
Multi-Omics Data Analysis Uncovers Molecular Networks and Gene Regulators for Metabolic Biomarkers


Abstract
:1. Introduction
2. Materials and Methods
2.1. GWAS Data for IGF-I and IR Phenotypes
2.2. Genotyping and IGF-I/IR Phenotypes
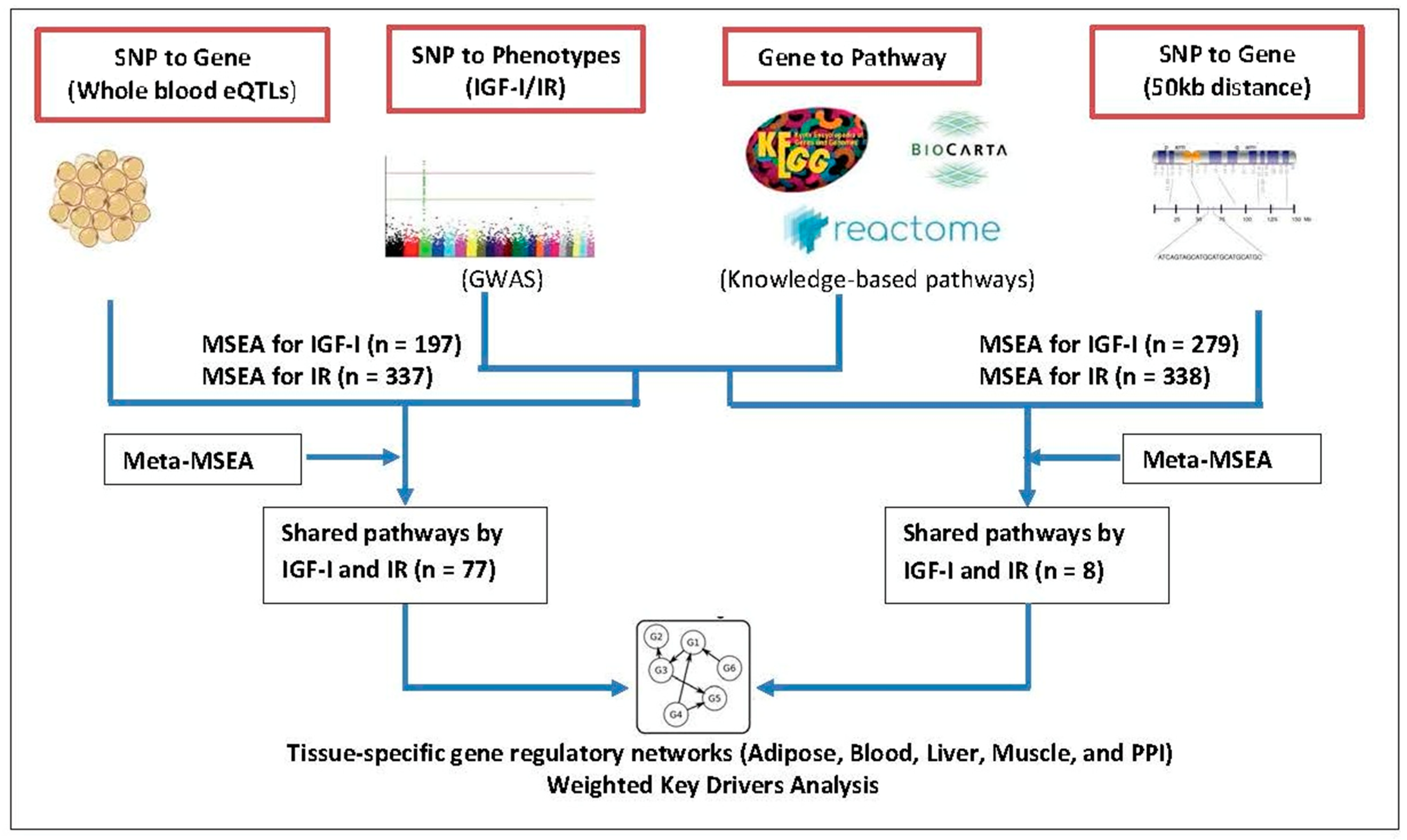
2.3. Mergeomics
2.3.1. Mapping SNPs to Genes
2.3.2. Marker-Set Enrichment Analysis (MSEA)
2.3.3. Tissue-Specific Gene Regulatory Networks and Weighted KD Analysis
3. Results
3.1. Phenotype-Specific and Common Pathways Shared by IGF-I and IR
3.2. Putative Key Regulatory Genes (i.e., KDs) for the IGF-I/IR–Associated Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belkina, A.C.; Denis, G.V. Obesity genes and insulin resistance. Curr. Opin. Endocrinol. Diabetes Obes 2010, 17, 472–477. [Google Scholar] [CrossRef] [Green Version]
- Hevener, A.L.; Febbraio, M.A. The 2009 stock conference report: Inflammation, obesity and metabolic disease. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2010, 11, 635–644. [Google Scholar] [CrossRef]
- Manning, A.K.; Hivert, M.F.; Scott, R.A.; Grimsby, J.L.; Bouatia-Naji, N.; Chen, H.; Rybin, D.; Liu, C.T.; Bielak, L.F.; Prokopenko, I.; et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012, 44, 659–669. [Google Scholar] [CrossRef]
- McCarthy, M.I. Genomics, type 2 diabetes, and obesity. N. Engl. J. Med. 2010, 363, 2339–2350. [Google Scholar] [CrossRef] [Green Version]
- Weichhaus, M.; Broom, J.; Wahle, K.; Bermano, G. A novel role for insulin resistance in the connection between obesity and postmenopausal breast cancer. Int. J. Oncol. 2012, 41, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, D.B. Insulin and cancer. Integr. Cancer Ther. 2003, 2, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.E.; Banerjee, I.; Murray, P.G.; Renehan, A.G. Growth hormone, the insulin-like growth factor axis, insulin and cancer risk. Nat. Rev. Endocrinol. 2011, 7, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Akker, M.; Güldiken, S.; Sipahi, T.; Palabıyık, O.; Tosunoğlu, A.; Çelik, Ö.; Tunçbilek, N.; Sezer, A.; Süt, N. Investigation of insulin resistance gene polymorphisms in patients with differentiated thyroid cancer. Mol. Biol. Rep. 2014, 41, 3541–3547. [Google Scholar] [CrossRef]
- Kabat, G.C.; Kim, M.Y.; Peters, U.; Stefanick, M.; Hou, L.; Wactawski-Wende, J.; Messina, C.; Shikany, J.M.; Rohan, T.E. A longitudinal study of the metabolic syndrome and risk of colorectal cancer in postmenopausal women. Eur. J. Cancer Prev. 2012, 21, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Howard, B.V.; Wylie-Rosett, J.; Anderson, G.L.; Ho, G.Y.; et al. Insulin, insulin-like growth factor-I, endogenous estradiol, and risk of colorectal cancer in postmenopausal women. Cancer Res. 2008, 68, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, N.; Thuesen, B.; Jørgensen, T.; Juul, A.; Spielhagen, C.; Wallaschofksi, H.; Linneberg, A. The association between IGF-I and insulin resistance: A general population study in Danish adults. Diabetes Care 2012, 35, 768–773. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, A.; Ma, H.; Xu, Y. Association between insulin receptor substrate 1 Gly972Arg polymorphism and cancer risk. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 2929–2936. [Google Scholar] [CrossRef]
- Ruan, Y.; Ma, J.; Xie, X. Association of IRS-1 and IRS-2 genes polymorphisms with polycystic ovary syndrome: A meta-analysis. Endocr. J. 2012, 59, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Carnero-Montoro, E.; van Dongen, J.; Lent, S.; Nedeljkovic, I.; Ligthart, S.; Tsai, P.C.; Martin, T.C.; Mandaviya, P.R.; Jansen, R.; et al. An integrative cross-omics analysis of DNA methylation sites of glucose and insulin homeostasis. Nat. Commun. 2019, 10, 2581. [Google Scholar] [CrossRef] [Green Version]
- Franks, P.W.; Mesa, J.L.; Harding, A.H.; Wareham, N.J. Gene-lifestyle interaction on risk of type 2 diabetes. Nutr. Metab. Cardiovasc. Diseases NMCD 2007, 17, 104–124. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Torres, R.; Johansson, M.; Gaborieau, V.; Haycock, P.C.; Wade, K.H.; Relton, C.L.; Martin, R.M.; Davey Smith, G.; Brennan, P. The Role of Obesity, Type 2 Diabetes, and Metabolic Factors in Pancreatic Cancer: A Mendelian Randomization Study. J. Natl. Cancer Inst. 2017, 109, djx012. [Google Scholar] [CrossRef] [Green Version]
- Shu, X.; Wu, L.; Khankari, N.K.; Shu, X.O.; Wang, T.J.; Michailidou, K.; Bolla, M.K.; Wang, Q.; Dennis, J.; Milne, R.L.; et al. Associations of obesity and circulating insulin and glucose with breast cancer risk: A Mendelian randomization analysis. Int. J. Epidemiol. 2019, 48, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MAGIC (The Meta-Analyses of Glucose and Insulin-Related Traits Consortium). MAGIC Publications. 2019. Available online: https://www.magicinvestigators.org/publications/ (accessed on 1 February 2021).
- Mohlke, K.L.; Boehnke, M. Recent advances in understanding the genetic architecture of type 2 diabetes. Hum. Mol. Genet. 2015, 24, R85–R92. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.S.; Arnett, D.K.; Lee, Y.C.; Shen, J.; Parnell, L.D.; Smith, C.E.; Richardson, K.; Li, D.; Borecki, I.B.; Ordovás, J.M.; et al. Genome-wide contribution of genotype by environment interaction to variation of diabetes-related traits. PLoS ONE 2013, 8, e77442. [Google Scholar] [CrossRef]
- Zhao, Y.; Jhamb, D.; Shu, L.; Arneson, D.; Rajpal, D.K.; Yang, X. Multi-omics integration reveals molecular networks and regulators of psoriasis. BMC Syst. Biol. 2019, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.K.; Huang, Y.T.; Meng, Q.; Wu, C.; Reiner, A.; Sobel, E.M.; Tinker, L.; Lusis, A.J.; Yang, X.; Liu, S. Shared molecular pathways and gene networks for cardiovascular disease and type 2 diabetes mellitus in women across diverse ethnicities. Circ. Cardiovasc. Genet. 2014, 7, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Yang, X.; Kaplan, L.M.; Molony, C.; Schadt, E.E. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am. J. Hum. Genet. 2010, 86, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Bucan, M. Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 2007, 81, 1278–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Beaulaurier, J.; Lum, P.Y.; Molony, C.; Yang, X.; MacNeil, D.J.; Weingarth, D.T.; Zhang, B.; Greenawalt, D.; Dobrin, R.; et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 2010, 6, e1000932. [Google Scholar] [CrossRef] [Green Version]
- Mäkinen, V.P.; Civelek, M.; Meng, Q.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.; Segrè, A.V.; Ghosh, S.; Vivar, J.; et al. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014, 10, e1004502. [Google Scholar] [CrossRef]
- Jung, S.Y.; Mancuso, N.; Yu, H.; Papp, J.; Sobel, E.; Zhang, Z.F. Genome-Wide Meta-analysis of Gene-Environmental Interaction for Insulin Resistance Phenotypes and Breast Cancer Risk in Postmenopausal Women. Cancer Prev. Res. 2019, 12, 31–42. [Google Scholar] [CrossRef] [Green Version]
- The Women’s Health Initiative Study Group. Design of the Women’s Health Initiative clinical trial and observational study. Control. Clin. Trials 1998, 19, 61–109. [Google Scholar] [CrossRef]
- NCBI: WHI Harmonized and Imputed GWAS Data. A Sub-Study of Women’s Health Initiative. Available online: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000746.v3.p3 (accessed on 1 February 2021).
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Shu, L.; Zhao, Y.; Kurt, Z.; Byars, S.G.; Tukiainen, T.; Kettunen, J.; Orozco, L.D.; Pellegrini, M.; Lusis, A.J.; Ripatti, S.; et al. Mergeomics: Multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genom. 2016, 17, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; D’Eustachio, P.; Schmidt, E.; de Bono, B.; Jassal, B.; Gopinath, G.R.; Wu, G.R. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33 (Suppl. 1), D428–D432. [Google Scholar] [CrossRef] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, K.C.; Kurt, Z.; Barrere-Cain, R.; Sabir, S.; Das, A.; Floyd, R.; Vergnes, L.; Zhao, Y.; Che, N.; Charugundla, S.; et al. Integration of Multi-omics Data from Mouse Diversity Panel Highlights Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 103–115.e7. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peri, S.; Navarro, J.D.; Amanchy, R.; Kristiansen, T.Z.; Jonnalagadda, C.K.; Surendranath, V.; Niranjan, V.; Muthusamy, B.; Gandhi, T.K.B.; Gronborg, M.; et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003, 13, 2363–2371. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S.; et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.M.; Zhang, B.; Yang, X.; Zhu, J.; Stepaniants, S.; Zhang, C.; Meng, Q.; Peters, M.; He, Y.; Ni, C.; et al. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol. 2012, 8, 594. [Google Scholar] [CrossRef]
- Segre, A.V.; Groop, L.; Mootha, V.K.; Daly, M.J.; Altshuler, D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010, 6, e1001058. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 843–854. [Google Scholar] [CrossRef]
- Schaffler, A.; Scholmerich, J. Innate immunity and adipose tissue biology. Trends Immunol. 2010, 31, 228–235. [Google Scholar] [CrossRef]
- Delle Bovi, R.J.; Kim, J.; Suresh, P.; London, E.; Miller, W.T. Sterol structure dependence of insulin receptor and insulin-like growth factor 1 receptor activation. Biochim. Biophys. Acta Biomembr. 2019, 1861, 819–826. [Google Scholar] [CrossRef]
- Palmqvist, R.; Hallmans, G.; Rinaldi, S.; Biessy, C.; Stenling, R.; Riboli, E.; Kaaks, R. The effects of recombinant human insulin-like growth factor-1/insulin-like growth factor binding protein-3 administration on lipid and carbohydrate metabolism in recreational athletes. Clin. Endocrinol. 2020, 50, 642–646. [Google Scholar]
- Kujawska-Luczak, M.; Szulinska, M.; Skrypnik, D.; Musialik, K.; Swora-Cwynar, E.; Kregielska-Narozna, M.; Markuszewski, L.; Grzymislawska, M.; Bogdanski, P. The influence of orlistat, metformin and diet on serum levels of insulin-like growth factor-1 in obeses women with and without insulin resistance. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2018, 69. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D. Insulin-like growth factor-1 activates different catalytic subunits p110 of PI3K in a cell-type-dependent manner to induce lipogenesis-dependent epithelial-mesenchymal transition through the regulation of ADAM10 and ADAM17. Mol. Cell. Biochem. 2018, 439, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Ferrières, J.; Lautsch, D.; Bramlage, P.; Horack, M.; Baxter, C.A.; Ambegaonkar, B.; Toth, P.P.; Poh, K.K.; De Ferrari, G.M.; Gitt, A.K. Lipid-lowering treatment and low-density lipoprotein cholesterol target achievement in patients with type 2 diabetes and acute coronary syndrome. Arch. Cardiovasc. Dis. 2020, 113, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Weeda, E.R.; Bishu, K.G.; Ward, R.; Axon, R.N.; Taber, D.J.; Gebregziabher, M. Joint effect of race/ethnicity or location of residence and sex on low density lipoprotein-cholesterol among veterans with type 2 diabetes: A 10-year retrospective cohort study. BMC Cardiovasc. Disord. 2020, 20, 449. [Google Scholar] [CrossRef]
- Maachi, H.; Fergusson, G.; Ethier, M.; Brill, G.N.; Katz, L.S.; Honig, L.B.; Metukuri, M.R.; Scott, D.K.; Ghislain, J.; Poitout, V. HB-EGF Signaling Is Required for Glucose-Induced Pancreatic beta-Cell Proliferation in Rats. Diabetes 2020, 69, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Cui, L.; Ding, N.; Zhu, L.; Qu, X.; Dong, W.; Du, J.; Liu, Q. Expressions of insulin-like growth factor receptor-1 and cezanne-1 in lung adenocarcinoma. Med. Oncol. 2017, 34, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, L.; Ma, K.; Zhao, Y.; Liu, X.; Wang, Y.; Liu, M.; Liang, S.; Zhu, H.; Xu, N. Polarization of macrophages in the tumor microenvironment is influenced by EGFR signaling within colon cancer cells. Oncotarget 2016, 7, 75366–75378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Park, S.; Kim, W.S.; Song, J.S.; Choi, C.M.; Sung, Y.H.; et al. Activation of the IGF1R pathway potentially mediates acquired resistance to mutant-selective 3rd-generation EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget 2016, 7, 22005–22015. [Google Scholar] [CrossRef] [Green Version]
- Iyer, G.; Price, J.; Bourgeois, S.; Armstrong, E.; Huang, S.; Harari, P.M. Insulin-like growth factor 1 receptor mediated tyrosine 845 phosphorylation of epidermal growth factor receptor in the presence of monoclonal antibody cetuximab. BMC Cancer 2016, 16, 773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleem, H.; Abdul, U.K.; Küçükosmanoglu, A.; Houweling, M.; Cornelissen, F.M.; Heiland, D.H.; Hegi, M.E.; Kouwenhoven, M.C.; Bailey, D.; Würdinger, T.; et al. The TICking clock of EGFR therapy resistance in glioblastoma: Target Independence or target Compensation. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2019, 43, 29–37. [Google Scholar] [CrossRef]
- Gene Card: Human Gene Database: ERBB4 Gene (Protein Coding) 2021. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=ERBB4&keywords=erbb4 (accessed on 1 February 2021).
- Genes & Expression—ERBB4 Gene 2021. Available online: https://www.ncbi.nlm.nih.gov/gene/2066 (accessed on 1 February 2021).
- Li, X.; Huang, Q.; Wang, S.; Huang, Z.; Yu, F.; Lin, J. HER4 promotes the growth and metastasis of osteosarcoma via the PI3K/AKT pathway. Acta Biochim. Biophys. Sin. 2020, 52, 345–362. [Google Scholar] [CrossRef]
- Sandholm, N.; Salem, R.M.; McKnight, A.J.; Brennan, E.P.; Forsblom, C.; Isakova, T.; McKay, G.J.; Williams, W.W.; Sadlier, D.M.; Mäkinen, V.P.; et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 2012, 8, e1002921. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Imamura, M.; Kurashige, M.; Araki, S.; Suzuki, D.; Babazono, T.; Uzu, T.; Umezono, T.; Toyoda, M.; Kawai, K.; et al. Replication study for the association of 3 SNP loci identified in a genome-wide association study for diabetic nephropathy in European type 1 diabetes with diabetic nephropathy in Japanese patients with type 2 diabetes. Clin. Exp. Nephrol. 2013, 17, 866–871. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011, 39 (Suppl. 1), D1035–D1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Card: Human Gene Database: AKT1 Gene (Protein Coding) 2021. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=AKT1&keywords=AKT1 (accessed on 1 February 2021).
- Genes & Expression—AKT1 Gene 2021. Available online: https://www.ncbi.nlm.nih.gov/gene/207 (accessed on 1 February 2021).
- Gene Card: Human Gene Database: HRAS Gene (Protein Coding) 2021. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=HRAS&keywords=HRAS (accessed on 1 February 2021).
- Genes & Expression—HRAS Gene 2021. Available online: https://www.ncbi.nlm.nih.gov/gene/3265 (accessed on 1 February 2021).
- Gene Card: Human Gene Database: JAK1 Gene (Protein Coding) 2021. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=JAK1&keywords=jak1 (accessed on 1 February 2021).
- Genes & Expression—JAK1 Gene 2021. Available online: https://www.ncbi.nlm.nih.gov/gene/3716 (accessed on 1 February 2021).
- Matsubara, A.; Wasson, J.C.; Donelan, S.S.; Welling, C.M.; Glaser, B.; Permutt, M.A. Isolation and characterization of the human AKT1 gene, identification of 13 single nucleotide polymorphisms (SNPs), and their lack of association with Type II diabetes. Diabetologia 2001, 44, 910–913. [Google Scholar] [PubMed]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabasi, A.L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Module Size of PPI (n of Genes) | Top 5 Key Drivers | ||||||
|---|---|---|---|---|---|---|---|
| Module | Description | Adipose | Blood | Liver | Muscle | PPI | |
| M19708 | Type 2 diabetes mellitus | 17 | N/A | N/A | N/A | N/A | IRS1 *, HRAS, JAK1, IGF1R, AKT1 |
| rctm0415 | Fatty acid, triacylglycerol, and ketone body metabolism | 46 | N/A | N/A | N/A | N/A | MED24 *, MED15 *, MED6 *, MED1, CDK8 |
| Module | Description | Module Size (n of Genes) | Top 5 Key Drivers | ||||
|---|---|---|---|---|---|---|---|
| Adipose | Blood | Liver | Muscle | PPI | |||
| M10462 | Adipocytokine signaling pathway | N/A **, N/A ¶, N/A ¥, N/A †, 33 § | N/A | N/A | N/A | N/A | GSK3B, FRAP1, HSP90AA2, PDPK1, IKBKB |
| M10792 | MAPK signaling pathway | N/A **, N/A ¶, N/A ¥, N/A †, 63 § | N/A | N/A | N/A | N/A | MAPK9 *, MAPK8 *, MAP2K1 *, MAP3K11 *, MAPK10 |
| M18155 | Insulin signaling pathway | N/A **, N/A ¶, N/A ¥, N/A †, 58 § | N/A | N/A | N/A | N/A | IRS1 *, HRAS *, RAC1, JAK1, RPS6KA3 |
| M699 | Fatty acid metabolism | 30 **, N/A ¶, 30 ¥, 28 †, N/A § | HADHB *, ACADVL *, ECHS1 *, ETFDH | N/A | HADH *, ACADM * | HADHB * | N/A |
| rctm0354 | EGFR downregulation | N/A **, N/A ¶, N/A ¥, N/A †, 15 § | N/A | N/A | N/A | N/A | EGF *, UBA52 *, EGFR, UBC, RPS27A |
| rctm0591 | Innate immune system | 251 **, N/A ¶, 252 ¥, 223 †, 282 § | LAT2 *, PTPN6, NCKAP1L, IL10RA, IRF5 | N/A | TYROBP *, NCKAP1L, RAC2, NCF2, IGSF6 | AK014135, COTL1 | GRB2 *, MAPKAPK2, RAP2A, FRK, C1QC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, S.Y. Multi-Omics Data Analysis Uncovers Molecular Networks and Gene Regulators for Metabolic Biomarkers. Biomolecules 2021, 11, 406. https://doi.org/10.3390/biom11030406
Jung SY. Multi-Omics Data Analysis Uncovers Molecular Networks and Gene Regulators for Metabolic Biomarkers. Biomolecules. 2021; 11(3):406. https://doi.org/10.3390/biom11030406
Chicago/Turabian StyleJung, Su Yon. 2021. "Multi-Omics Data Analysis Uncovers Molecular Networks and Gene Regulators for Metabolic Biomarkers" Biomolecules 11, no. 3: 406. https://doi.org/10.3390/biom11030406
APA StyleJung, S. Y. (2021). Multi-Omics Data Analysis Uncovers Molecular Networks and Gene Regulators for Metabolic Biomarkers. Biomolecules, 11(3), 406. https://doi.org/10.3390/biom11030406