
Identification of Potential Kinase Inhibitors within the PI3K/AKT Pathway of Leishmania Species

 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. List of Kinases Detected in Different Leishmania Species
2.2. Modelling, Ligandibility and Druggability Prediction
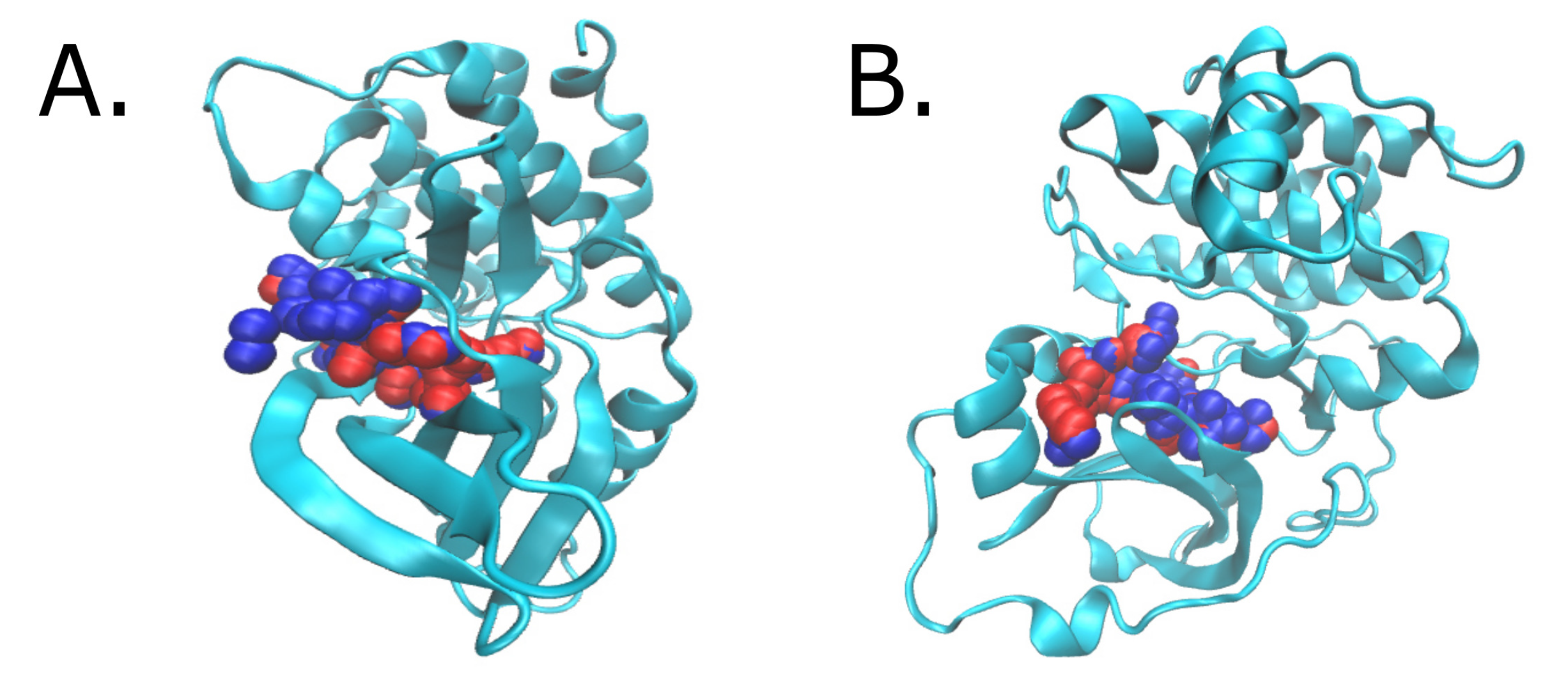
2.3. Hits Found by Molecular Docking
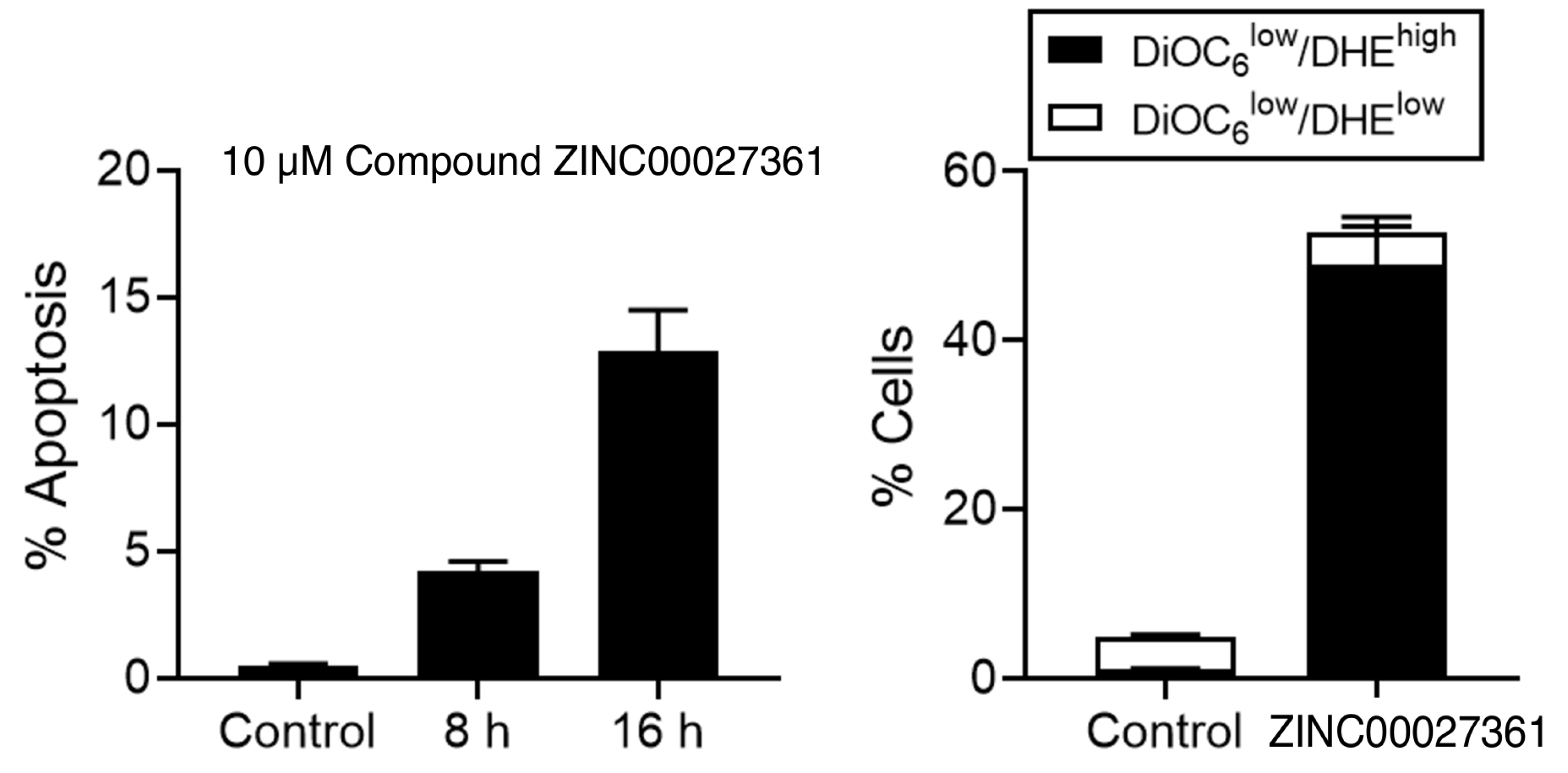
2.4. Induction of Cell Death by GSK3 Inhibitor
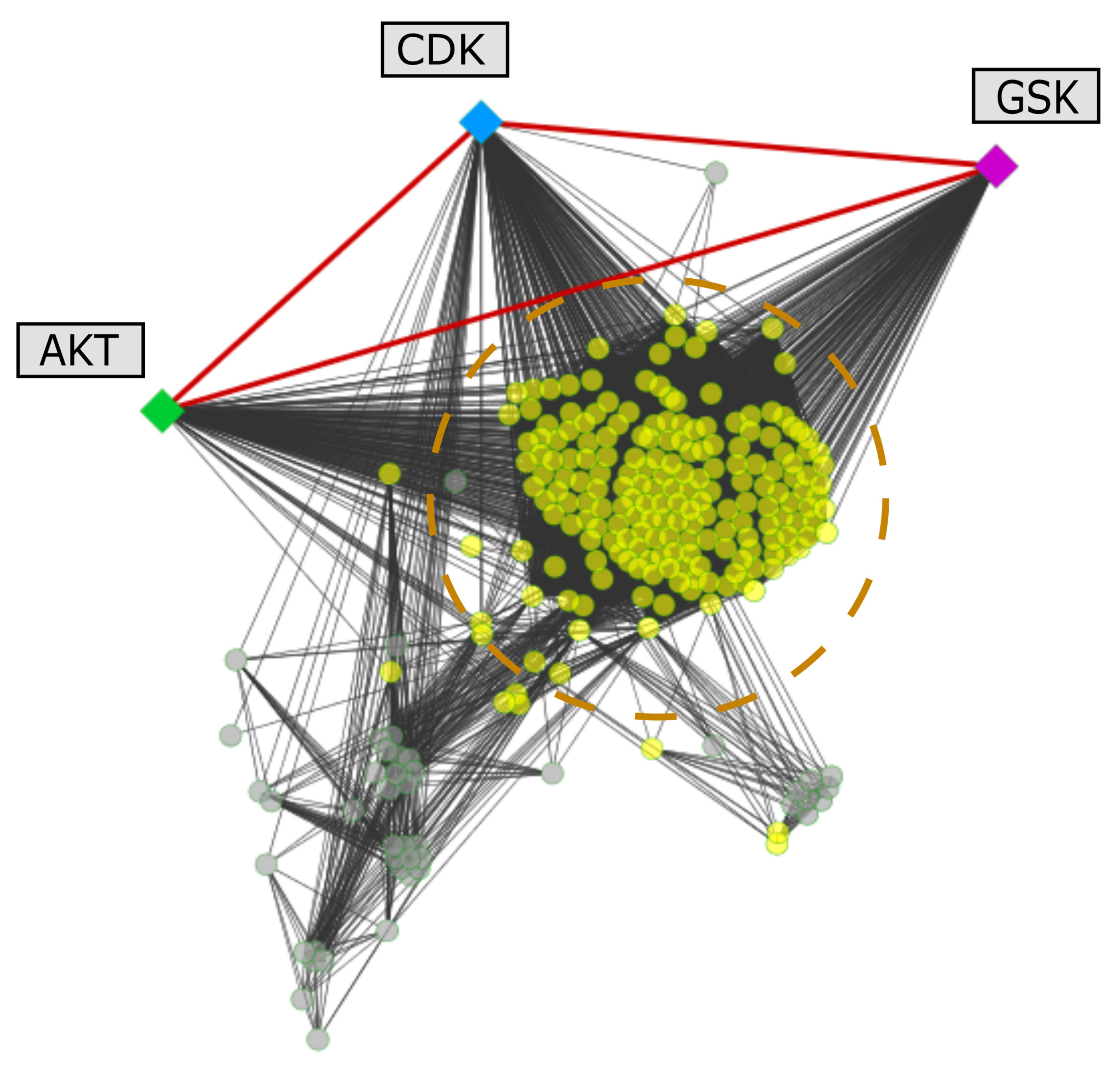
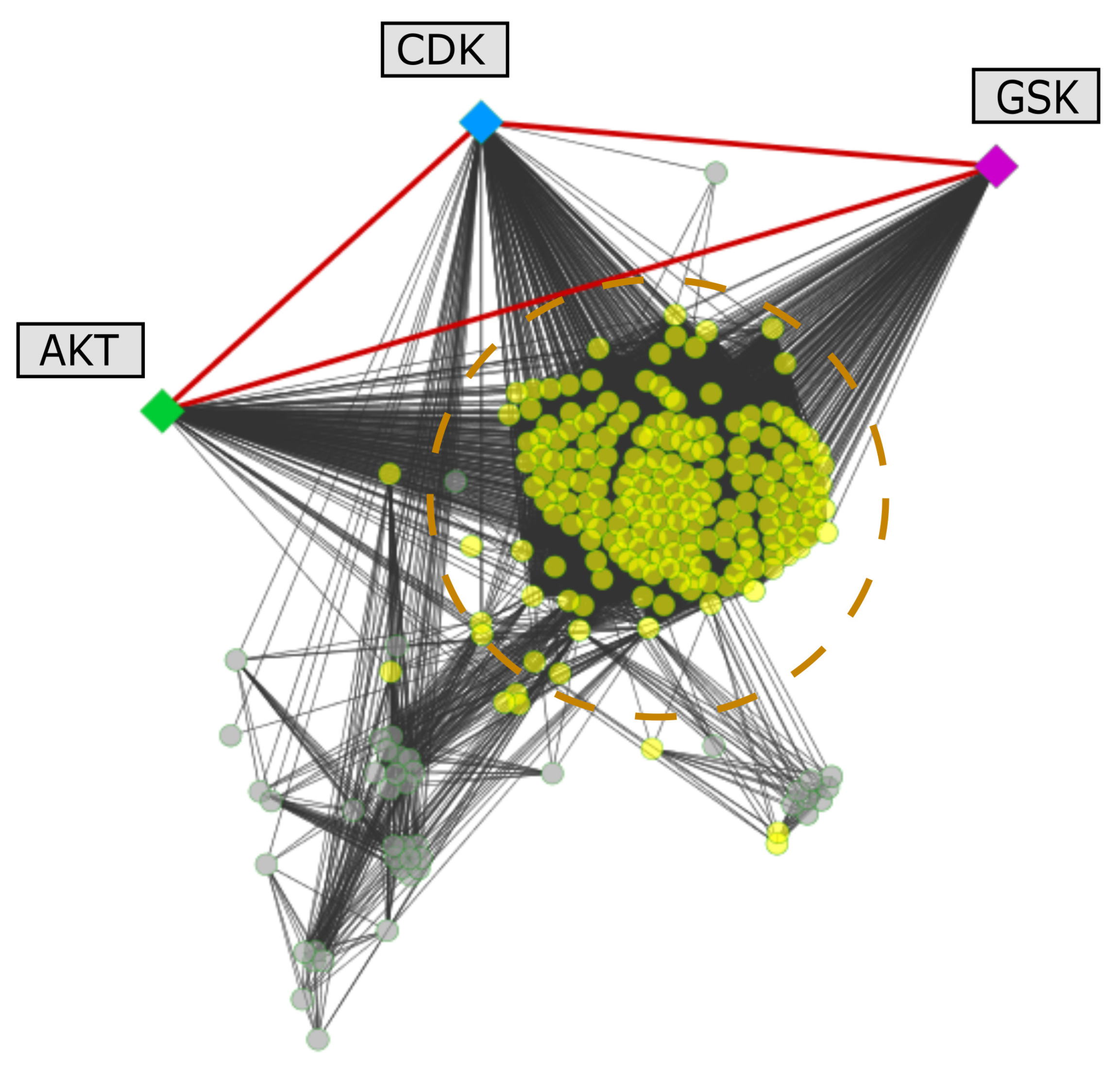
2.5. Protein Interactions Associated with the Selected Kinases
2.6. Literature Search of the Selected Kinases
3. Materials and Methods
3.1. Selection and Comparison of Human and Leishmania Kinase Sequences
3.2. Prediction of Ligandibility Metrics for the Leishmania Kinases
3.3. Generation of Structural Homology-Based Models and Druggability Assessment
3.4. Molecular Docking Screening
3.5. Analysis of Apoptosis-Like Cell Death by Flow Cytometry Using the GSK3 Inhibitor
3.6. Mapping of Selected Leishmania spp. Proteins on Protein-Protein Interaction Networks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mott, K.E.; Desjeux, P.; Moncayo, A.; Ranque, P.; de Raadt, P. Parasitic diseases and urban development. Bull. World Health Organ. 1990, 68, 691–698. [Google Scholar]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- Jain, K.; Jain, N.K. Vaccines for visceral leishmaniasis: A review. J. Immunol. Methods 2015, 422, 1–12. [Google Scholar] [CrossRef]
- Khamesipour, A. Therapeutic vaccines for leishmaniasis. Expert Opin. Biol. Ther. 2014, 14, 1641–1649. [Google Scholar] [CrossRef]
- Thakur, C.P.; Kanyok, T.P.; Pandey, A.K.; Sinha, G.P.; Messick, C.; Olliaro, P. Treatment of visceral leishmaniasis with injectable paromomycin (aminosidine). An open-label randomized phase-II clinical study. Trans. R. Soc. Trop. Med. Hyg. 2000, 94, 432–433. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J. Paromomycin in the treatment of leishmaniasis. Expert Opin. Investig. Drugs 2008, 17, 787–794. [Google Scholar] [CrossRef]
- Meyerhoff, A. U.S. Food and Drug Administration approval of AmBisome (Liposomal amphotericin B) for treatment of visceral leishmaniasis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1999, 28, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Engel, J.; Sindermann, H.; Fischer, C.; Junge, K.; Bryceson, A.; Berman, J. Oral miltefosine for Indian visceral leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desjeux, P. Leishmaniasis: Current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 305–318. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.M.; Myler, P.J.; Blandin, G.; Berriman, M.; Crabtree, J.; Aggarwal, G.; Caler, E.; Renauld, H.; Worthey, E.A.; Hertz-Fowler, C.; et al. Comparative genomics of trypanosomatid parasitic protozoa. Science (N. Y.) 2005, 309, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Ash, C.; Jasny, B.R. Trypanosomatid genomes. Introduction. Science (N. Y.) 2005, 309, 399. [Google Scholar] [CrossRef] [Green Version]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.A.; Adlem, E.; Aert, R.; et al. The genome of the kinetoplastid parasite, Leishmania major. Science (N. Y.) 2005, 309, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Gonzalez, R.; Kuhlmann, F.M.; Galan-Rodriguez, C.; Madeira da Silva, L.; Saldivia, M.; Karver, C.E.; Rodriguez, A.; Beverley, S.M.; Navarro, M.; Pollastri, M.P. The susceptibility of trypanosomatid pathogens to PI3/mTOR kinase inhibitors affords a new opportunity for drug repurposing. PLoS Negl. Trop. Dis. 2011, 5, e1297. [Google Scholar] [CrossRef] [Green Version]
- Smirlis, D.; Duszenko, M.; Ruiz, A.J.; Scoulica, E.; Bastien, P.; Fasel, N.; Soteriadou, K. Targeting essential pathways in trypanosomatids gives insights into protozoan mechanisms of cell death. Parasites Vectors 2010, 3, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, M.E.; Del Portillo, H.A.; Milder, R.V.; Balanco, J.M.; Barcinski, M.A. Heat shock induction of apoptosis in promastigotes of the unicellular organism Leishmania (Leishmania) amazonensis. J. Cell. Physiol. 1996, 167, 305–313. [Google Scholar] [CrossRef]
- Arnoult, D.; Akarid, K.; Grodet, A.; Petit, P.X.; Estaquier, J.; Ameisen, J.C. On the evolution of programmed cell death: Apoptosis of the unicellular eukaryote Leishmania major involves cysteine proteinase activation and mitochondrion permeabilization. Cell Death Differ. 2002, 9, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Sereno, D.; Holzmuller, P.; Mangot, I.; Cuny, G.; Ouaissi, A.; Lemesre, J.L. Antimonial-mediated DNA fragmentation in Leishmania infantum amastigotes. Antimicrob. Agents Chemother. 2001, 45, 2064–2069. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Ochoa, R.; García, E.; Robledo, S.M.; Cardona, G.W. Virtual and experimental screening of phenylfuranchalcones as potential anti-Leishmania candidates. J. Mol. Graph. Model. 2019, 91, 164–171. [Google Scholar] [CrossRef]
- Ochoa, R.; Watowich, S.J.; Flórez, A.; Mesa, C.V.; Robledo, S.M.; Muskus, C. Drug search for leishmaniasis: A virtual screening approach by grid computing. J. Comput. Aided Mol. Des. 2016, 30, 541–552. [Google Scholar] [CrossRef]
- Varela-M, R.E.; Ochoa, R.; Muskus, C.E.; Muro, A.; Mollinedo, F. Identification of a RAC/AKT-like gene in Leishmania parasites as a putative therapeutic target in leishmaniasis. Parasites Vectors 2017, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tirado-Duarte, D.; Marín-Villa, M.; Ochoa, R.; Blandón-Fuentes, G.; Soares, M.J.; Robledo, S.M.; Varela-Miranda, R.E. The Akt-like kinase of Leishmania panamensis: As a new molecular target for drug discovery. Acta Trop. 2018, 177, 171–178. [Google Scholar] [CrossRef]
- Ochoa, R.; Rocha-Roa, C.; Marín-Villa, M.; Robledo, S.; Varela-M, R. Search of Allosteric Inhibitors and Associated Proteins of an AKT-like Kinase from Trypanosoma cruzi. Int. J. Mol. Sci. 2018, 19, 3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flórez, A.F.; Park, D.; Bhak, J.; Kim, B.C.; Kuchinsky, A.; Morris, J.H.; Espinosa, J.; Muskus, C. Protein network prediction and topological analysis in Leishmania major as a tool for drug target selection. BMC Bioinform. 2010, 11, 484. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Bustamante, C.; Ochoa, R.; Asela, C.; Muskus, C. Repurposing of known drugs for leishmaniasis treatment using bioinformatic predictions, in vitro validations and pharmacokinetic simulations. J. Comput. Aided Mol. Des. 2019, 33, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Koehler, D.; Shah, Z.A.; Williams, F.E. The GSK3β inhibitor, TDZD-8, rescues cognition in a zebrafish model of okadaic acid-induced Alzheimer’s disease. Neurochem. Int. 2019, 122, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, H.; Turlova, E.; Abussaud, A.; Ji, X.; Britto, L.R.; Miller, S.P.; Martinez, A.; Sun, H.S.; Feng, Z.P. GSK-3β inhibitor TDZD-8 reduces neonatal hypoxic-ischemic brain injury in mice. CNS Neurosci. Ther. 2017, 23, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela-M, R.E.; Villa-Pulgarin, J.A.; Yepes, E.; Müller, I.; Modolell, M.; Munoz, D.L.; Robledo, S.M.; Muskus, C.E.; Lopez-Aban, J.; Muro, A.; et al. In vitro and in vivo efficacy of ether lipid edelfosine against Leishmania spp. and SbV-resistant parasites. PLoS Negl. Trop. Dis. 2012, 6, e1612. [Google Scholar] [CrossRef] [Green Version]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Rastrojo, A.; Carrasco-Ramiro, F.; Martín, D.; Crespillo, A.; Reguera, R.M.; Aguado, B.; Requena, J.M. The transcriptome of Leishmania major in the axenic promastigote stage: Transcript annotation and relative expression levels by RNA-seq. BMC Genom. 2013, 14, 223. [Google Scholar] [CrossRef] [Green Version]
- Naula, C.; Parsons, M.; Mottram, J.C. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2005, 1754, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, A.C.; Trotman, L.C. Turning off AKT: PHLPP as a drug target. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 537–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Fontanillo, C. Protein–Protein Interactions Essentials: Key Concepts to Building and Analyzing Interactome Networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [Green Version]
- Ihle, N.T.; Powis, G. Take your PIK: Phosphatidylinositol 3-kinase inhibitors race through the clinic and toward cancer therapy. Mol. Cancer Ther. 2009, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Marone, R.; Cmiljanovic, V.; Giese, B.; Wymann, M.P. Targeting phosphoinositide 3-kinase: Moving towards therapy. Biochim. Biophys. Acta 2008, 1784, 159–185. [Google Scholar] [CrossRef]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science (N. Y.) 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Abeyrathna, P.; Su, Y. The critical role of Akt in cardiovascular function. Vasc. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [Green Version]
- Duronio, V. The life of a cell: Apoptosis regulation by the PI3K/PKB pathway. Biochem. J. 2008, 415, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [Green Version]
- Elledge, S.J. Cell cycle checkpoints: Preventing an identity crisis. Science (N. Y.) 1996, 274, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Hassan, P.; Fergusson, D.; Grant, K.M.; Mottram, J.C. The CRK3 protein kinase is essential for cell cycle progression of Leishmania mexicana. Mol. Biochem. Parasitol. 2001, 113, 189–198. [Google Scholar] [CrossRef]
- Banerjee, S.; Banerjee, R.; Das, R.; Duttagupta, S.; Saha, P. Isolation, characterization and expression of a cyclin from Leishmania donovani. FEMS Microbiol. Lett. 2003, 226, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Grant, K.M.; Dunion, M.H.; Yardley, V.; Skaltsounis, A.L.; Marko, D.; Eisenbrand, G.; Croft, S.L.; Meijer, L.; Mottram, J.C. Inhibitors of Leishmania mexicana CRK3 cyclin-dependent kinase: Chemical library screen and antileishmanial activity. Antimicrob. Agents Chemother. 2004, 48, 3033–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleghorn, L.A.T.; Woodland, A.; Collie, I.T.; Torrie, L.S.; Norcross, N.; Luksch, T.; Mpamhanga, C.; Walker, R.G.; Mottram, J.C.; Brenk, R.; et al. Identification of inhibitors of the Leishmania cdc2-related protein kinase CRK3. ChemMedChem 2011, 6, 2214–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira da Silva, L.; Beverley, S.M. Expansion of the target of rapamycin (TOR) kinase family and function in Leishmania shows that TOR3 is required for acidocalcisome biogenesis and animal infectivity. Proc. Natl. Acad. Sci. USA 2010, 107, 11965–11970. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Evers, B.M. mTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/mTOR signaling pathway as a therapeutic target for ovarian cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [Green Version]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R. The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for cancer prevention and treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R. Glycogen synthase kinase 3 beta: Can it be a target for oral cancer. Mol. Cancer 2010, 9, 144. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [Green Version]
- Xingi, E.; Smirlis, D.; Myrianthopoulos, V.; Magiatis, P.; Grant, K.M.; Meijer, L.; Mikros, E.; Skaltsounis, A.L.; Soteriadou, K. 6-Br-5methylindirubin-3’oxime (5-Me-6-BIO) targeting the leishmanial glycogen synthase kinase-3 (GSK-3) short form affects cell-cycle progression and induces apoptosis-like death: Exploitation of GSK-3 for treating leishmaniasis. Int. J. Parasitol. 2009, 39, 1289–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osolodkin, D.I.; Zakharevich, N.V.; Palyulin, V.a.; Danilenko, V.N.; Zefirov, N.S. Bioinformatic analysis of glycogen synthase kinase 3: Human versus parasite kinases. Parasitology 2011, 138, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wu, C.H.; Apweiler, R.; Bairoch, A.; Natale, D.A.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; et al. The Universal Protein Resource (UniProt): An expanding universe of protein information. Nucleic Acids Res. 2006, 34, D187–D191. [Google Scholar] [CrossRef]
- Myler, P.J. Searching the Tritryp genomes for drug targets. Adv. Exp. Med. Biol. 2008, 625, 133–140. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Vukovic, S.; Huggins, D.J. Quantitative metrics for drug–target ligandability. Drug Discov. Today 2018, 23, 1258–1266. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radusky, L.; Defelipe, L.A.; Lanzarotti, E.; Luque, J.; Barril, X.; Marti, M.A.; Turjanski, A.G. TuberQ: A Mycobacterium tuberculosis protein druggability database. Database 2014, 2014, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Tosatto, S.C.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins Struct. Funct. Genet. 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef]
- Schmidtke, P.; Barril, X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.I.P.; Fernández Do Porto, D.; Lanzarotti, E.; Sosa, E.J.; Burguener, G.; Pardo, A.M.; Klein, C.C.; Sagot, M.F.; De Vasconcelos, A.T.R.; Gales, A.C.; et al. An integrative, multi-omics approach towards the prioritization of Klebsiella pneumoniae drug targets. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Sosa, E.J.; Burguener, G.; Lanzarotti, E.; Defelipe, L.; Radusky, L.; Pardo, A.M.; Marti, M.; Turjanski, A.G.; Fernández Do Porto, D. Target-Pathogen: A structural bioinformatic approach to prioritize drug targets in pathogens. Nucleic Acids Res. 2018, 46, D413–D418. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butina, D. Unsupervised data base clustering based on daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Jiang, T.; Girke, T. A maximum common substructure-based algorithm for searching and predicting drug-like compounds. Bioinformatics 2008, 24, i366–i374. [Google Scholar] [CrossRef] [Green Version]
- Radusky, L.; Ruiz-Carmona, S.; Modenutti, C.; Barril, X.; Turjanski, A.G.; Martí, M.A. LigQ: A Webserver to Select and Prepare Ligands for Virtual Screening. J. Chem. Inf. Model. 2017, 57, 1741–1746. [Google Scholar] [CrossRef]
- Urrea, D.A.; Duitama, J.; Imamura, H.; Alzate, J.F.; Gil, J.; Munoz, N.; Villa, J.A.; Dujardin, J.C.; Ramirez-Pineda, J.R.; Triana-Chavez, O. Genomic Analysis of Colombian Leishmania panamensis strains with different level of virulence. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Villa-Pulgarín, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela-M, R.E.; Cuesta-Marbán, Á.; Müller, I.; Modolell, M.; Revuelta, J.L.; et al. Mitochondria and lipid raft-located FOF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Gene KEGG | L. mexicana ID | L. major ID | L. braziliensis ID | L. infantum ID | L. donovani ID |
|---|---|---|---|---|---|
| AKT | E9ARP5 | Q27687 | A4H9L8 | A4HXY2 | E9BDT9 |
| GSK3 | E9ARG4 | Q4QE15 | A4H9D1 | A4HXQ3 | E9BDK8 |
| AMPK | E9ALM1 | E9AE64 | A4HHK1 | A4I4Q9 | E9BL11 |
| mTOR | E9AU66 | Q4Q0C8 | A4HBF9 | A4IE36 | E9BV14 |
| CDK | E9ASH4 | O96526 | A4HNR5 | A4ICT0 | E9BTB9 |
| Kinase | PDB Template | Lig | QMEAN | DS |
|---|---|---|---|---|
| CDK | 6GU6.A | 0.654 | −1.71 | 0.727 |
| AKT | 4WB5.A | 0.741 | −0.44 | 0.509 |
| mTOR | 4JSN.A | 0.791 | −3.76 | 0.533 |
| AMPK | 5EZV.A | 0.524 | −1.07 | 0.571 |
| GSK3 | 3E3P.A | 0.516 | 0.56 | 0.625 |
| ZINC ID—Docking Results | ChEMBL ID—ligQ Results | Similarity |
|---|---|---|
| ZINC00027361 | CHEMBL284861 | 1.000 |
| ZINC00135232 | CHEMBL1989856 | 0.583 |
| ZINC00135232 | CHEMBL2000433 | 0.625 |
| ZINC19835187 | CHEMBL1569442 | 0.515 |
| Kinase | Degree | Bottleneck |
|---|---|---|
| CDK | 159 | 45676 |
| AKT | 140 | 29858 |
| mTOR | – | – |
| AMPK | 170 | 41 |
| GSK3 | 132 | 31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochoa, R.; Ortega-Pajares, A.; Castello, F.A.; Serral, F.; Fernández Do Porto, D.; Villa-Pulgarin, J.A.; Varela-M, R.E.; Muskus, C. Identification of Potential Kinase Inhibitors within the PI3K/AKT Pathway of Leishmania Species. Biomolecules 2021, 11, 1037. https://doi.org/10.3390/biom11071037
Ochoa R, Ortega-Pajares A, Castello FA, Serral F, Fernández Do Porto D, Villa-Pulgarin JA, Varela-M RE, Muskus C. Identification of Potential Kinase Inhibitors within the PI3K/AKT Pathway of Leishmania Species. Biomolecules. 2021; 11(7):1037. https://doi.org/10.3390/biom11071037
Chicago/Turabian StyleOchoa, Rodrigo, Amaya Ortega-Pajares, Florencia A. Castello, Federico Serral, Darío Fernández Do Porto, Janny A. Villa-Pulgarin, Rubén E. Varela-M, and Carlos Muskus. 2021. "Identification of Potential Kinase Inhibitors within the PI3K/AKT Pathway of Leishmania Species" Biomolecules 11, no. 7: 1037. https://doi.org/10.3390/biom11071037