 6GJ8 (BI-2852), 6GJ7, 6ZL5 (BI-2852), 7ACA (BI-5747) | |
| Trimer | α4–α5α (interface 1) | D47, Q131, I142, E143, Q150, G151, D153, D154, Y157, T158, R161 ‡ | [64] | 3GFT, 5OCO, 5OCT, 6F76, 6FA1, 6FA2, 6FA3, 6FA4, 6GOG, 6GOM, 6GQT, 6GQW, 6GQX, 6GQY, 5MLB, 5O2S, 5WHE (peptide), 5KYK |
| α1 switch 1 (interface 2) | Q25, N26, H27, F28, D30, K147, T148 ‡ | |||
| Protein Condition and Methodology | KRAS Dimer KD | References |
|---|---|---|
| In solution; ITC and MTS | ~1 μM | [58] |
| On nanodiscs; NMR | 530 μM GTP, 610 μM GDP | [57] |
| In solution; MD | ~5 μM (α3–α4), ~107 μM (α4–α5) | [62] |
| In solution; MD | ~870 μM (β2) | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van, Q.N.; Prakash, P.; Shrestha, R.; Balius, T.E.; Turbyville, T.J.; Stephen, A.G. RAS Nanoclusters: Dynamic Signaling Platforms Amenable to Therapeutic Intervention. Biomolecules 2021, 11, 377. https://doi.org/10.3390/biom11030377
Van QN, Prakash P, Shrestha R, Balius TE, Turbyville TJ, Stephen AG. RAS Nanoclusters: Dynamic Signaling Platforms Amenable to Therapeutic Intervention. Biomolecules. 2021; 11(3):377. https://doi.org/10.3390/biom11030377
Chicago/Turabian StyleVan, Que N., Priyanka Prakash, Rebika Shrestha, Trent E. Balius, Thomas J. Turbyville, and Andrew G. Stephen. 2021. "RAS Nanoclusters: Dynamic Signaling Platforms Amenable to Therapeutic Intervention" Biomolecules 11, no. 3: 377. https://doi.org/10.3390/biom11030377