
In Search of a Role for Extracellular Purine Enzymes in Bone Function

 and
and
Abstract
:1. Introduction
2. Brief Overview on the Activity of Purines and Related Receptors in Bone Function
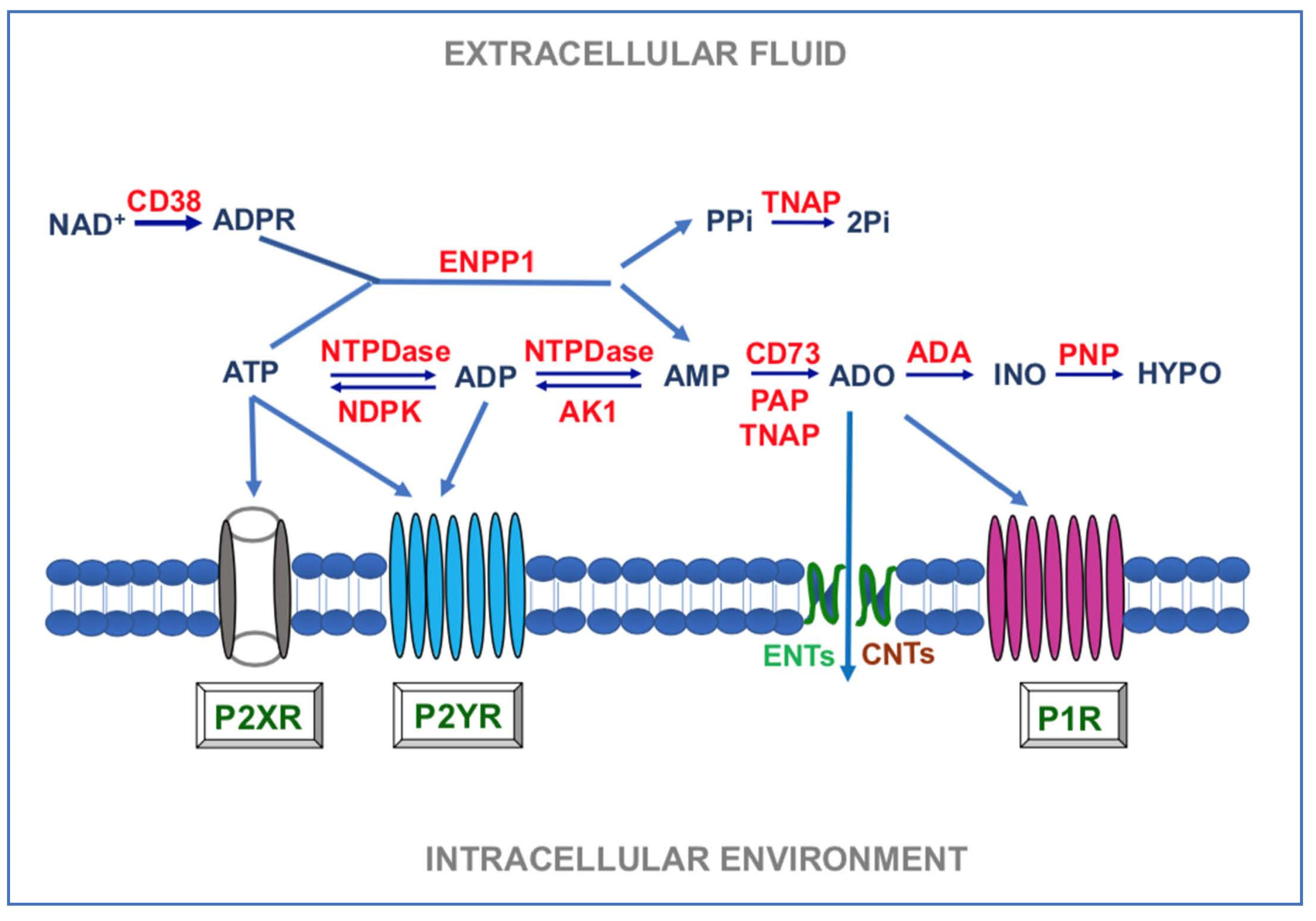
3. Enzymes and Other Mechanisms Generally Involved in the Turnover of Extracellular Purines
4. Role of Enzymes Deputed to Purine Metabolism in Bone Function and Diseases
5. Role of Purine Enzymes in Bone Inflammatory Conditions
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADO | adenosine |
| ADSCs | adipose-derived mesenchymal stromal cells |
| AK1 | adenylate kinase isoenzyme 1 |
| ANK | ankylosis protein |
| AR-A1R-A2AR-A2BR-A3R | Adenosine, A1, A2A, A2B, A3 receptors |
| BzATP | 2’(3’)-O-(4-Benzoylbenzoyl)adenosine-5’-triphosphate |
| CD38 | ADP-ribosyl cyclase 1 |
| CD73 | ecto-5′nucleotidase |
| CDK | chronic kidney disease |
| DAMPs | damage-associated molecular patterns |
| e-ADA | extracellular adenosine deaminase |
| ECM | extracellular matrix |
| ENPP | ectonucleotide pyrophosphatase/phosphodiesterase |
| HA | hydroxyapatite |
| HPP | hypophosphatasia |
| KO | knockout |
| MSCs | mesenchymal stem cells |
| MVs | matrix vesicles |
| NAD+ | nicotinamide adenine dinucleotide |
| NDPK | nucleoside diphosphokinase |
| NTPDases | ectonucleoside triphosphate diphosphohydrolases |
| P2 × 1R-P2 × 7R-P2Y2R-P2Y6R, P2Y13R, P2Y14R | P2 × 1, P2 × 7, P2Y2, P2Y6, P2Y13, P2Y14 receptors |
| PAP | prostatic acid phosphatase |
| Pi | inorganic phosphate |
| PNP | purine nucleoside phosphorylase |
| PPi | inorganic pyrophosphate |
| RA | rheumatoid arthritis |
| RANKL | receptor activator of nuclear factor kB ligand |
| SCI | severe combined immunodeficiency |
| TNAP | tissue nonspecific alkaline phosphatase |
| VNUT | vesicular nucleotide transporter |
References
- Oldknow, K.J.; Macrae, V.E.; Farquharson, C. Endocrine role of bone: Recent and emerging perspectives beyond osteocalcin. J. Endocrinol. 2015, 225, R1–R19. [Google Scholar] [CrossRef] [Green Version]
- Karkache, I.Y.; Damodaran, J.R.; Molstad, D.H.; Bradley, E.W. Serine/threonine phosphatases in osteoclastogenesis and bone resorption. Gene 2021, 771, 145362. [Google Scholar] [CrossRef]
- Intemann, J.; De Gorter, D.J.; Naylor, A.J.; Dankbar, B.; Wehmeyer, C. Importance of osteocyte-mediated regulation of bone remodelling in inflammatory bone disease. Swiss Med. Wkly. 2020, 150, w20187. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.d.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [Green Version]
- El Sayed, S.A.; Nezwek, T.A.; Varacallo, M. Physiology, Bone. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Agas, D.; Lacava, G.; Sabbieti, M.G. Bone and bone marrow disruption by endocrine-active substances. J. Cell. Physiol. 2019, 234, 192–213. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Jørgensen, N.R. Extracellular purines and bone homeostasis. Biochem. Pharmacol. 2021, 114425. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, M.P.; Middlemiss, P.J.; Gysbers, J.W.; Andrew, C.; Herman, M.A.; Reed, J.K.; Ciccarelli, R.; Di Iorio, P.; Caciagli, F. Trophic effects of purines in neurons and glial cells. Prog. Neurobiol. 1999, 59, 663–690. [Google Scholar] [CrossRef]
- Camici, M.; Garcia-Gil, M.; Tozzi, M.G. The Inside Story of Adenosine. Int. J. Mol. Sci. 2018, 19, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Mbalaviele, G. Role of APD-Ribosylation in Bone Health and Disease. Cells 2019, 8, 1201. [Google Scholar] [CrossRef] [Green Version]
- Lazarowski, E.R.; Sesma, J.I.; Seminario-Vidal, L.; Kreda, S.M. Molecular Mechanisms of Purine and Pyrimidine Nucleotide Release. Adv. Pharmacol. 2011, 61, 221–261. [Google Scholar] [CrossRef]
- Burnstock, G. Introduction to Purinergic Signalling in the Brain. Adv. Exp. Med. Biol. 2020, 1202, 1–12. [Google Scholar] [CrossRef]
- Di Liberto, V.; Mudò, G.; Garozzo, R.; Frinchi, M.; Fernandez-Dueñas, V.; Di Iorio, P.; Ciccarelli, R.; Caciagli, F.; Condorelli, D.F.; Ciruela, F.; et al. The Guanine-Based Purinergic System: The Tale of an Orphan Neuromodulation. Front. Pharmacol. 2016, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharma-cology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Kennedy, C. The P2Y/P2X divide: How it began. Biochem. Pharmacol. 2021, 114408. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Nakao-Kuroishi, K.; Kometani-Gunjigake, K.; Mizuhara, M.; Shirakawa, T.; Ito-Sago, M.; Yasuda, K.; Nakatomi, M.; Matsubara, T.; Tada-Shigeyama, Y.; et al. VNUT/SLC17A9, a vesicular nucleotide transporter, regulates osteoblast differentiation. FEBS Open Bio 2020, 10, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Song, L.; Zhang, Y.; Zhang, T.; Li, J.; Tao, C. Purinergic Signaling Mediates PTH and Fluid Flow-Induced Osteoblast Proliferation. BioMed Res. Int. 2021, 2021, 6674570. [Google Scholar] [CrossRef] [PubMed]
- Mikolajewicz, N.; Zimmermann, E.A.; Willie, B.M.; Komarova, S.V. Mechanically stimulated ATP release from murine bone cells is regulated by a balance of injury and repair. eLife 2018, 7, 37812. [Google Scholar] [CrossRef]
- Mikolajewicz, N.; Komarova, S.V. Role of UDP-Sugar Receptor P2Y14 in Murine Osteoblasts. Int. J. Mol. Sci. 2020, 21, 2747. [Google Scholar] [CrossRef]
- Mikolajewicz, N.; Sehayek, S.; Wiseman, P.W.; Komarova, S.V. Transmission of Mechanical Information by Purinergic Signaling. Biophys. J. 2019, 116, 2009–2022. [Google Scholar] [CrossRef]
- Sindhavajiva, P.R.; Sastravaha, P.; Arksornnukit, M.; Pavasant, P. Intermittent compressive force induces human mandibu-lar-derived osteoblast differentiation via WNT/β-catenin signaling. J. Cell Biochem. 2018, 119, 3474–3485. [Google Scholar] [CrossRef]
- Sindhavajiva, P.R.; Sastravaha, P.; Arksornnukit, M.; Pavasant, P. Purinergic 2X7 receptor activation regulates WNT signal-ing in human mandibular-derived osteoblasts. Arch. Oral Biol. 2017, 81, 167–174. [Google Scholar] [CrossRef]
- Carluccio, M.; Zuccarini, M.; Ziberi, S.; Giuliani, P.; Morabito, C.; Mariggiò, M.A.; Lonardo, M.T.; Adinolfi, E.; Orioli, E.; Di Iorio, P.; et al. Involvement of P2X7 Receptors in the Osteogenic Differentiation of Mesenchymal Stromal/Stem Cells Derived from Human Subcutaneous Adipose Tissue. Stem Cell Rev. Rep. 2019, 15, 574–589. [Google Scholar] [CrossRef]
- Bratengeier, C.; Bakker, A.D.; Fahlgren, A. Mechanical loading releases osteoclastogenesis-modulating factors through stimulation of the P2X7 receptor in hematopoietic progenitor cells. J. Cell. Physiol. 2019, 234, 13057–13067. [Google Scholar] [CrossRef]
- Binderman, I.; Gadban, N.; Yaffe, A. Extracellular ATP is a key modulator of alveolar bone loss in periodontitis. Arch. Oral Biol. 2017, 81, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Orriss, I.R.; Guneri, D.; Hajjawi, M.O.R.; Shaw, K.; Patel, J.J.; Arnett, T.R. Activation of the P2Y2 receptor regulates bone cell function by enhancing ATP release. J. Endocrinol. 2017, 233, 341–356. [Google Scholar] [CrossRef]
- Eisenstein, A.; Chitalia, S.V.; Ravid, K. Bone Marrow and Adipose Tissue Adenosine Receptors Effect on Osteogenesis and Adipogenesis. Int. J. Mol. Sci. 2020, 21, 7470. [Google Scholar] [CrossRef] [PubMed]
- Gharibi, B.; Abraham, A.A.; Ham, J.; Evans, B.A.J. Adenosine receptor subtype expression and activation influence the dif-ferentiation of mesenchymal stem cells to osteoblasts and adipocytes. J. Bone Miner. Res. 2011, 2, 2112–2124. [Google Scholar] [CrossRef] [Green Version]
- Carroll, S.H.; Wigner, N.A.; Kulkarni, N.; Johnston-Cox, H.; Gerstenfeld, L.C.; Ravid, K. A2B Adenosine Receptor Promotes Mesenchymal Stem Cell Differentiation to Osteoblasts and Bone Formation In Vivo. J. Biol. Chem. 2012, 287, 15718–15727. [Google Scholar] [CrossRef] [Green Version]
- Ciciarello, M.; Zini, R.; Rossi, L.; Salvestrini, V.; Ferrari, D.; Manfredini, R.; Lemoli, R.M. Extracellular Purines Promote the Differentiation of Human Bone Marrow-Derived Mesenchymal Stem Cells to the Osteogenic and Adipogenic Lineages. Stem Cells Dev. 2013, 22, 1097–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajjawi, M.O.R.; Patel, J.J.; Corcelli, M.; Arnett, T.; Orriss, I.R. Lack of effect of adenosine on the function of rodent osteo-blasts and osteoclasts in vitro. Purinergic Signal. 2016, 12, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Mediero, A.; Cronstein, B.N. Adenosine and bone metabolism. Trends Endocrinol. Metab. 2013, 24, 290–300. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Webb, N.E.; Song, Y.; Tuan, R.S. Identification and Functional Analysis of Candidate Genes Regulating Mesenchymal Stem Cell Self-Renewal and Multipotency. Stem Cells 2006, 24, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Kukulski, F.; Lévesque, S.A.; Sévigny, J. Impact of Ectoenzymes on P2 and P1 Receptor Signaling. Adv. Pharmacol. 2011, 61, 263–299. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Ye, W.; Chen, H.; Kuang, X.; Guo, J.; Xiang, M.; Peng, C.; Chen, X.; Liu, H. Role of purines in regulation of metabolic reprogramming. Purinergic Signal. 2019, 15, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, H. Ectonucleotidases: Some recent developments and a note on nomenclature. Drug Dev. Res. 2001, 52, 44–56. [Google Scholar] [CrossRef]
- Patel, J.J.; Zhu, D.; Opdebeeck, B.; D’Haese, P.; Millán, J.L.; Bourne, L.E.; Wheeler-Jones, C.P.; Arnett, T.R.; Macrae, V.E.; Orriss, I.R. Inhibition of arterial medial calcification and bone mineralization by extracellular nucleotides: The same functional effect mediated by different cellular mechanisms. J. Cell. Physiol. 2018, 233, 3230–3243. [Google Scholar] [CrossRef]
- Millán, J.L. Alkaline Phosphatases: Structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006, 2, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, H. Prostatic acid phosphatase, a neglected ectonucleotidase. Purinergic Signal. 2009, 5, 273–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losenkova, K.; Zuccarini, M.; Karikoski, M.; Laurila, J.; Boison, D.; Jalkanen, S.; Yegutkin, G.G. Compartmentalization of adenosine metabolism in cancer cells and its modulation during acute hypoxia. J. Cell Sci. 2020, 133, jcs241463. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, J.; Loukovaara, S.; Losenkova, K.; Zuccarini, M.; Korhonen, A.M.; Lehti, K.; Kauppinen, A.; Kaarniranta, K.; Müller, C.E.; Jalkanen, S.; et al. Soluble and membrane-bound adenylate kinase and nucleotidases augment ATP-mediated inflammation in diabetic retinopathy eyes with vitreous hemorrhage. J. Mol. Med. 2019, 97, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Minor, M.; Alcedo, K.P.; Battaglia, R.A.; Snider, N.T. Cell type- and tissue-specific functions of ecto-5′-nucleotidase (CD73). Am. J. Physiol. Physiol. 2019, 317, C1079–C1092. [Google Scholar] [CrossRef]
- Longhi, M.S.; Robson, S.C.; Bernstein, S.H.; Serra, S.; Deaglio, S. Biological functions of ecto-enzymes in regulating extra-cellular adenosine levels in neoplastic and inflammatory disease states. J. Mol. Med. 2013, 91, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yegutkin, G.G. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: Functional implications and measurement of activities. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 473–497. [Google Scholar] [CrossRef]
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Pacher, P.; Haskó, G. Adenosine signaling and the immune system: When a lot could be too much. Immunol. Lett. 2019, 205, 9–15. [Google Scholar] [CrossRef]
- Camici, M.; Garcia-Gil, M.; Pesi, R.; Allegrini, S.; Tozzi, M.G. Purine-Metabolising Enzymes and Apoptosis in Cancer. Cancers 2019, 11, 1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor-Anglada, M.; Pérez-Torras, S. Nucleoside transporter proteins as biomarkers of drug responsiveness and drug targets. Front. Pharmacol. 2015, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Kutryb-Zajac, B.; Mierzejewska, P.; Slominska, E.M.; Smolenski, R.T. Therapeutic Perspectives of Adenosine Deaminase Inhibition in Cardiovascular Diseases. Molecules 2020, 25, 4652. [Google Scholar] [CrossRef]
- Inoue, K. Molecular Basis of Nucleobase Transport Systems in Mammals. Biol. Pharm. Bull. 2017, 40, 1130–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, J.L.; Lawson, K.V.; Powers, J.P. Targeting Metabolism of Extracellular Nucleotides via Inhibition of Ectonucleotidases CD73 and CD39. J. Med. Chem. 2020, 63, 13444–13465. [Google Scholar] [CrossRef]
- Zimmermann, H. Ectonucleoside triphosphate diphosphohydrolases and ecto-5′-nucleotidase in purinergic signaling: How the field developed and where we are now. Purinergic Signal. 2021, 17, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Bussiere, D.E. Targeting the Purinome. Methods Mol. Biol. 2009, 575, 47–92. [Google Scholar] [CrossRef]
- Demenis, M.A.; Furriel, R.P.M.; Leone, F.A. Characterization of an ectonucleoside triphosphate diphosphohydrolase 1 activity in alkaline phosphatase-depleted rat osseous plate membranes: Possible functional involvement in the calcification process. Biochim. Biophys. Acta 2003, 1646, 216–225. [Google Scholar] [CrossRef]
- Noronha-Matos, J.B.; Costa, M.A.C.; Magalhães-Cardoso, M.; Ferreirinha, F.; Pelletier, J.; Freitas, R.; Neves, J.; Sévigny, J.; Correia-De-Sá, P.; Magalhães-Cardoso, T. Role of ecto-NTPDases on UDP-sensitive P2Y6receptor activation during osteogenic differentiation of primary bone marrow stromal cells from postmenopausal women. J. Cell. Physiol. 2011, 227, 2694–2709. [Google Scholar] [CrossRef]
- Wu, W.; Xiao, Z.; Chen, Y.; Deng, Y.; Zeng, D.; Liu, Y.; Huang, F.; Wang, J.; Liu, Y.; Bellanti, J.A.; et al. CD39 Produced from Human GMSCs Regulates the Balance of Osteoclasts and Osteoblasts through the Wnt/β-Catenin Pathway in Osteoporosis. Mol. Ther. 2020, 28, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wu, W.; Gu, J.; Zhang, X.; Dang, J.; Wang, J.; Zheng, Y.; Huang, F.; Yuan, J.; Xue, Y.; et al. Human gingival tissue-derived MSC suppress osteoclastogenesis and bone erosion via CD39-adenosine signal pathway in autoimmune arthritis. EBioMedicine 2019, 43, 620–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Q.; Li, C.; Lu, Y.; Geng, R.; Wei, J.; Hu, J. Adipose-derived mesenchymal stromal cells suppress osteoclastogenesis and bone erosion in collagen-induced arthritis. Scand. J. Immunol. 2020, 92, e12877. [Google Scholar] [CrossRef]
- Giachelli, C. Inducers and inhibitors of biomineralization: Lessons from pathological calcification. Orthod. Craniofac. Res. 2005, 8, 229–231. [Google Scholar] [CrossRef]
- Terkeltaub, R.A. Inorganic pyrophosphate generation and disposition in pathophysiology. Am. J. Physiol. Physiol. 2001, 281, C1–C11. [Google Scholar] [CrossRef] [PubMed]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G.G. Pyrophosphate: A key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orriss, I.R. Extracellular pyrophosphate: The body’s “water softener”. Bone 2020, 134, 115243. [Google Scholar] [CrossRef]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ciancaglini, P.; Yadav, M.C.; Simão, A.M.S.; Narisawa, S.; Pizauro, J.M.; Farquharson, C.; Hoylaerts, M.F.; Millán, J.L. Kinetic Analysis of Substrate Utilization by Native and TNAP-, NPP1- or PHOSPHO1-Deficient Matrix Vesicles. J. Bone Miner. Res. 2009, 25, 716–737. [Google Scholar] [CrossRef]
- Simão, A.M.S.; Bolean, M.; Favarin, B.Z.; Veschi, E.A.; Tovani, C.B.; Ramos, A.P.; Bottini, M.; Buchet, R.; Millán, J.L.; Ciancaglini, P. Lipid microenvironment affects the ability of proteoliposomes harboring TNAP to induce mineralization without nucleators. J. Bone Miner. Metab. 2018, 37, 607–613. [Google Scholar] [CrossRef]
- Vaingankar, S.M.; Fitzpatrick, T.A.; Johnson, K.; Goding, J.W.; Maurice, M.; Terkeltaub, R. Subcellular targeting and function of osteoblast nucleotide pyrophosphatase phosphodiesterase 1. Am. J. Physiol. Physiol. 2004, 286, C1177–C1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millán, J.L. Concerted Regulation of Inorganic Pyrophosphate and Osteopontin by Akp2, Enpp1, and Ank, an integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 2004, 164, 1199–1209. [Google Scholar] [CrossRef]
- Anderson, H.C.; Harmey, D.; Camacho, N.P.; Garimella, R.; Sipe, J.B.; Tague, S.; Bi, X.; Johnson, K.; Terkeltaub, R.; Millán, J.L. Sustained Osteomalacia of Long Bones Despite Major Improvement in Other Hypophosphatasia-Related Mineral Deficits in Tissue Nonspecific Alkaline Phosphatase/Nucleotide Pyrophosphatase Phosphodiesterase 1 Double-Deficient Mice. Am. J. Pathol. 2005, 166, 1711–1720. [Google Scholar] [CrossRef] [Green Version]
- Brandao-Burch, A.; Utting, J.C.; Orriss, I.R.; Arnett, T.R. Acidosis Inhibits Bone Formation by Osteoblasts In Vitro by Preventing Mineralization. Calcif. Tissue Int. 2005, 77, 167–174. [Google Scholar] [CrossRef]
- Orriss, I.R.; Key, M.L.; Hajjawi, M.O.; Millán, J.L.; Arnett, T.R. Acidosis is a key regulator of osteoblast ecto-nucleotidase pyrophosphatase/phosphodiesterase 1 (NPP1) expression and activity. J. Cell Physiol. 2015, 230, 49–56. [Google Scholar] [CrossRef]
- Wolf, M.; Ao, M.; Chavez, M.; Kolli, T.; Thumbigere-Math, V.; Becker, K.; Chu, E.; Jäger, A.; Somerman, M.; Foster, B. Reduced Orthodontic Tooth Movement in Enpp1 Mutant Mice with Hypercementosis. J. Dent. Res. 2018, 97, 937–945. [Google Scholar] [CrossRef]
- Takedachi, M.; Oohara, H.; Smith, B.J.; Iyama, M.; Kobashi, M.; Maeda, K.; Long, C.L.; Humphrey, M.B.; Stoecker, B.J.; Toyosawa, S.; et al. CD73-generated adenosine promotes osteoblast differentiation. J. Cell. Physiol. 2012, 227, 2622–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradaschia-Correa, V.; Josephson, A.M.; Egol, A.J.; Mizrahi, M.M.; Leclerc, K.; Huo, J.; Cronstein, B.N.; Leucht, P. Ecto-5′-nucleotidase (CD73) regulates bone formation and remodeling during intramembranous bone repair in aging mice. Tissue Cell 2017, 49, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-R.V.; Liu, M.; Kwon, S.K.; Iida, M.; Gong, Y.; Sangaj, N.; Varghese, S. Dysregulation of ectonucleotidase-mediated extracellular adenosine during postmenopausal bone loss. Sci. Adv. 2019, 5, eaax1387. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Mazumder, A.; Wilder, T.; Cronstein, B.N. Adenosine regulates bone metabolism via A 1, A 2A, and A 2B receptors in bone marrow cells from normal humans and patients with multiple myeloma. FASEB J. 2013, 27, 3446–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falahati-Nini, A.; Riggs, B.L.; Atkinson, E.J.; O’Fallon, W.M.; Eastell, R.; Khosla, S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J. Clin. Investig. 2000, 106, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Graser, S.; Liedtke, D.; Jakob, F. TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. Int. J. Mol. Sci. 2021, 22, 919. [Google Scholar] [CrossRef]
- Chrobak, P.; Charlebois, R.; Rejtar, P.; El Bikai, R.; Allard, B.; Stagg, J. CD73 Plays a Protective Role in Collagen-Induced Arthritis. J. Immunol. 2015, 194, 2487–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joolharzadeh, P.; Hilaire, C.S. CD73 (Cluster of Differentiation 73) and the Differences between Mice and Humans. Arter. Thromb. Vasc. Biol. 2019, 39, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Jaques, J.A.D.S.; Becker, L.V.; Souza, V.D.C.G.; Leal, C.A.M.; Bertoldo, T.M.D.; Pinheiro, K.D.V.; Morsch, V.M.; Schetinger, M.R.C.; Leal, D.B.R. Activities of enzymes that hydrolyze adenine nucleotides in lymphocytes from patients with rheumatoid arthritis. Cell Biochem. Funct. 2013, 31, 395–399. [Google Scholar] [CrossRef] [PubMed]
- da Silveira, K.L.; da Silveira, L.L.; Thorstenberg, M.L.P.; Cabral, F.L.; Castilhos, L.G.; Rezer, J.F.P.; de Andrade, D.F.; Beck, R.C.R.; Palma, H.E.; de Andrade, C.M.; et al. Free and nanoencapsulated vitamin D3: Effects on E-NTPDase and E-ADA activities in an animal model with induced arthritis. Cell Biochem. Funct. 2016, 34, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Saccol, R.D.S.P.; Da Silveira, K.L.; Adefegha, S.A.; Manzoni, A.G.; Da Silveira, L.L.; Coelho, A.P.V.; Castilhos, L.G.; Abdalla, F.H.; Becker, L.V.; Martins, N.M.B.; et al. Effect of quercetin on E-NTPDase/E-ADA activities and cytokine secretion of complete Freund adjuvant–induced arthritic rats. Cell Biochem. Funct. 2019, 37, 474–485. [Google Scholar] [CrossRef]
- Kohn, D.B.; Hershfield, M.S.; Puck, J.M.; Aiuti, A.; Blincoe, A.; Gaspar, H.B.; Notarangelo, L.D.; Grunebaum, E. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2019, 143, 852–863. [Google Scholar] [CrossRef]
- Kendall, J.L.; Springer, J.M. The Many Faces of a Monogenic Autoinflammatory Disease: Adenosine Deaminase 2 Deficiency. Curr. Rheumatol. Rep. 2020, 22, 64. [Google Scholar] [CrossRef]
- Fardellone, P.; Salawati, E.; Le Monnier, L.; Goëb, V. Bone Loss, Osteoporosis, and Fractures in Patients with Rheumatoid Arthritis: A Review. J. Clin. Med. 2020, 9, 3361. [Google Scholar] [CrossRef]
- Yang, D.-H.; Yang, M.-Y. The Role of Macrophage in the Pathogenesis of Osteoporosis. Int. J. Mol. Sci. 2019, 20, 2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitzmann, M.N. T-cells and B-cells in osteoporosis. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.C.; Vullings, J.; van de Loo, F.A.J. Osteoporosis and osteoarthritis are two sides of the same coin paid for obesity. Nutrition 2020, 70, 110486. [Google Scholar] [CrossRef] [PubMed]
- Shahen, V.A.; Gerbaix, M.; Koeppenkastrop, S.; Lim, S.; McFarlane, K.; Nguyen, A.N.; Peng, X.; Weiss, N.; Brennan-Speranza, T. Multifactorial effects of hyperglycaemia, hyperinsulinemia and inflammation on bone remodelling in type 2 diabetes mellitus. Cytokine Growth Factor Rev. 2020, 55, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, S.; De Martini, N.; Rotondi, S.; Tartaglione, L.; Ureña-Torres, P.; Bover, J.; Pasquali, M.; ERA-EDTA Working Group on CKD-MBD. Bone, inflammation and chronic kidney disease. Clin. Chim. Acta 2020, 506, 236–240. [Google Scholar] [CrossRef]
- Compston, J.E.; McClung, M.R.; Leslie, W.D. Osteoporosis. Lancet 2019, 393, 364–376. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, S.; Leng, S.X. Chronic Low-grade Inflammatory Phenotype (CLIP) and Senescent Immune Dysregulation. Clin. Ther. 2019, 41, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, J.L.; Passos, D.F.; Bernardes, V.M.; Leal, D.B. ATP and adenosine: Role in the immunopathogenesis of rheumatoid arthritis. Immunol. Lett. 2019, 214, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.; Ceruti, S. Adenosine Signaling in Autoimmune Disorders. Pharmaceuticals 2020, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.L.; Sarti, A.C.; Di Virgilio, F. Ectonucleotidases in Acute and Chronic Inflammation. Front. Pharmacol. 2021, 11, 619458. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, B.A.; Elford, C.; Pexa, A.; Francis, K.; Hughes, A.C.; Deussen, A.; Ham, J. Human Osteoblast Precursors Produce Extracellular Adenosine, Which Modulates Their Secretion of IL-6 and Osteoprotegerin. J. Bone Miner. Res. 2005, 21, 228–236. [Google Scholar] [CrossRef]
- Lopez, C.D.; Bekisz, J.M.; Corciulo, C.; Mediero, A.; Coelho, P.G.; Witek, L.; Flores, R.L.; Cronstein, B.N. Local delivery of adenosine receptor agonists to promote bone regeneration and defect healing. Adv. Drug Deliv. Rev. 2019, 146, 240–247. [Google Scholar] [CrossRef]
- Witek, L.; Alifarag, A.M.; Tovar, N.; Lopez, C.D.; Cronstein, B.N.; Rodriguez, E.D.; Coelho, P.G. Repair of Critical-Sized Long Bone Defects Using Dipyridamole-Augmented 3D-Printed Bioactive Ceramic Scaffolds. J. Orthop. Res. 2019, 37, 2499–2507. [Google Scholar] [CrossRef] [PubMed]
- Diemar, S.S.; Lylloff, L.; Rønne, M.S.; Møllehave, L.T.; Heidemann, M.; Thuesen, B.H.; Johannesen, J.; Schou, A.J.; Husby, S.; Wedderkopp, N.; et al. Reference intervals in Danish children and adolescents for bone turnover markers carboxy-terminal cross-linked telopeptide of type I collagen (β-CTX), pro-collagen type I N-terminal propeptide (PINP), osteocalcin (OC) and bone-specific alkaline phosphatase (bone ALP). Bone 2021, 146, 115879. [Google Scholar] [CrossRef]
- Bourne, L.E.; Wheeler-Jones, C.P.; Orriss, I.R. Regulation of mineralisation in bone and vascular tissue: A comparative review. J. Endocrinol. 2021, 248, R51–R65. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Orriss, I.R.; Neven, E.; D’Haese, P.C.; Verhulst, A. Extracellular Nucleotides Regulate Arterial Calcification by Activating Both Independent and Dependent Purinergic Receptor Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 7636. [Google Scholar] [CrossRef] [PubMed]
- Goettsch, C.; Strzelecka-Kiliszek, A.; Bessueille, L.; Quillard, T.; Mechtouff, L.; Pikula, S.; Canet-Soulas, E.; Millan, J.L.; Fonta, C.; Magne, D. TNAP as a therapeutic target for cardiovascular calcification: A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2020, 299. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, P.; Ciccarelli, R. Adenine-Based Purines and Related Metabolizing Enzymes: Evidence for Their Impact on Tumor Extracellular Vesicle Activities. Cells 2021, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Braganhol, E.; Wink, M.R.; Lenz, G.; Battastini, A.M.O. Purinergic Signaling in Glioma Progression. Adv. Exp. Med. Biol. 2020, 1202, 87–108. [Google Scholar] [CrossRef]
- Vaisitti, T.; Arruga, F.; Guerra, G.; Deaglio, S. Ectonucleotidases in Blood Malignancies: A Tale of Surface Markers and Therapeutic Targets. Front. Immunol. 2019, 10, 2301. [Google Scholar] [CrossRef]
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 42, 101304. [Google Scholar] [CrossRef]
- Campos-Contreras, A.D.R.; Díaz-Muñoz, M.; Vázquez-Cuevas, F.G. Purinergic Signaling in the Hallmarks of Cancer. Cells 2020, 9, 1612. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Gao, Z.-W.; Zhang, H.-Z. The role of adenosinergic pathway in human autoimmune diseases. Immunol. Res. 2016, 64, 1133–1141. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Type of Enzyme | Experimental Model | Activity in Bone | References |
|---|---|---|---|
| CD39 | Alkaline phosphatase-depleted rat osseous plate membranes | Involvement in calcification process | [55] |
| Human bone marrow MSC (postmenopausal women) | Together with NTPDases 2 and 3 regulation of osteoblast proliferation or differentiation caused by UDP activity on P2Y6R | [56] | |
| Mouse gingiva MSCs | Promotion of ADO availability and its reparative osteogenesis in ovariectomized or arthritic mice | [57,58] | |
| Human ADSCs | Inhibition of RANKL-induced genesis of mice/human osteoclasts | [59] | |
| NPP1 | Different mammalian tissues | Inhibition of ECM mineralization | [61,62,63] |
| NPP1 (Enpp1−/−) KO mice | Development of soft tissue calcification as the result of reduced production or transport of PPi | [68] | |
| Rat osteoblasts | Acidosis/inflammation upregulate its expression. | [71] | |
| Rat osteoblasts/osteoclasts | NPP1 loss of function might directly affect cell function/recruitment, indirectly altering periodontal remodeling | [72] | |
| TNAP | Mouse osteoblast-derived MVs | Assures an adequate Pi/PPi ratio for normal bone mineralization. | [65] |
| Mice deficient in TNAP function (Akp2−/−) | Induction of rickets and osteomalacia due to blockade of HA crystals formation caused by an increase in extracellular PP1 concentrations | [69] | |
| Rat osteoblasts | Acidosis/inflammation downregulate its expression together with bone mineralization. | [70,71] | |
| Human deficiency | Various bone disorders | [78] | |
| CD73 | Human CD73 deficiency | Vascular calcification, arterio-megaly and tortuosity as well as calcification in small joints, without specific bone alterations | [44] |
| CD73 KO mice | Impaired osteoblast differentiation and decreased bone formation with development of osteopenia | [73] | |
| Osteoblasts and osteoclasts | Its expression and activity together with that of CD39 controlled by estrogens in mice and man. | [75,77] | |
| CD73 KO mice | Development of spontaneous arthritis and inflammatory symptoms | [79] | |
| Human and mouse tissues | Differences in enzyme activity | [80] | |
| ADA | Mouse model of RA | Decreased activity in arthritic mouse to preserve ADO levels and its anti-inflammatory and immuno-suppressive properties | [81] |
| Animal model of induced arthritis | Activity altered by vitamin D3 given as an alternative treatment for chronic arthritis | [82] | |
| Arthritic rats | Activity attenuated by Quercetin together that of IFN-gamma and IL-4 | [83] | |
| Human SCI | Bony dysplasia | [84] | |
| Human selective ADA2 defi-ciency | No reported bone alterations; vasculitis, hematological disease and immuno-deficiency were present | [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuccarini, M.; Giuliani, P.; Caciagli, F.; Ciccarelli, R.; Di Iorio, P. In Search of a Role for Extracellular Purine Enzymes in Bone Function. Biomolecules 2021, 11, 679. https://doi.org/10.3390/biom11050679
Zuccarini M, Giuliani P, Caciagli F, Ciccarelli R, Di Iorio P. In Search of a Role for Extracellular Purine Enzymes in Bone Function. Biomolecules. 2021; 11(5):679. https://doi.org/10.3390/biom11050679
Chicago/Turabian StyleZuccarini, Mariachiara, Patricia Giuliani, Francesco Caciagli, Renata Ciccarelli, and Patrizia Di Iorio. 2021. "In Search of a Role for Extracellular Purine Enzymes in Bone Function" Biomolecules 11, no. 5: 679. https://doi.org/10.3390/biom11050679
APA StyleZuccarini, M., Giuliani, P., Caciagli, F., Ciccarelli, R., & Di Iorio, P. (2021). In Search of a Role for Extracellular Purine Enzymes in Bone Function. Biomolecules, 11(5), 679. https://doi.org/10.3390/biom11050679