
Molecular Dynamics Simulations of Human FOXO3 Reveal Intrinsically Disordered Regions Spread Spatially by Intramolecular Electrostatic Repulsion


Abstract
: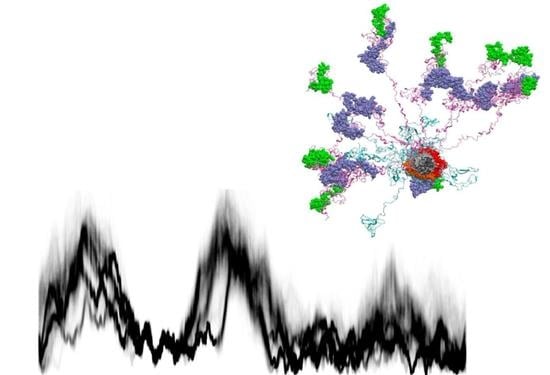
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
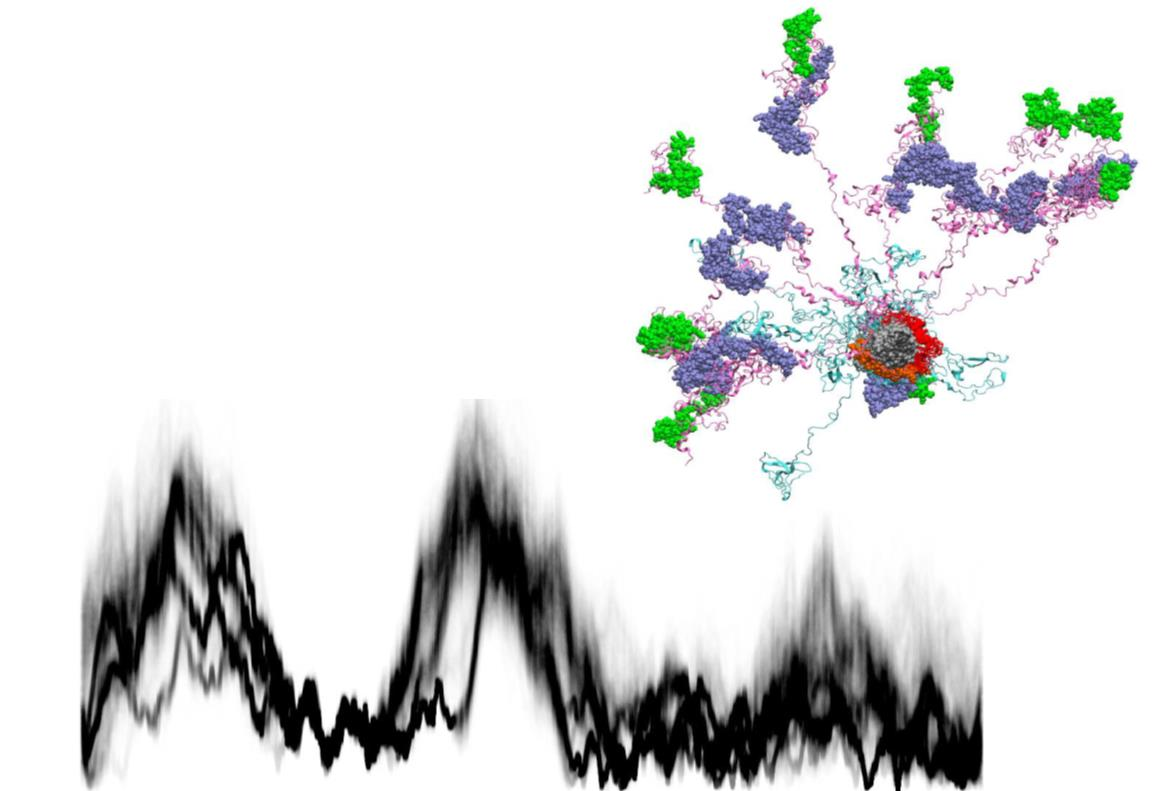
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analysis of Primary Sequence
2.2. Creation of the Starting Structure for MD Simulations
2.3. Implicit MD Simulations (gb8_md#1—gb8_md#10)
2.4. Accelerated Molecular Dynamics of FOXO120−530 in TIP4P-D Water
2.5. Trajectory Visualization and Analysis Methods
3. Results
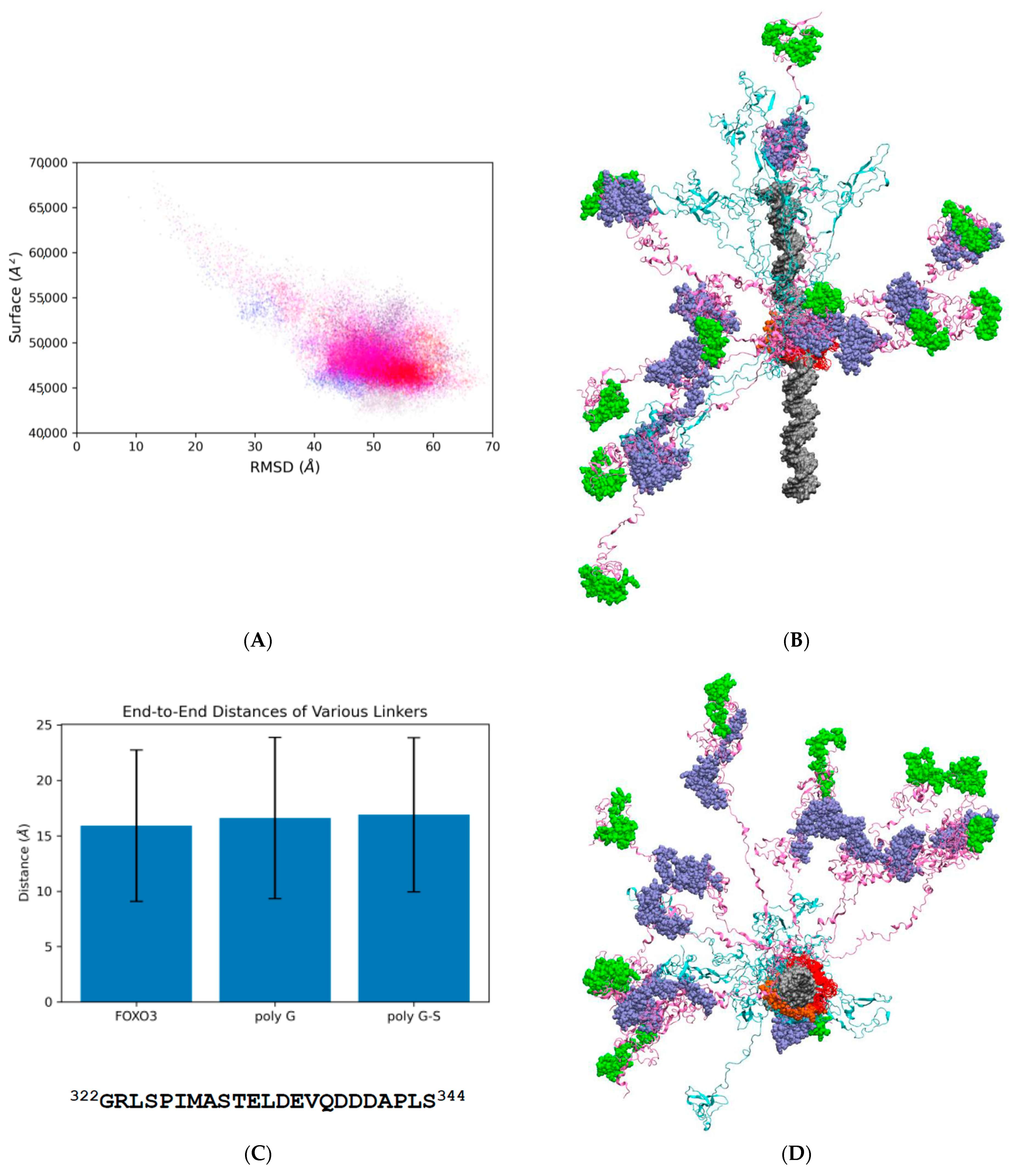
3.1. FOXO3 Is Highly Conserved in Evolution, but Represents an Unusually Extensively Disordered IDP
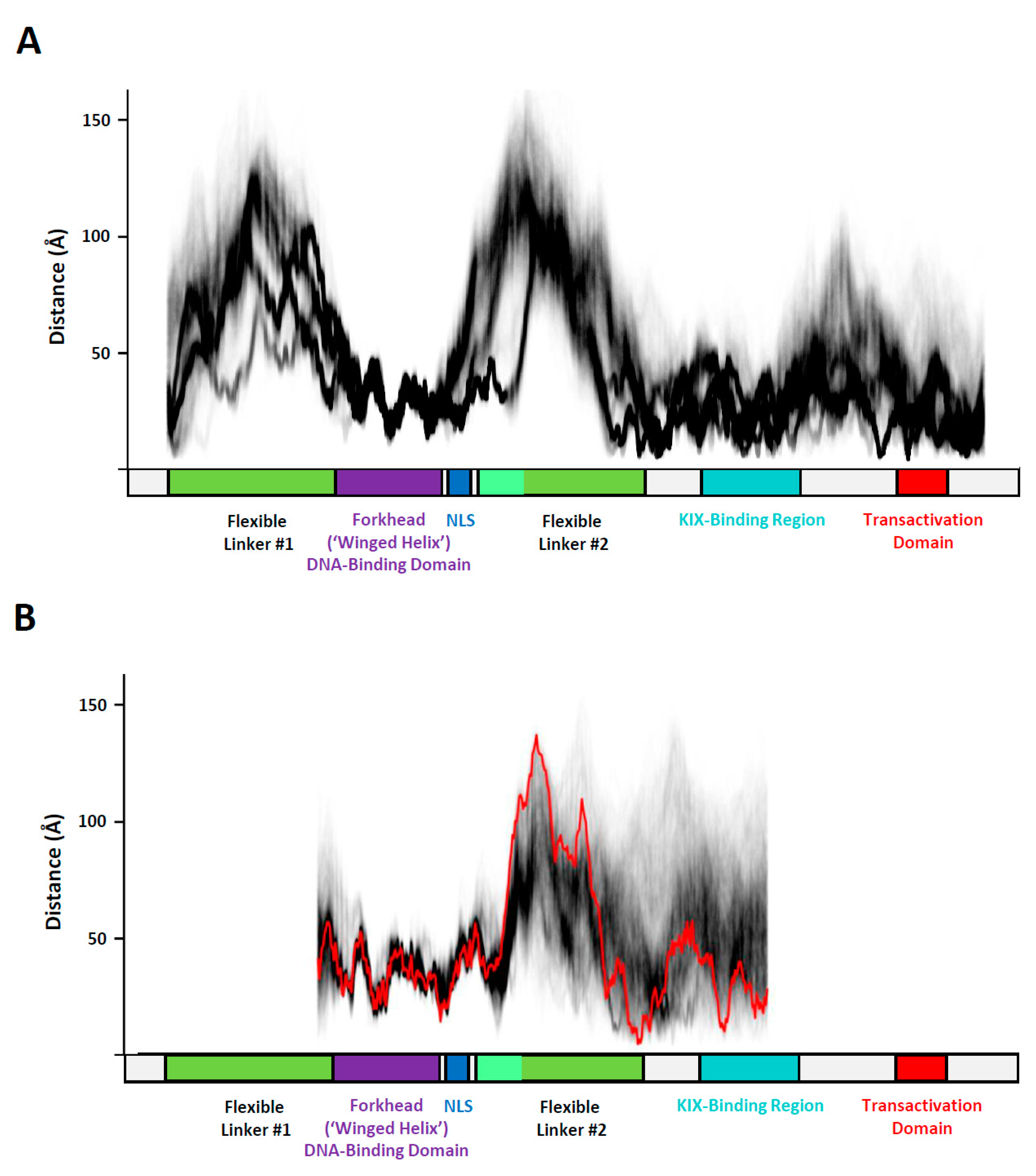
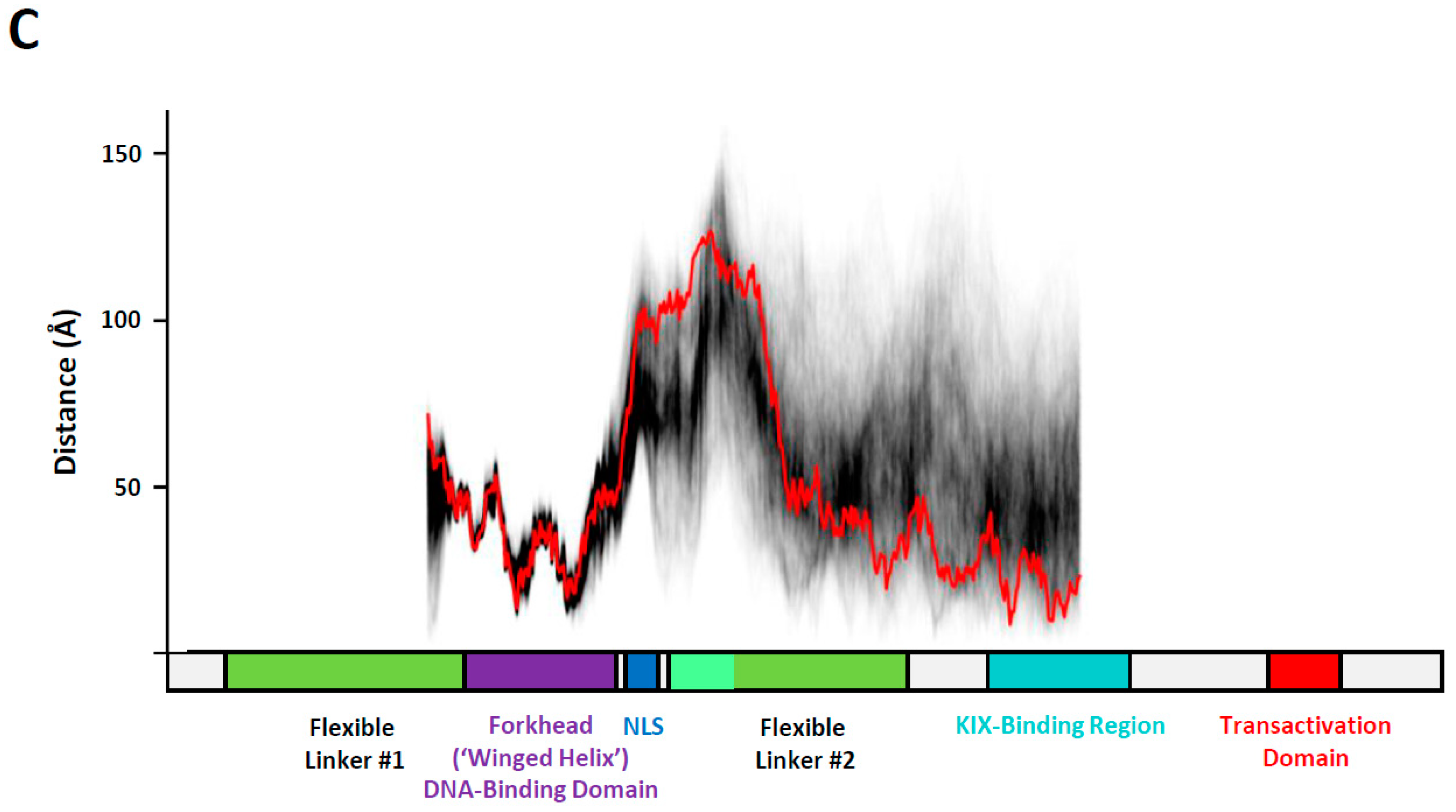
3.2. Confirmation of Electrostatic Repulsion Effect in Explicit Solvent Models
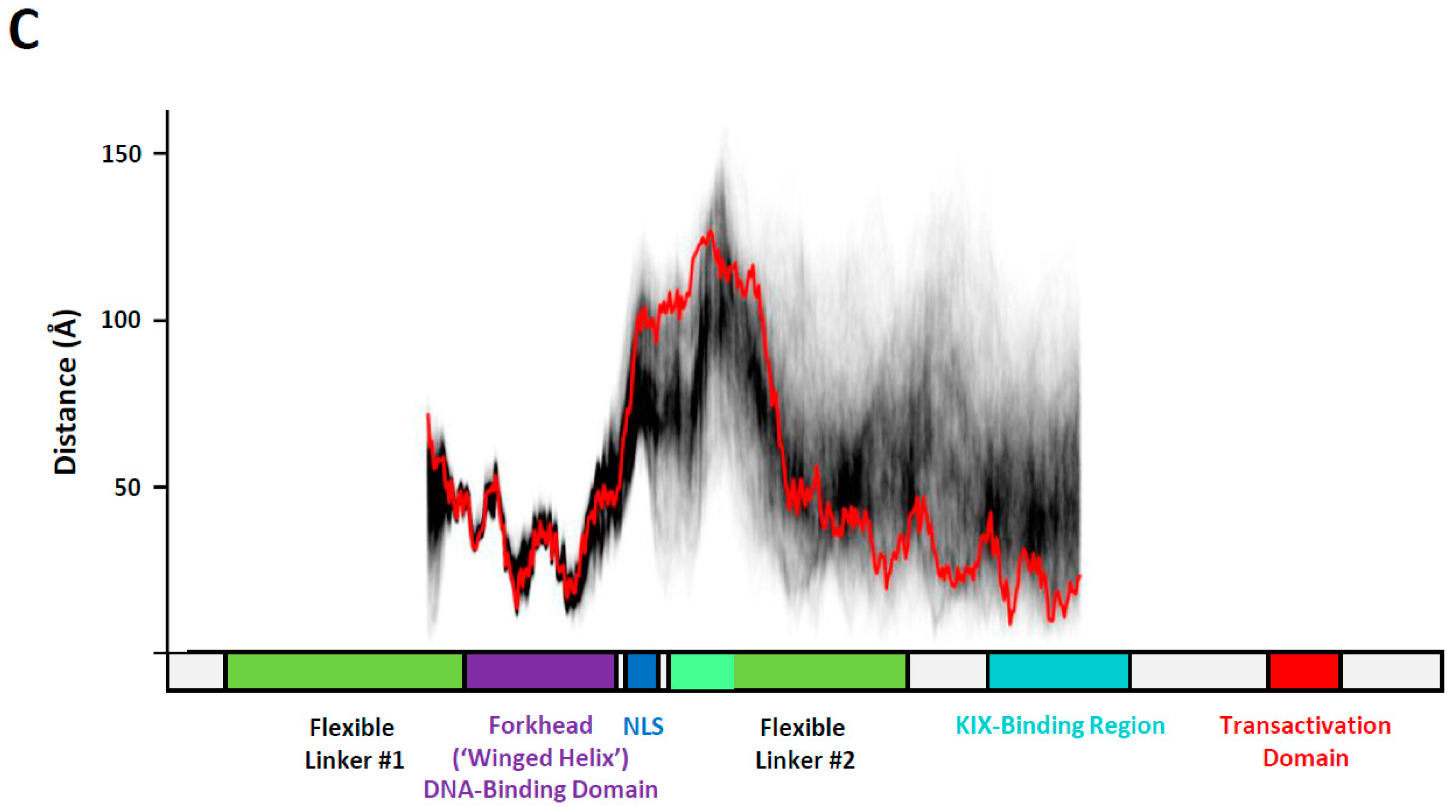
3.3. Conformational Variability in the KIX-Binding Region and Transactivation Domain
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Eijkelenboom, A.P.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Hornsveld, M.; Dansen, T.; Derksen, P.; Burgering, B. Re-evaluating the role of FOXOs in cancer. Semin. Cancer Biol. 2018, 50, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Calissi, G.; Lam, E.W.-F.; Link, W. Therapeutic strategies targeting FOXO transcription factors. Nat. Rev. Drug Discov. 2021, 20, 21–38. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chauhan, A.S.; Zhuang, L.; Gan, B. FoxO transcription factors in cancer metabolism. Semin. Cancer Biol. 2018, 50, 65–76. [Google Scholar] [CrossRef]
- Coomans de Brachene, A.; Demoulin, J.B. FOXO transcription factors in cancer development and therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heide, L.P.; Hoekman, M.F.; Smidt, M.P. The ins and outs of FoxO shuttling: Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, E.M. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Obsil, T.; Obšilová, V. Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene 2008, 27, 2263–2275. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.L.; Sun, Y.J.; Huang, C.Y.; Yang, J.Y.; Hung, M.C.; Hsiao, C.D. Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res. 2007, 35, 6984–6994. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Caburet, S.; Veitia, R.A. Forkhead transcription factors: Key players in health and disease. Trends Genet. 2011, 27, 224–232. [Google Scholar] [CrossRef]
- Ho, K.K.; Myatt, S.S.; Lam, E.W.-F. Many forks in the path: Cycling with FoxO. Oncogene 2008, 27, 2300–2311. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Marshall, C.B.; Ikura, M. Forkhead followed by disordered tail: The intrinsically disordered regions of FOXO3a. Intrinsically Disord. Proteins 2015, 3, e1056906. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z. Prediction of protein disorder based on IUPred. Protein Sci. 2018, 27, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, D.; Necci, M.; Escobedo, N.; Monzon, A.M.; Hatos, A.; Mičetić, I.; Quaglia, F.; Paladin, L.; Ramasamy, P.; Dosztányi, Z.; et al. MobiDB: Intrinsically disordered proteins in 2021. Nucleic Acids Res. 2021, 49, D361–D367. [Google Scholar] [CrossRef]
- So, C.W.; Cleary, M.L. MLL-AFX Requires the Transcriptional Effector Domains of AFX To Transform Myeloid Progenitors and Transdominantly Interfere with Forkhead Protein Function. Mol. Cell. Biol. 2002, 22, 6542–6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Marshall, C.B.; Yamamoto, K.; Li, G.-Y.; Gasmi-Seabrook, G.M.C.; Okada, H.; Mak, T.W.; Ikura, M. Structures of KIX domain of CBP in complex with two FOXO3a transactivation domains reveal promiscuity and plasticity in coactivator recruitment. Proc. Natl. Acad. Sci. USA 2012, 109, 6078–6083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef]
- Mercadante, D.; Milles, S.; Fuertes, G.; Svergun, D.I.; Lemke, E.A.; Gräter, F. Kirkwood–Buff Approach Rescues Overcollapse of a Disordered Protein in Canonical Protein Force Fields. J. Phys. Chem. B 2015, 119, 7975–7984. [Google Scholar] [CrossRef] [PubMed]
- Henriques, J.; Skepö, M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: On the Accuracy of the TIP4P-D Water Model and the Representativeness of Protein Disorder Models. J. Chem. Theory Comput. 2016, 12, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Lin, X. Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins. Biomolecules 2019, 9, 146. [Google Scholar] [CrossRef] [Green Version]
- Andresen, C.; Helander, S.; Lemak, A.; Farès, C.; Csizmok, V.; Carlsson, J.; Penn, L.Z.; Forman-Kay, J.D.; Arrowsmith, C.; Lundström, P.; et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012, 40, 6353–6366. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, S.S.; Weinzierl, R.O.J. Optimization of Molecular Dynamics Simulations of c-MYC(1-88)-An Intrinsically Disordered System. Life 2020, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, W.; Frellsen, J.; Harder, T.; Bottaro, S.; Johansson, K.E.; Tian, P.; Stovgaard, K.; Andreetta, C.; Olsson, S.; Valentin, J.B.; et al. PHAISTOS: A framework for Markov chain Monte Carlo simulation and inference of protein structure. J. Comput. Chem. 2013, 34, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Pierce, L.C.; Salomon-Ferrer, R.; De Oliveira, C.A.F.; McCammon, J.A.; Walker, R.C. Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Bioinform. 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Toto, A.; Malagrinò, F.; Visconti, L.; Troilo, F.; Pagano, L.; Brunori, M.; Jemth, P.; Gianni, S. Templated folding of intrinsically disordered proteins. J. Biol. Chem. 2020, 295, 6586–6593. [Google Scholar] [CrossRef] [Green Version]
- Toto, A.; Camilloni, C.; Giri, R.; Brunori, M.; Vendruscolo, M.; Gianni, S. Molecular Recognition by Templated Folding of an Intrinsically Disordered Protein. Sci. Rep. 2016, 6, 21994. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Heikaus, C.C.; Kisselev, L.; Vernon, R.; Herbig, E.; Pacheco, D.; Warfield, L.; Littlefield, P.; Baker, D.; Klevit, R.E.; et al. The Acidic Transcription Activator Gcn4 Binds the Mediator Subunit Gal11/Med15 Using a Simple Protein Interface Forming a Fuzzy Complex. Mol. Cell 2011, 44, 942–953. [Google Scholar] [CrossRef] [Green Version]
- Scholes, N.S.; Weinzierl, R.O.J. Molecular Dynamics of “Fuzzy” Transcriptional Activator-Coactivator Interactions. PLoS Comput. Biol. 2016, 12, e1004935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Li, E.-M.; Xu, L.-Y. From start to end: Phase separation and transcriptional regulation. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2020, 1863, 194641. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, J.; Tikhanovich, I.; Kuravi, S.; Helzberg, J.; Dorko, K.; Roberts, B.; Kumer, S.; A Weinman, S. Serine 574 phosphorylation alters transcriptional programming of FOXO3 by selectively enhancing apoptotic gene expression. Cell Death Differ. 2015, 23, 583–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hu, S.; Liu, L. Phosphorylation and acetylation modifications of FOXO3a: Independently or synergistically? Oncol. Lett. 2017, 13, 2867–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavel, S.; Siffroi-Fernandez, S.; Coldefy, A.S.; Boulukos, K.; Pisani, D.F.; Dérijard, B. Regulation of the Intracellular Localization of Foxo3a by Stress-Activated Protein Kinase Signaling Pathways in Skeletal Muscle Cells. Mol. Cell. Biol. 2009, 30, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinzierl, R.O.J. Molecular Dynamics Simulations of Human FOXO3 Reveal Intrinsically Disordered Regions Spread Spatially by Intramolecular Electrostatic Repulsion. Biomolecules 2021, 11, 856. https://doi.org/10.3390/biom11060856
Weinzierl ROJ. Molecular Dynamics Simulations of Human FOXO3 Reveal Intrinsically Disordered Regions Spread Spatially by Intramolecular Electrostatic Repulsion. Biomolecules. 2021; 11(6):856. https://doi.org/10.3390/biom11060856
Chicago/Turabian StyleWeinzierl, Robert O.J. 2021. "Molecular Dynamics Simulations of Human FOXO3 Reveal Intrinsically Disordered Regions Spread Spatially by Intramolecular Electrostatic Repulsion" Biomolecules 11, no. 6: 856. https://doi.org/10.3390/biom11060856