
The ‘Shape-Shifter’ Peptide from the Disulphide Isomerase PmScsC Shows Context-Dependent Conformational Preferences


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. NMR Spectroscopy and NMR Data Analysis
2.3. MD Simulations
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, W.L.; Kinch, L.N.; Karplus, P.A.; Grishin, N.V. ChSeq: A database of chameleon sequences. Protein Sci. 2015, 24, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. On the use of sequence homologies to predict protein-structure—identical pentapeptides can have completely different conformations. Proc. Nat. Acad. Sci. USA 1984, 81, 1075–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.T.; Jaromczyk, J.W.; Xu, Y. Analysis of chameleon sequences and their implications in biological processes. Proteins 2007, 67, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Gendoo, D.M.A.; Harrison, P.M. Discordant and chameleon sequences: Their distribution and implications for amyloidogenicity. Protein Sci. 2011, 20, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Bahramali, G.; Goliaei, B.; Minuchehr, Z.; Salari, A. Chameleon sequences in neurodegenerative diseases. Biochem. Biophys. Res. Comm. 2016, 472, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Chockalingam, K.; Blenner, M.; Banta, S. Design and application of stimulus-responsive peptide systems. Protein Eng. Des. Sel. 2007, 20, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Bioch. Biophys. Acta Proteins Proteom. 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [Green Version]
- Hsu, W.L.; Oldfield, C.J.; Xue, B.; Meng, J.W.; Huang, F.; Romero, P.; Uversky, V.N.; Dunker, A.K. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013, 22, 258–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Furlong, E.J.; Lo, A.W.; Kurth, F.; Premkumar, L.; Totsika, M.; Achard, M.E.S.; Halili, M.A.; Heras, B.; Whitten, A.E.; Choudhury, H.G.; et al. A shape-shifting redox foldase contributes to Proteus mirabilis copper resistance. Nat. Commun. 2017, 8, 16065. [Google Scholar] [CrossRef] [PubMed]
- Furlong, E.J.; Kurth, F.; Premkumar, L.; Whitten, A.E.; Martin, J.L. Engineered variants provide new insight into the structural properties important for activity of the highly dynamic, trimeric protein disulfide isomerase ScsC from Proteus mirabilis. Acta Cryst. D 2019, 75, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe—A multidimensional spectral processing system based on Unix Pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, P.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Marsh, J.A.; Singh, V.K.; Jia, Z.C.; Forman-Kay, J.D. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: Implications for fibrillation. Protein Sci. 2006, 15, 2795–2804. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.4.0; Schrödinger, LLC: New York, NY, USA, 2015; Available online: https://pymol.org/2/support.html?#citing (accessed on 26 April 2021).
- Schmid, N.; Christ, C.D.; Christen, M.; Eichenberger, A.P.; van Gunsteren, W.F. Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comput. Phys. Comm. 2012, 183, 890–903. [Google Scholar] [CrossRef]
- Gromos. Available online: http://www.gromos.net (accessed on 1 October 2018).
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. Biophys. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; Pullman, B., Ed.; Reidel: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of cartesian equations of motion of a system with constraints—molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Tironi, I.G.; Sperb, R.; Smith, P.E.; van Gunsteren, W.F. A generalised reaction field method for molecular dynamics simulations. J. Chem. Phys. 1995, 102, 5451–5459. [Google Scholar] [CrossRef]
- Daura, X.; van Gunsteren, W.F.; Mark, A.E. Folding-unfolding thermodynamics of a beta-heptapeptide from equilibrium simulations. Proteins 1999, 34, 269–280. [Google Scholar] [CrossRef]
- Munoz, V.; Serrano, L. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1994, 1, 399–409. [Google Scholar] [CrossRef]
- Penkett, C.J.; Redfield, C.; Dodd, I.; Hubbard, J.; McBay, D.L.; Mossakowska, D.E.; Smith, R.A.G.; Dobson, C.M.; Smith, L.J. NMR analysis of main-chain conformational preferences in an unfolded fibronectin-binding protein. J. Mol. Biol. 1997, 274, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M. Trifluoroethanol and colleagues: Cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 1998, 31, 297–355. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Bliven, S.; Lafita, A.; Parker, A.; Capitani, G.; Duarte, J.M. Automated evaluation of quaternary structures from protein crystals. PLoS Comp. Biol. 2018, 14, e1006104. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.J.; van Gunsteren, W.F.; Hansen, N. On the Use of Side-Chain NMR Relaxation Data to Derive Structural and Dynamical Information on Proteins: A Case Study Using Hen Lysozyme. ChemBioChem 2021, 22, 1049–1064. [Google Scholar] [CrossRef]
- Fandrich, M.; Fletcher, M.A.; Dobson, C.M. Amyloid fibrils from muscle myoglobin—Even an ordinary globular protein can assume a rogue guise if conditions are right. Nature 2001, 410, 165–166. [Google Scholar] [CrossRef]
- Pertinhez, T.A.; Bouchard, M.L.; Tomlinson, E.J.; Wain, R.; Ferguson, S.J.; Dobson, C.M.; Smith, L.J. Amyloid fibril formation by a helical cytochrome. FEBS Let. 2001, 495, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Webster, P.; Taddei, N.; Clark, A.; Stefani, M.; Ramponi, G.; Dobson, C.M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Nat. Acad. Sci. USA 1999, 96, 3590–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, Y.S.; Bugg, C.E.; Cook, W.J. Structure of calmodulin refined at 2.2 Ǻ resolution. J. Mol. Biol. 1988, 204, 191–204. [Google Scholar] [CrossRef]
- Taylor, D.A.; Sack, J.S.; Maune, J.F.; Beckingham, K.; Quiocho, F.A. Structure of a recombinant calmodulin from Drosophila-Melanogaster refined at 2.2-Ǻ resolution. J. Biol. Chem. 1991, 266, 21375–21380. [Google Scholar] [CrossRef]
- Barbato, G.; Ikura, M.; Kay, L.E.; Pastor, R.W.; Bax, A. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected 2-Dimensional NMR-spectroscopy—the central helix is flexible. Biochemistry 1992, 31, 5269–5278. [Google Scholar] [CrossRef]
- Horstmann, M.; Ehses, P.; Schweimer, K.; Steinert, M.; Kamphausen, T.; Fischer, G.; Hacker, J.; Rosch, P.; Faber, C. Domain motions of the Mip protein from Legionella pneumophila. Biochemistry 2006, 45, 12303–12311. [Google Scholar] [CrossRef]
- Wahl, M.C.; Bourenkov, G.P.; Bartunik, H.D.; Huber, R. Flexibility, conformational diversity and two dimerization modes in complexes of ribosomal protein L12. EMBO J. 2000, 19, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocharov, E.V.; Sobol, A.G.; Pavlov, K.V.; Korzhnev, D.M.; Jaravine, V.A.; Gudkov, A.T.; Arseniev, A.S. From structure and dynamics of protein L7/L12 to molecular switching in ribosome. J. Biol. Chem. 2004, 279, 17697–17706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liljas, A.; Sanyal, S. The enigmatic ribosomal stalk. Q. Rev. Biophys. 2018, 51, e12. [Google Scholar] [CrossRef] [Green Version]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikura, M.; Clore, G.M.; Gronenborn, A.M.; Zhu, G.; Klee, C.B.; Bax, A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 1992, 256, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Walden, P.M.; Whitten, A.E.; Premkumar, L.; Halili, M.A.; Heras, B.; King, G.J.; Martin, J.L. The atypical thiol-disulfide exchange protein alpha-DsbA2 from Wolbachia pipientis is a homotrimeric disulfide isomerase. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 283–295. [Google Scholar] [CrossRef] [Green Version]
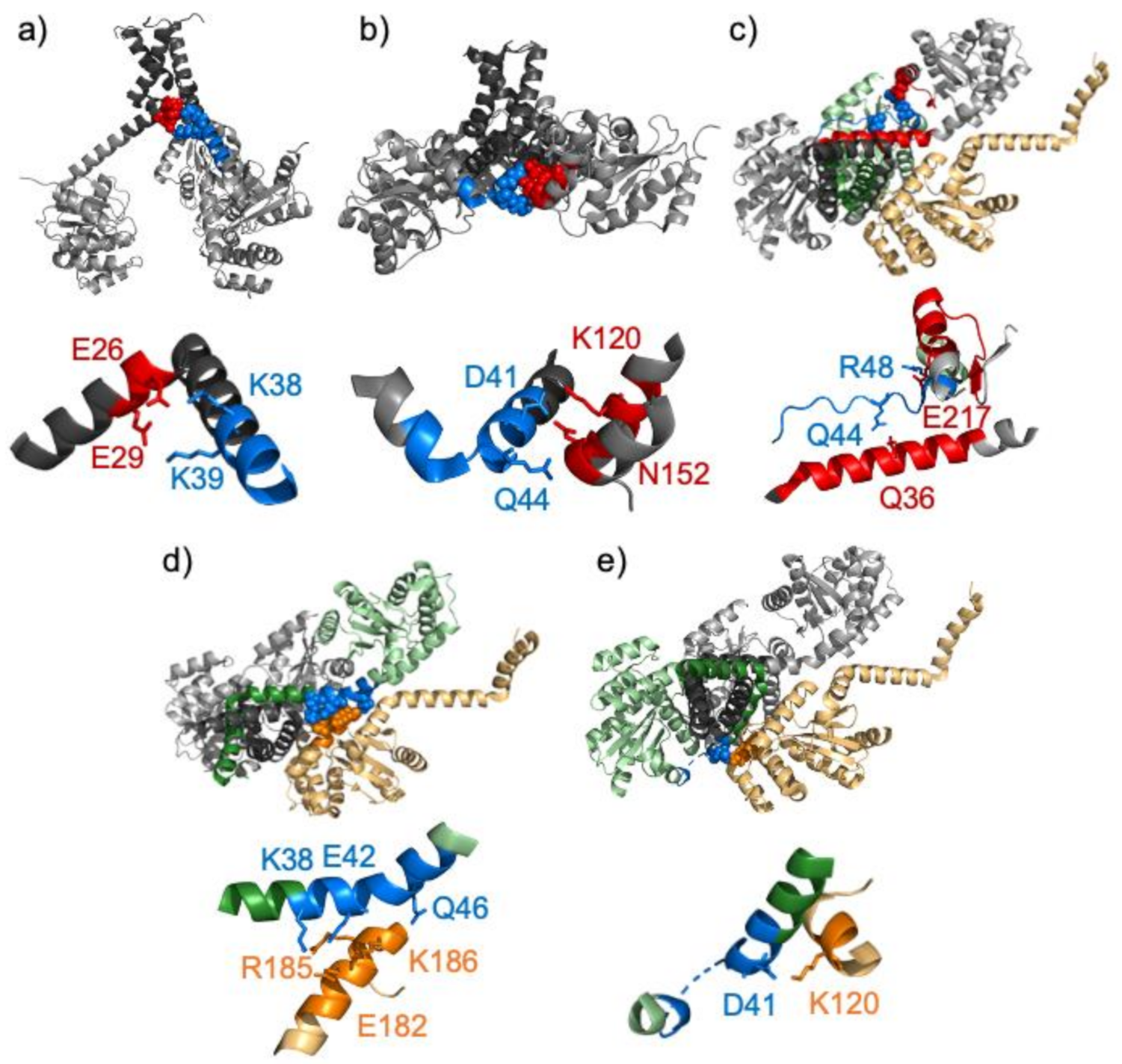
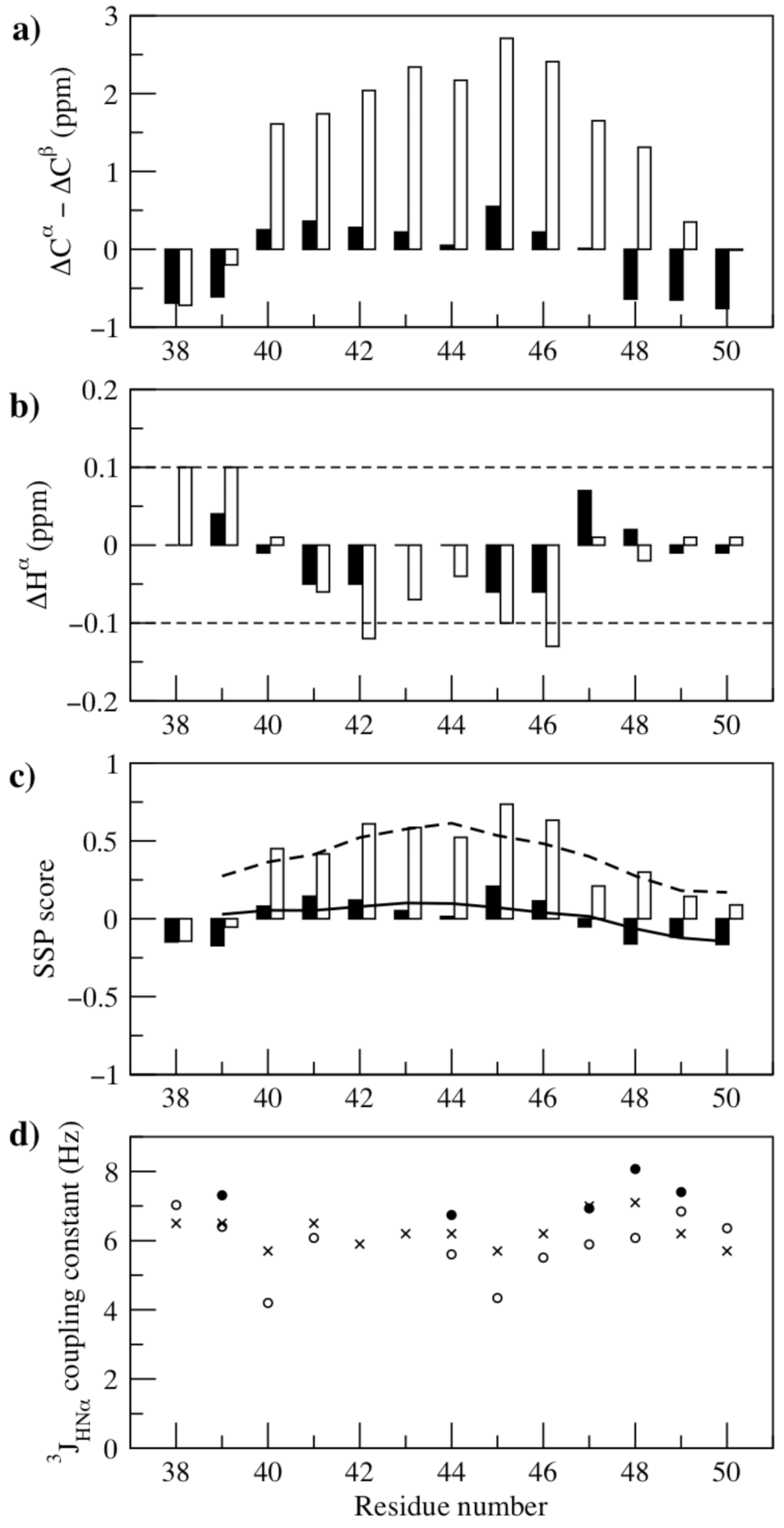
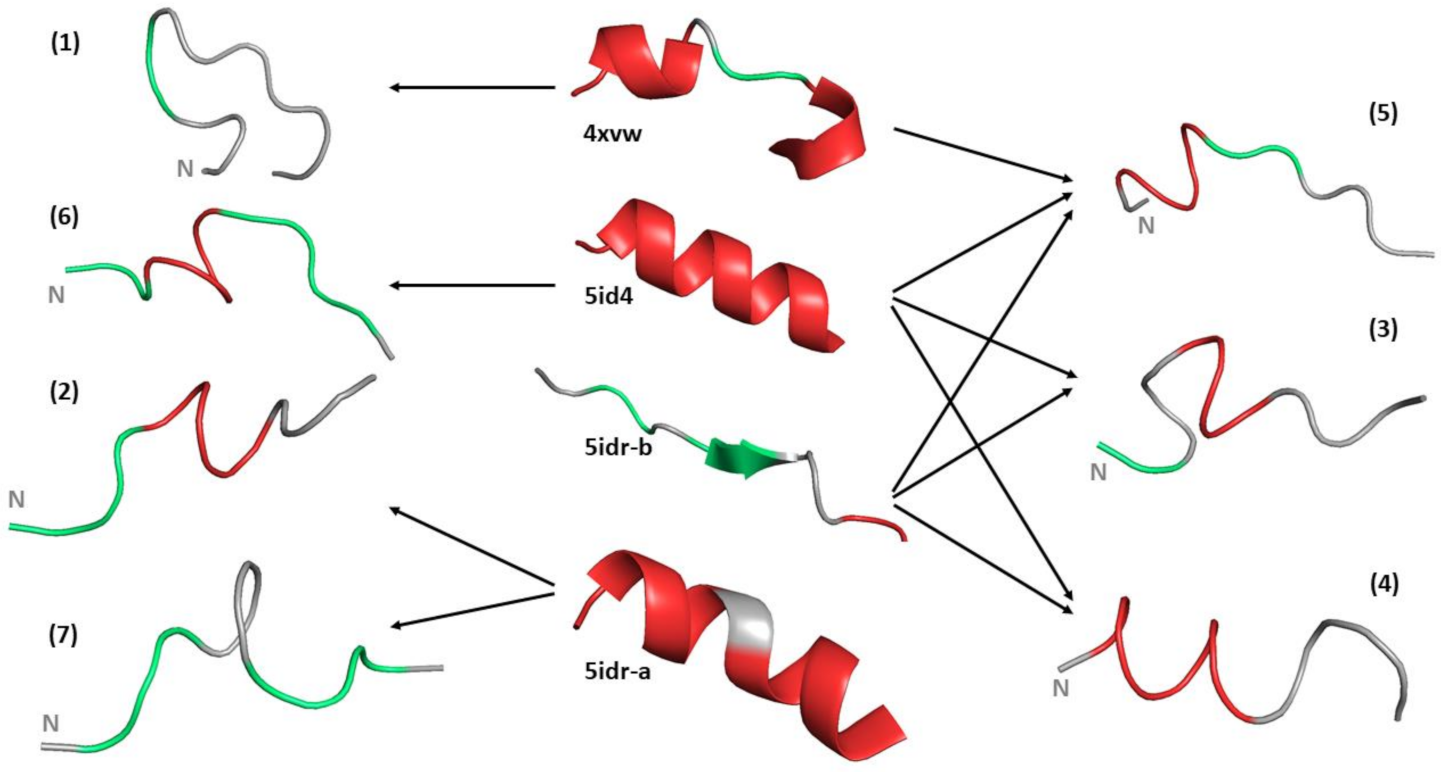
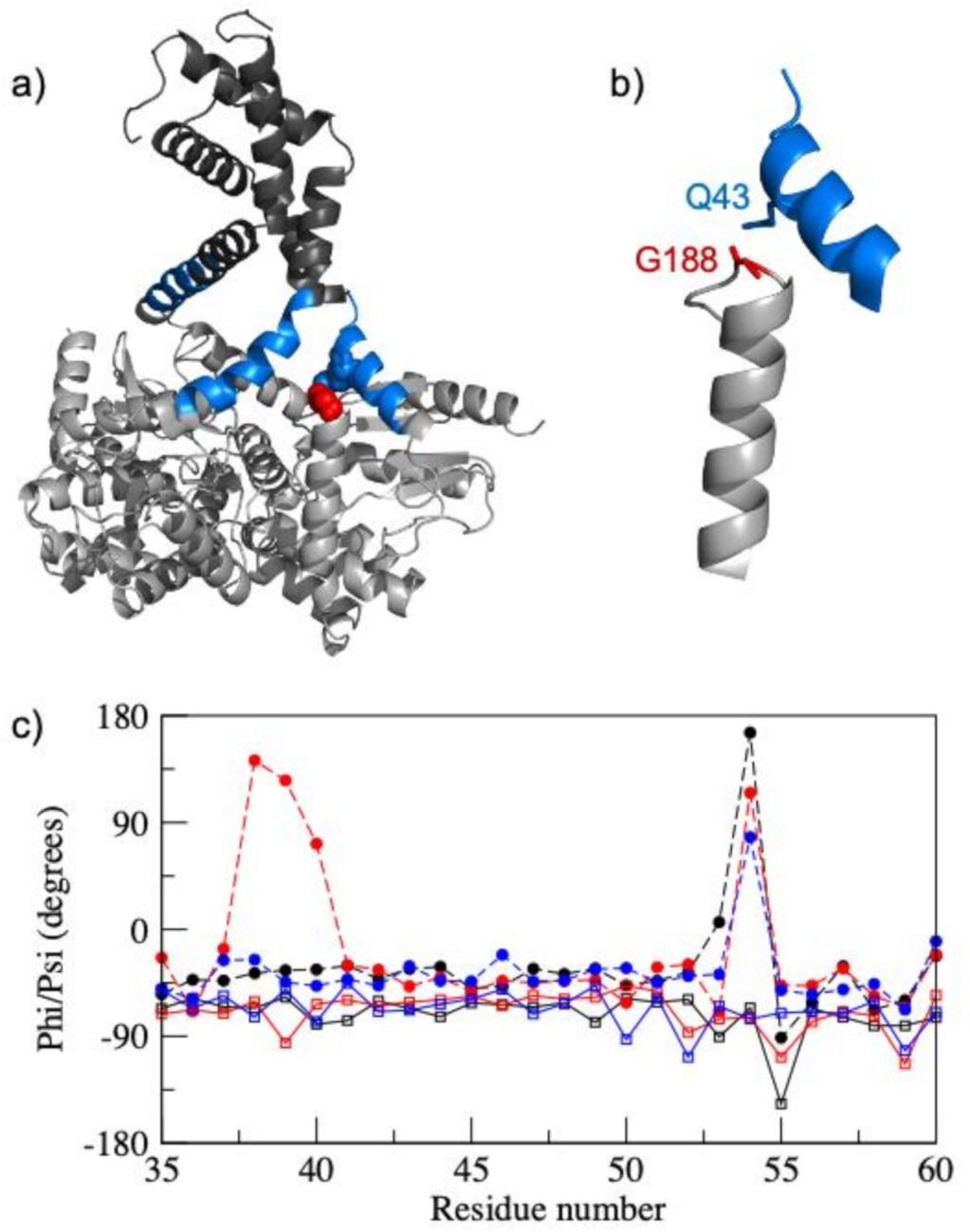

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogen Bond | 5idr_A | 5idr_B | 5id4 | 4vxw | Trajectory (%) |
|---|---|---|---|---|---|
| 40 NH-38 O | x | 1.5 | |||
| 40 NH-49 O | 20.0 | ||||
| 42 NH-38 O | x | x | x | 5.5 | |
| 42 NH-39 O | 20.0 | ||||
| 43 NH-39 O | x | x | x | 16.4 | |
| 44 NH-40 O | x | x | 19.2 | ||
| 44 NH-201 O | x | - | |||
| 45 NH-41 O | x | 33.8 | |||
| 45 NH-42 O | x | 6.6 | |||
| 46 NHE22-40 O | x | 8.1 | |||
| 46 NH-42 O | x | 36.1 | |||
| 46 NH-46 OE1 | x | 4.9 | |||
| 47 NH-43 O | x | 29.3 | |||
| 48 NH-44 O | x | x | 20.4 | ||
| 48 NH11-41 OD1 | x | 0.1 | |||
| 49 NH-45 O | x | 17.0 | |||
| 50 NH-46 O | x | x | 8.1 | ||
| 50 NH-47 O | x | x | 2.1 | ||
| 201 NH-44 O | x | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, L.J.; Green, C.W.; Redfield, C. The ‘Shape-Shifter’ Peptide from the Disulphide Isomerase PmScsC Shows Context-Dependent Conformational Preferences. Biomolecules 2021, 11, 642. https://doi.org/10.3390/biom11050642
Smith LJ, Green CW, Redfield C. The ‘Shape-Shifter’ Peptide from the Disulphide Isomerase PmScsC Shows Context-Dependent Conformational Preferences. Biomolecules. 2021; 11(5):642. https://doi.org/10.3390/biom11050642
Chicago/Turabian StyleSmith, Lorna J., Chloe W. Green, and Christina Redfield. 2021. "The ‘Shape-Shifter’ Peptide from the Disulphide Isomerase PmScsC Shows Context-Dependent Conformational Preferences" Biomolecules 11, no. 5: 642. https://doi.org/10.3390/biom11050642
APA StyleSmith, L. J., Green, C. W., & Redfield, C. (2021). The ‘Shape-Shifter’ Peptide from the Disulphide Isomerase PmScsC Shows Context-Dependent Conformational Preferences. Biomolecules, 11(5), 642. https://doi.org/10.3390/biom11050642