
Fibrinogen and a Triad of Thrombosis, Inflammation, and the Renin-Angiotensin System in Premature Coronary Artery Disease in Women: A New Insight into Sex-Related Differences in the Pathogenesis of the Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
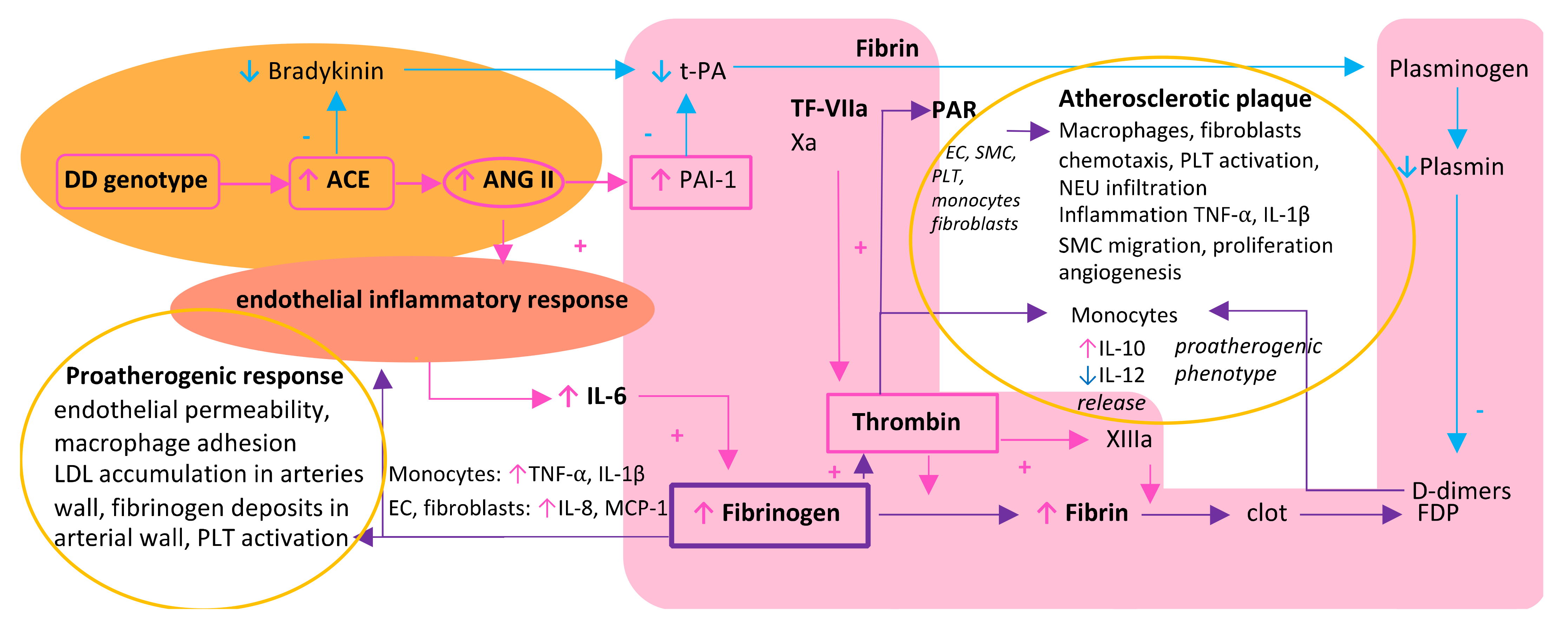
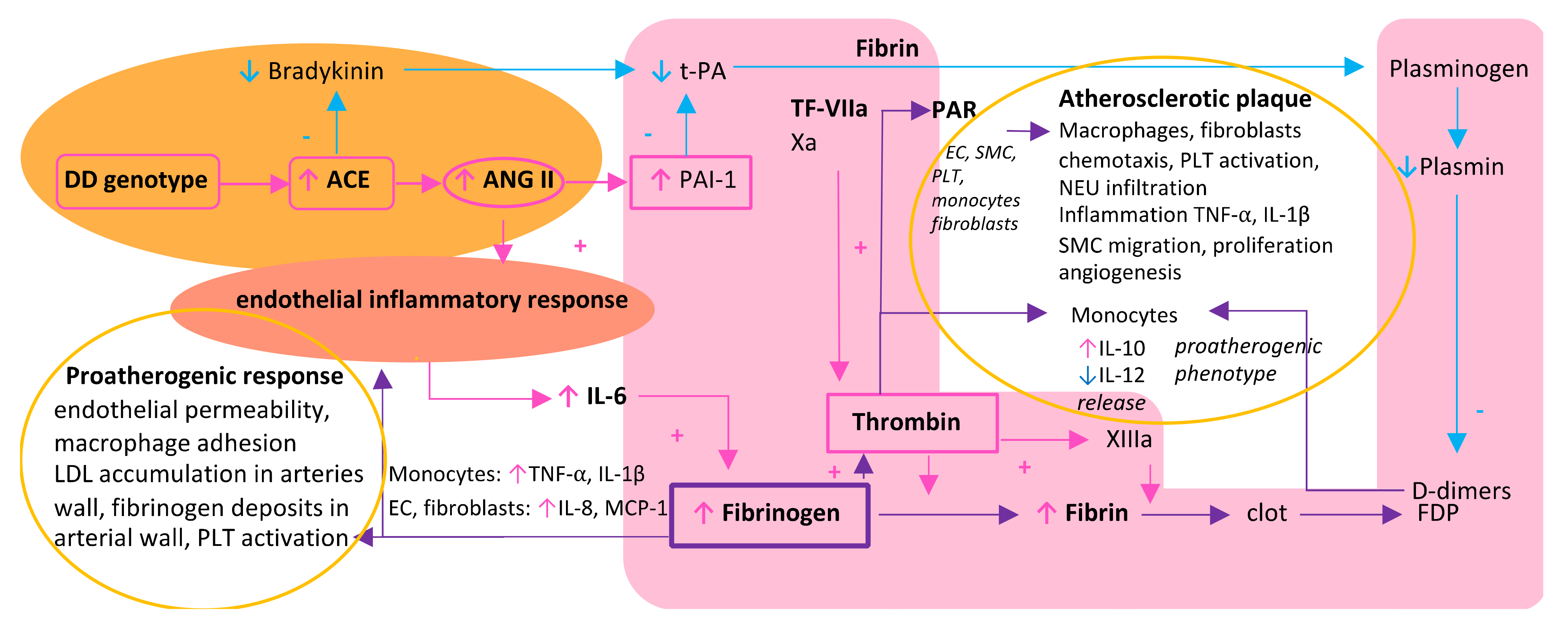
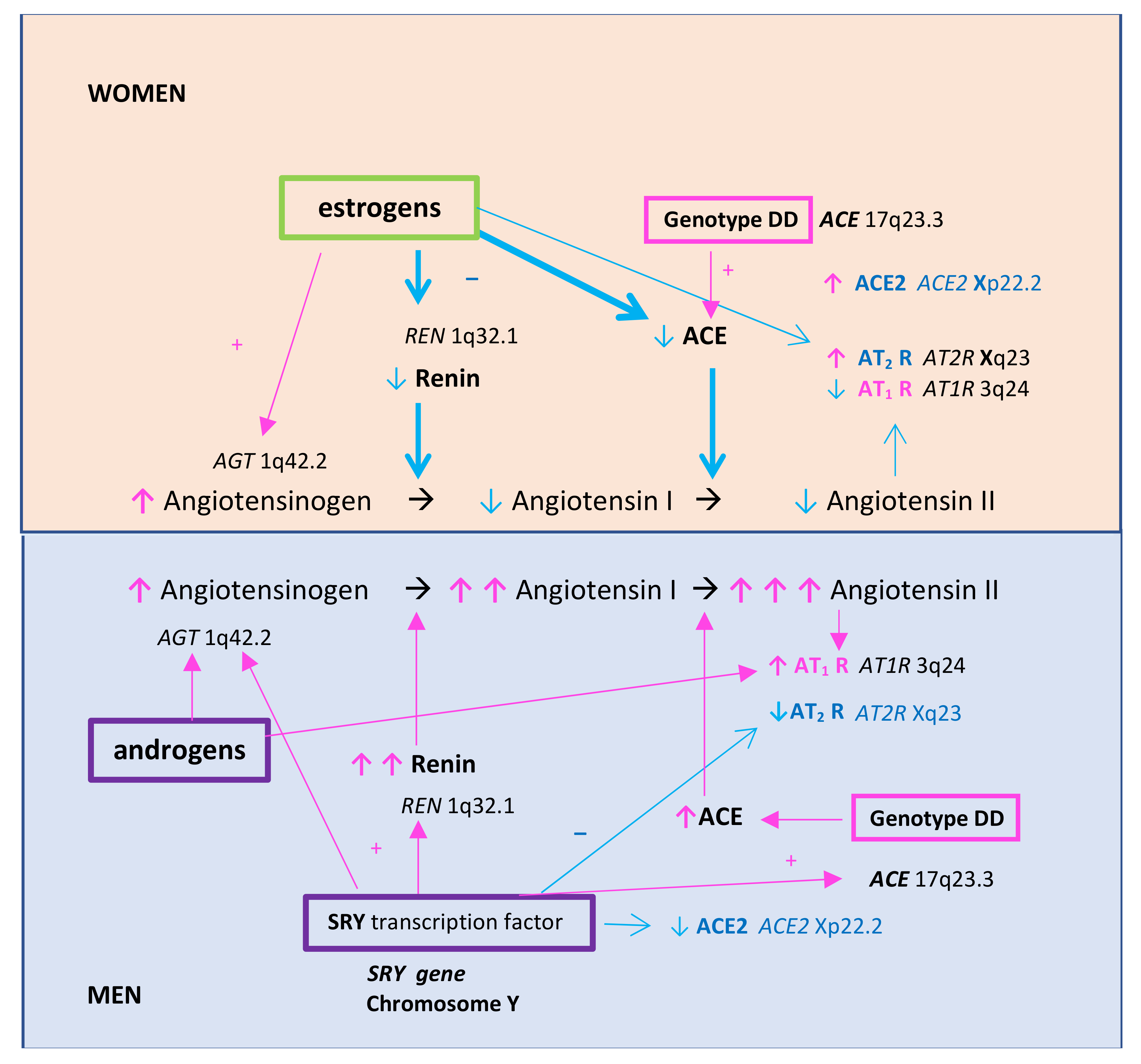
2. A Triad of Thrombosis, Inflammation, and Renin–Angiotensin System in the Vicious Circle of Atherosclerosis Formation—Differences between Women and Men
3. Fibrinogen as a Resultant and the Central Biomolecule in a Triad of Thrombosis, Inflammation, and Renin–Angiotensin System
3.1. Fibrinogen and Its Central Role in Thrombosis
3.2. Fibrinogen as Both an Effector and Stimulator of Inflammatory Reaction
3.3. Fibrinogen-Induced Atherosclerosis Formation
4. Distinct Clinical Impact of Fibrinogen on Coronary Artery Disease in Women and Men
4.1. Fibrinogen as an Independent Cardiovascular Risk Factor
4.2. Relationship between Fibrinogen and CAD in Women and Men
4.3. Smoking as a Modifier of Fibrinogen Impact on CAD
4.4. Fibrinogen and Sex Differences in Atherosclerotic Plaque Morphology and Clot Lysability
4.4.1. The Role of Estrogens
4.4.2. Stronger Impact of Smoking and Diabetes on Atherosclerosis in Women
4.4.3. Different Fibrinogen Levels and Plaque Composition in Women and Men
5. Therapeutic Implications of Fibrinogen Lowering on Residual Vascular Risk
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hajar, R. Framingham Contribution to Cardiovascular Disease. Heart Views 2016, 17, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Puymirat, E.; Simon, T.; Steg, P.G.; Schiele, F.; Guéret, P.; Blanchard, D.; Khalife, K.; Goldstein, P.; Cattan, S.; Vaur, L.; et al. Association of changes in clinical characteristics and management with improvement in survival among patients with ST-elevation myocardial infarction. JAMA 2012, 308, 998–1006. [Google Scholar] [CrossRef]
- Ford, E.S.; Capewell, S. Coronary heart disease mortality among young adults in the US from 1980 through 2002. Concealed leveling of mortality rates. J. Am. Coll. Cardiol. 2007, 50, 2128–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Jones, D.M.; Hong, Y.; Labarthe, D.; Mozaffarian, D.; Appel, L.J.; van Horn, L.; Greenlund, K.; Daniels, S.; Nichol, G.; Tomaselli, G.F.; et al. Defining and setting national goals for cardiovascular health promotion and disease reduction: The American Heart Association’s strategic Impact Goal through 2020 and beyond. Circulation 2010, 121, 586–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Cogswell, M.E.; Flanders, W.D.; Hong, Y.; Zhang, Z.; Loustalot, F.; Gillespie, C.; Merritt, R.; Hu, F.B. Trends in cardiovascular health metrics and associations with all-cause and CVD mortality among US adults. JAMA 2012, 307, 1273–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Njolstad, I.; Arnesen, E.; Lund-Larsen, P.G. Smoking, serum lipids, blood pressure, and sex differences in myocardial infarction. A 12-year follow-up of the Finnmark study. Circulation 1996, 93, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Record, N.B.; Onion, D.K.; Prior, R.E.; Record, S.S.; Cayer, G.R.; Amos, C.I.; Pearson, T.A. Community-wide cardiovascular disease prevention programs and health outcomes in a rural county, 1970–2010. JAMA 2015, 313, 147–155. [Google Scholar] [CrossRef]
- Trzos, E.; Uznańska, B.; Rechciński, T.; Krzemińska-Pakuła, M.; Bugała, M.; Kurpesa, M. Myocardial infarction in young people. Cardiol. J. 2009, 16, 307–311. [Google Scholar]
- Huxley, R.; Barzi, F.; Woodward, M. Excess risk of fatal coronary heart disease associated with diabetes in men and women: Meta-analysis of 37 prospective cohort studies. BMJ Clin. Res. 2006, 332, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chester, M.; Kaski, J.C. Clinical factors and angiographic features associated with premature coronary artery disease. Chest 1995, 108, 364–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, H.C., Jr.; McMahan, C.A.; Herderick, E.E.; Zieske, A.W.; Malcom, G.T.; Tracy, R.E.; Strong, J.P. Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation 2002, 105, 2712–2718. [Google Scholar] [CrossRef]
- De Backer, G.; Ambrosioni, E.; Borch-Johnsen, K.; Brotons, C.; Cifkova, R.; Dallongeville, J.; Ebrahim, S.; Faergeman, O.; Graham, I.; Mancia, G.; et al. Third Joint Task Force of European and other Societies on Cardiovascular Diseases Prevention in Clinical Practice. European guidelines on cardiovascular diseases prevention in clinical practice. Eur. Heart J. 2003, 24, 1601–1610. [Google Scholar] [CrossRef]
- Hamelin, B.A.; Zakrzewski-Jakubiak, M.; Robitaille, N.M.; Bogaty, P.; Labbé, L.; Turgeon, J. Increased risk of myocardial infarction associated with angiotensin-converting enzyme gene polymorphism is age dependent. J. Clin. Pharmacol. 2011, 51, 1286–1292. [Google Scholar] [CrossRef]
- Trisvetova, E. Likely features of female coronary artery disease. EJ Cardiol. Pract. 2014, 12, 22. [Google Scholar]
- Kryczka, K.E.; Płoski, R.; Księżycka, E.; Kruk, M.; Kostrzewa, G.; Kowalik, I.; Demkow, M.; Lubiszewska, B. The association between the insertion/deletion polymorphism of the angiotensin-converting enzyme gene and the plasma fibrinogen level in women and men with premature coronary artery atherosclerosis. Pol. Arch. Intern. Med. 2020, 130, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Ariens, R.A.S. Fibrin Clot Structure and Function. A Role in the Pathophysiology of Arterial and Venous Thromboembolic Diseases. Arter. Thromb. Vasc. Biol. 2011, 31, e88–e99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safdar, B.; Spatz, E.S.; Dreyer, P.R.; Beltrame, J.F.; Lichtman, J.H.; Spertus, J.A.; Reynolds, H.R.; Geda, M.; Bueno, H.; Dziura, J.D.; et al. Presentation, Clinical Profile, and Prognosis of Young Patients with Myocardial Infarction With Nonobstructive Coronary Arteries (MINOCA): Results From the VIRGO Study. J. Am. Heart Assoc. 2018, 7, e009174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R. Atherosclerosis—An inflammatory disease. New Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Chaudhary, R.; Sukhi, A.; Chaudhary, R.; Jindal, M.; Vyas, A.; Rout, A.; Bliden, K.; Tantry, U.; Gurbel, P. Gender differences in thrombogenicity among patients with angina and non-obstructive coronary artery disease. J. Thromb. Thrombolysis 2019, 48, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Qasim, A.N.; Budharaju, V.; Mehta, N.N.; St. Clair, C.; Farouk, S.; Braunstein, S.; Schutta, M.; Iqbal, N.; Rader, D.J.; Reilly, M.P. Gender Differences in the Association of C-Reactive Protein with Coronary Artery Calcium in Type-2 Diabetes. Clin. Endocrinol. 2011, 74, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Man, J.J.; Beckman, J.A.; Jaffe, I.Z. Sex as a Biological Variable in Atherosclerosis. Circ. Res. 2020, 126, 1297–1319. [Google Scholar] [CrossRef]
- Hilliard, L.M.; Sampson, A.K.; Brown, R.D.; Denton, K.M. The “his and hers” of the renin-angiotensin system. Curr. Hypertens. Rep. 2013, 15, 71–79. [Google Scholar] [CrossRef]
- Spronk, H.M.H.; Padro, T.; Siland, J.E.; Winters, J.; van der Wal, A.C.; Posthuma, J.J.; Lowe, G.; d’Alessandro, E.; Wenzel, P.; Coenen, D.M.; et al. Atherothrombosis and Thromboembolism: Position Paper from the Second Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2018, 118, 229–250. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, W.H. Fibrinogen—Marker or mediator of vascular disease? Vasc. Med. 2003, 8, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dielis, A.W.; Castoldi, E.; Spronk, H.M.; van Oerle, R.; Hamulyák, K.; Ten Cate, H.; Rosing, J. Coagulation factors and the protein C system as determinants of thrombin generation in a normal population. J. Thromb. Haemost. 2008, 6, 125–131. [Google Scholar] [CrossRef]
- Yahagi, K.; Davis, H.R.; Arbustini, E.; Virmani, R. Sex differences in coronary artery disease: Pathological observations. Atherosclerosis 2015, 239, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Gram, J.B.; Sidelmann, J.J.; Dey, D.; Kusk, M.W.; Nørgaard, B.L.; Sand, N.P.R. Sex difference in fibrin clot lysability: Association with coronary plaque composition. Thromb. Res. 2019, 174, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Farb, A.; Malcom, G.; Liang, Y.; Smialek, J.; Virmani, R. Effect of menopause on plaque morphologic characteristics in coronary atherosclerosis. Am. Heart J. 2001, 141, S58–S62. [Google Scholar] [CrossRef] [PubMed]
- Nasir, K.; Gopal, A.; Blankstein, R.; Ahmadi, N.; Pal, R.; Khosa, F.; Shaw, L.J.; Blumenthal, R.S.; Budoff, M.J. Noninvasive assessment of gender differences in coronary plaque composition with multidetector computed tomographic angiography. Am. J. Cardiol. 2010, 105, 453–458. [Google Scholar] [CrossRef]
- Alzahrani, S.H.; Hess, K.; Price, J.F.; Strachan, M.; Baxter, P.D.; Cubbon, R.; Phoenix, F.; Gamlen, T.; Ariëns, R.A.; Grant, P.J.; et al. Gender-specific alterations in fibrin structure function in type 2 diabetes: Associations with cardiometabolic and vascular markers. J. Clin. Endocrinol. Metab. 2012, 97, E2282–E2287. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, F.; Peng, R.; Pei, S.; Hou, Z.; Lu, B.; Cong, X.; Chen, X. Sex-related differences in the association between plasma fibrinogen and non-calcified or mixed coronary atherosclerotic plaques. Biol. Sex Differ. 2018, 9, 51. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T.; Büller, H.R. Bidirectional relation between inflammation and coagulation. Circulation 2004, 109, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, D.E.; Lazos, S.; Tong, K. Angiotensin II regulates the expression of plasminogen activator inhibitor-type1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J. Clin. Investig. 1995, 95, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Brull, D.J.; Humphries, S.E.; Montgomery, H.E. Genetics of inflammation and risk of coronary artery disease: The central role of interleukin-6. Eur. Heart J. 2000, 21, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Green, F.; Humphries, S. Control of plasma fibrinogen levels. Baillieres Clin. Haematol. 1989, 2, 945–959. [Google Scholar] [CrossRef]
- Vitale, C.; Mendelsohn, M.E.; Rosano, G.M.C. Gender differences in the cardiovascular effect of sex hormones. Nat. Rev. Cardiol. 2009, 6, 532–542. [Google Scholar] [CrossRef]
- Rigat, B.; Hubert, C.; Alhenc Gelas, F.; Cambien, F.; Corvol, P.; Soubrier, F. An insertion/deletion poly morphism in the angiotensin I converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Investig. 1990, 86, 1343–1346. [Google Scholar] [CrossRef] [Green Version]
- Behague, I.; Poirier, O.; Nicaud, V.; Evans, A.; Arveiler, D.; Luc, G.; Cambou, J.P.; Scarabin, P.Y.; Bara, L.; Green, F.; et al. Beta fibrinogen gene polymor phisms are associated with plasma fibrinogen and coronary artery disease in patients with myocardial infarction. The ECTIM Study. Etude Cas Temoins sur l’Infarctus du Myocarde. Circulation 1996, 93, 440–449. [Google Scholar] [CrossRef]
- Khramtsova, E.A.; Davis, L.K.; Stranger, B.E. The role of sex in the genomics of human complex traits. Nature 2019, 20, 173–190. [Google Scholar] [CrossRef]
- Tennent, G.A.; Brennan, S.O.; Stangou, A.J.; O’Grady, J.; Hawkins, P.N.; Pepys, M.B. Human plasma fibrinogen is synthesized in the liver. Blood 2007, 109, 1971–1974. [Google Scholar] [CrossRef]
- Lee, A.J.; Fowkes, F.G.; Lowe, G.D.; Connor, J.M.; Rumley, A. Fibrinogen, factor VII and PAI-1 genotypes and the risk of coronary and peripheral atherosclerosis: Edinburgh Artery Study. Thromb. Haemost. 1999, 81, 553–560. [Google Scholar] [CrossRef]
- Mannila, M.N.; Lovely, R.S.; Kazmierczak, S.C.; Eriksson, P.; Samnegard, A.; Farrell, D.H.; Hamsten, A.; Silviera, A. Elevated plasma fibrinogen gamma’ concentration is associated with myocardial infarction: Effects of variation in fibrinogen genes and environmental factors. J. Thromb. Haemost. 2007, 5, 766–773. [Google Scholar] [CrossRef]
- Koenig, W. Haemostatic risk factors for cardiovascular diseases. Eur. Heart J. 1998, 19 (Suppl. C), C39–C43. [Google Scholar] [PubMed]
- Koenig, W.; Enst, E. Exercise and thrombosis. Coron. Artery Dis. 2000, 11, 123–127. [Google Scholar] [CrossRef]
- McDonagh, J.; Lee, M.H. How does hyperfibrinogenemia lead to thrombosis? Fibrinol. Proteol. 1997, 11 (Suppl. S1), 13–17. [Google Scholar] [CrossRef]
- Resch, K.L.; Ernst, E.; Matrai, A.; Paulsen, H.F. Fibrinogen and viscosity as risk factors for subsequent cardiovascular events in stroke survivors. Ann. Intern. Med. 1992, 117, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Brandts, A.; van Hylckama Vlieg, A.; Rosing, J.; Baglin, T.P.; Rosendaal, F.R. The risk of venous thrombosis associated with a high endogenous thrombin potential in the absence and presence of activated protein C. J. Thromb. Haemost. 2007, 5, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Ariens, R.A.S. Fibrin(ogen) and thrombotic disease. J. Thromb. Haemost. 2013, 11, 294–305. [Google Scholar] [CrossRef]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through Toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsakadze, N.L.; Zhao, Z.; D’Souza, S.E. Interactions of intercellular adhesion molecule-1 with fibrinogen. Trends Cardiovasc. Med. 2002, 12, 101–108. [Google Scholar] [CrossRef]
- Hicks, R.C.; Golledge, J.; Mir-Hasseine, R.; Powell, J.T. Vasoactive effects of fibrinogen on saphenous vein. Nature 1996, 379, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Retzinger, G.S.; DeAngelis, A.P.; Patuto, S.J. Adsorption of fibrinogen to droplets of liquid hydrophobic phases. Functionality of the bound protein and biological implications. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1948–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Springer, T.A. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J. Clin. Investig. 1997, 100, 2085–2093. [Google Scholar] [CrossRef]
- Kannel, W.B.; Wolf, P.A.; Castelli, W.P.; D’Agostino, R.B. Fibrinogen and Risk of Cardiovascular Disease. The Framingham Study. JAMA 1987, 258, 1183–1186. [Google Scholar] [CrossRef]
- Handa, K.; Kono, S.; Saku, K.; Sasaki, J.; Kawano, T.; Sasaki, Y.; Hiroki, T.; Arakawa, K. Plasma fibrinogen levels as an independent indicator of severity of coronary atherosclerosis. Atherosclerosis 1989, 77, 209–213. [Google Scholar] [CrossRef]
- Danesh, J.; Levington, S.; Thompson, S.G.; Lowe, G.D.; Collins, R.; Kostis, J.B.; Wilson, A.C.; Folsom, A.R.; Wu, K.; Benderly, M.; et al. Plasma Fibrinogen Level and the Risk of Major Cardiovascular Diseases and Nonvascular Mortality. An Individual Participant Meta-analysis Fibrinogen Studies. JAMA 2005, 294, 1799–1809. [Google Scholar] [PubMed]
- Vorster, H.H. Fibrinogen and women’s health. Thromb. Res. 1999, 95, 137–154. [Google Scholar] [CrossRef]
- Giansante, C.; Fiotti, N.; Cattin, L.; Da Col, P.G.; Calabrese, S. Fibrinogen, D-Dimer and Thrombin-Antithrombin Complexes in a Random Population Sample: Relationships with Other Cardiovascular Risk Factors. Thromb. Haemost. 1994, 7, 581–586. [Google Scholar] [CrossRef]
- The Emerging Risk Factors Collaboration. C-reactive protein, fibrinogen, and cardiovascular disease prediction. New Engl. J. Med. 2012, 367, 1310–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, E.; Koenig, W. Fibrinogen and cardiovascular risk. Vasc. Med. 1997, 2, 115–125. [Google Scholar] [CrossRef]
- Heinrich, J.; Balleisen, L.; Schulte, H.; Assmann, G.; van de Loo, J. Fibrinogen and factor VII in the prediction of coronary risk. Results from the PROCAM study in healthy men. Arterioscler. Thromb. 1994, 14, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Ernst, E. Plasma fibrinogen-an independent cardiovascular risk factor. J. Intern. Med. 1990, 227, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Meade, T.W.; North, W.R.; Chakrabarti, R.; Stirling, Y.; Haines, A.P.; Thompson, S.G.; Brozovié, M. Haemostatic function and cardiovascular death: Early resuts of a prospective study. Lancet 1980, 1, 1050–1054. [Google Scholar] [CrossRef]
- Meade, T.W.; Mellows, S.; Brozovic, M.; Miller, G.J.; Chakrabarti, R.R.; North, W.R.; Haines, A.P.; Stirling, Y.; Imeson, J.D.; Thompson, S.G. Haemostatic function and ischaemic heart disease: Principal results of the Northwick Park Heart Study. Lancet 1986, 8506, 533–537. [Google Scholar] [CrossRef]
- Lassé, M.; Pilbrow, A.P.; Kleffmann, T.; Andersson Överström, E.; von Zychlinski, A.; Frampton, C.M.A.; Poppe, K.K.; Troughton, R.W.; Lewis, L.K.; Prickett, T.C.R.; et al. Fibrinogen and hemoglobin predict near future cardiovascular events in asymptomatic individuals. Sci. Rep. 2021, 11, 4605. [Google Scholar] [CrossRef] [PubMed]
- De Stavola, B.L.; Meade, T.W. Long-term effects of hemostatic variables on fatal coronary heart disease: 30-year results from the first prospective Northwick Park Heart Study (NPHS-I). J. Thromb. Haemost. 2007, 5, 461–471. [Google Scholar] [CrossRef]
- The Health Consequences of Smoking—50 Years of Progress. A Report of the Surgeon General 2014. Available online: https://www.hhs.gov/sites/default/files/consequences-smoking-exec-summary.pdf (accessed on 20 March 2021).
- Kryczka, K.E.; Kruk, M.; Piotrowski, W.; Księżycka, E.; Pracoń, R.; Witkowski, A.; Demkow, M.; Lubiszewska, B. Menopause improves the predictive value of common cardiovascular risk scores in women with premature coronary artery disease. Menopause 2018, 25, 408–414. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Stefan Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. ESC Scientific Document Group, 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar]
- Dahm, A.; van Hylckama Vlieg, A.; Bendz, B.; Bendz, B.; Rosendaal, F.; Bertina, R.M.; Sandset, P.M. Low levels of tissue factor pathway inhibitor (TFPI) increase the risk of venous thrombosis. Blood 2003, 101, 4387–4392. [Google Scholar] [CrossRef] [Green Version]
- Rosing, J.; Middeldorp, S.; Curvers, J.; Christella, M.; Thomassen, L.G.; Nicolaes, G.A.; Meijers, J.C.; Bouma, B.N.; Büller, H.R.; Prins, M.H.; et al. Low-dose oral contraceptives and acquired resistance to activated protein C: A randomised cross-over study. Lancet 1999, 354, 2036–2040. [Google Scholar] [CrossRef]
- Harris, G.M.; Stendt, C.L.; Vollenhoven, B.J.; Gan, T.E.; Tipping, P.G. Decreased plasma tissue factor pathway inhibitor in women taking combined oral contraceptives. Am. J. Hematol. 1999, 60, 175–180. [Google Scholar] [CrossRef]
- Kluft, C. Effects on haemostasis variables by second and third generation combined oral contraceptives: A review of directly comparative studies. Curr. Med. Chem. 2000, 7, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Kryczka, K.E.; Kruk, M.; Lubiszewska, B. Regarding Article, “Arterial Stiffness Accelerates Within 1 Year of the Final Menstrual Period: The SWAN Heart Study”. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e238–e239. [Google Scholar] [CrossRef]
- Koh, K.K.; Ahn, J.U.; Kim, D.S.; Han, S.H.; Shin, M.S.; Ryu, W.S.; Park, G.S.; Ahn, T.H.; Choi, I.S.; Shin, E.K. Effect of hormone replacement therapy on tissue factor activity, C-reactive protein, and tissue factor pathway inhibitor. Am. J. Cardiol. 2003, 91, 371–373. [Google Scholar] [CrossRef]
- Neergaard-Petersen, S.; Hvas, A.M.; Kristensen, S.D.; Grove, E.L.; Larsen, S.B.; Phoenix, F.; Kurdee, Z.; Grant, P.J.; Ajjan, R.A. The influence of type 2 diabetes on fibrin clot properties in patients with coronary artery disease. Thromb. Haemost. 2014, 112, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Lieb, W.; Enserro, D.M.; Larson, M.G.; Vasan, R.S. Residual cardiovascular risk in individuals on lipid-lowering treatment: Quantifying absolute and relative risk in the community. Open Heart 2018, 5, e000722. [Google Scholar] [CrossRef]
- Fruchart, J.-C.; Sacks, F.M.; Hermans, M.P.; Assmann, G.; Brown, W.V.; Ceska, R.; Chapman, M.J.; Dodson, P.M.; Fioretto, P.; Ginsberg, H.N.; et al. The Residual Risk Reduction Initiative: A call to action to reduce residual vascular risk in dyslipidaemic patients. Diabetes Vasc. Dis. Res. 2008, 5, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Avanzini, F.; Marzona, I.; Baviera, M.; Barlera, S.; Milani, V.; Caimi, V.; Longoni, P.; Tombesi, M.; Silletta, M.G.; Tognoni, G.; et al. Risk and Prevention Study Collaborative Group. Improving cardiovascular prevention in general practice: Results of a comprehensive personalized strategy in subjects at high risk. Eur. J. Prev. Cardiol. 2016, 23, 947–955. [Google Scholar] [CrossRef]
- Ernst, E.; Resch, K.L. Therapeutic interventions to lower plasma fibrinogen concentration. Eur. Heart J. 1995, 16 (Suppl. A), 47–53. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Fruchart, J.C.; Staels, B. Pleiotropic effects of fibrates. Curr. Atheroscler. Rep. 2005, 7, 396–401. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, P.K.; Cooper, J.A.; Rumley, A.; Lowe, G.D.; Meade, T.W. Effect of bezafibrate on plasma homocysteine concentration in men with lower extremity arterial disease. J. Thromb. Haemost. 2004, 2, 364–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kryczka, K.E.; Kruk, M.; Demkow, M.; Lubiszewska, B. Fibrinogen and a Triad of Thrombosis, Inflammation, and the Renin-Angiotensin System in Premature Coronary Artery Disease in Women: A New Insight into Sex-Related Differences in the Pathogenesis of the Disease. Biomolecules 2021, 11, 1036. https://doi.org/10.3390/biom11071036
Kryczka KE, Kruk M, Demkow M, Lubiszewska B. Fibrinogen and a Triad of Thrombosis, Inflammation, and the Renin-Angiotensin System in Premature Coronary Artery Disease in Women: A New Insight into Sex-Related Differences in the Pathogenesis of the Disease. Biomolecules. 2021; 11(7):1036. https://doi.org/10.3390/biom11071036
Chicago/Turabian StyleKryczka, Karolina E., Mariusz Kruk, Marcin Demkow, and Barbara Lubiszewska. 2021. "Fibrinogen and a Triad of Thrombosis, Inflammation, and the Renin-Angiotensin System in Premature Coronary Artery Disease in Women: A New Insight into Sex-Related Differences in the Pathogenesis of the Disease" Biomolecules 11, no. 7: 1036. https://doi.org/10.3390/biom11071036
APA StyleKryczka, K. E., Kruk, M., Demkow, M., & Lubiszewska, B. (2021). Fibrinogen and a Triad of Thrombosis, Inflammation, and the Renin-Angiotensin System in Premature Coronary Artery Disease in Women: A New Insight into Sex-Related Differences in the Pathogenesis of the Disease. Biomolecules, 11(7), 1036. https://doi.org/10.3390/biom11071036