
The Cx43 Carboxyl-Terminal Mimetic Peptide αCT1 Protects Endothelial Barrier Function in a ZO1 Binding-Competent Manner


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Test Reagents
2.2. FITC-Dextran Extravasation
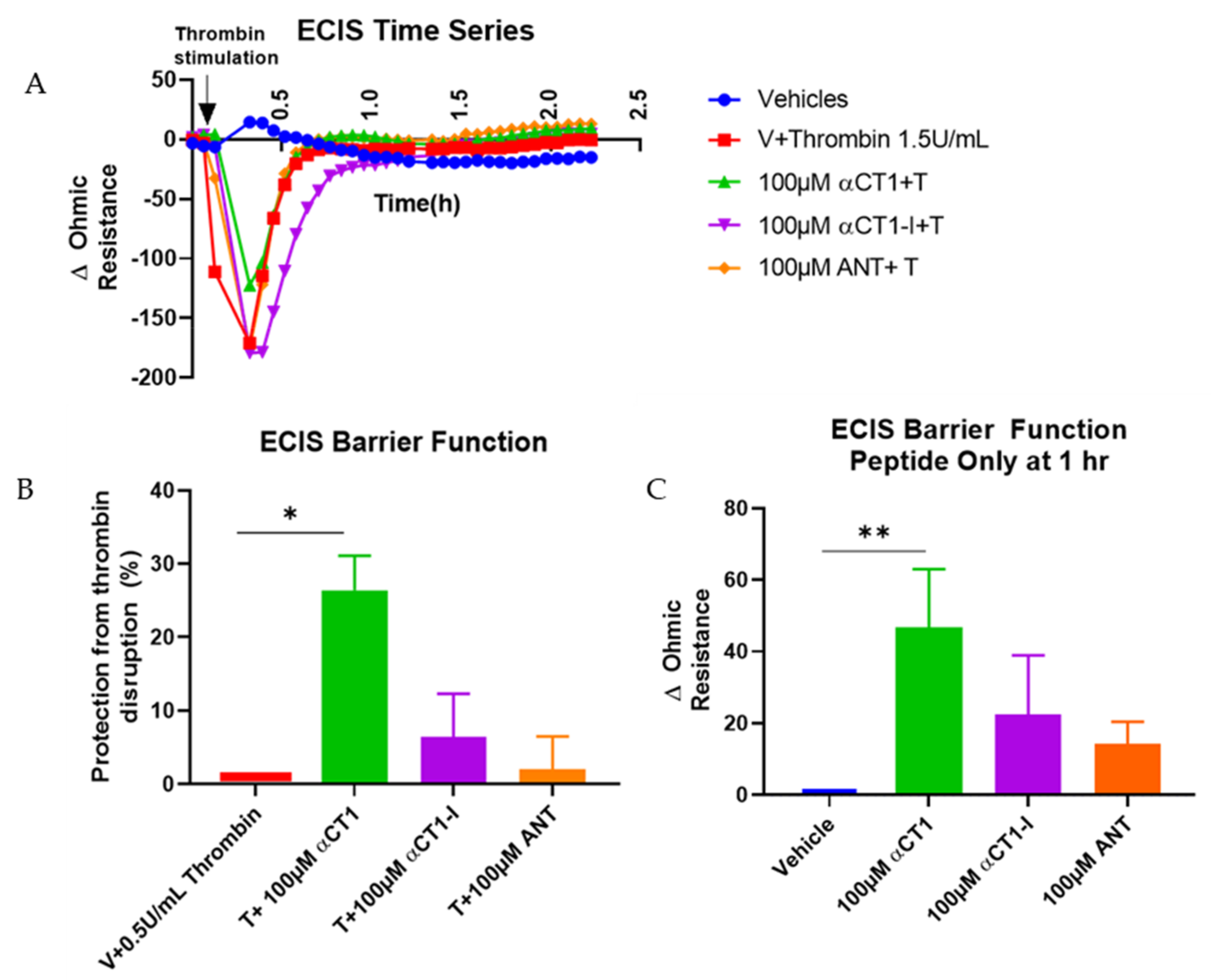
2.3. Impedance Measurement Using ECIS
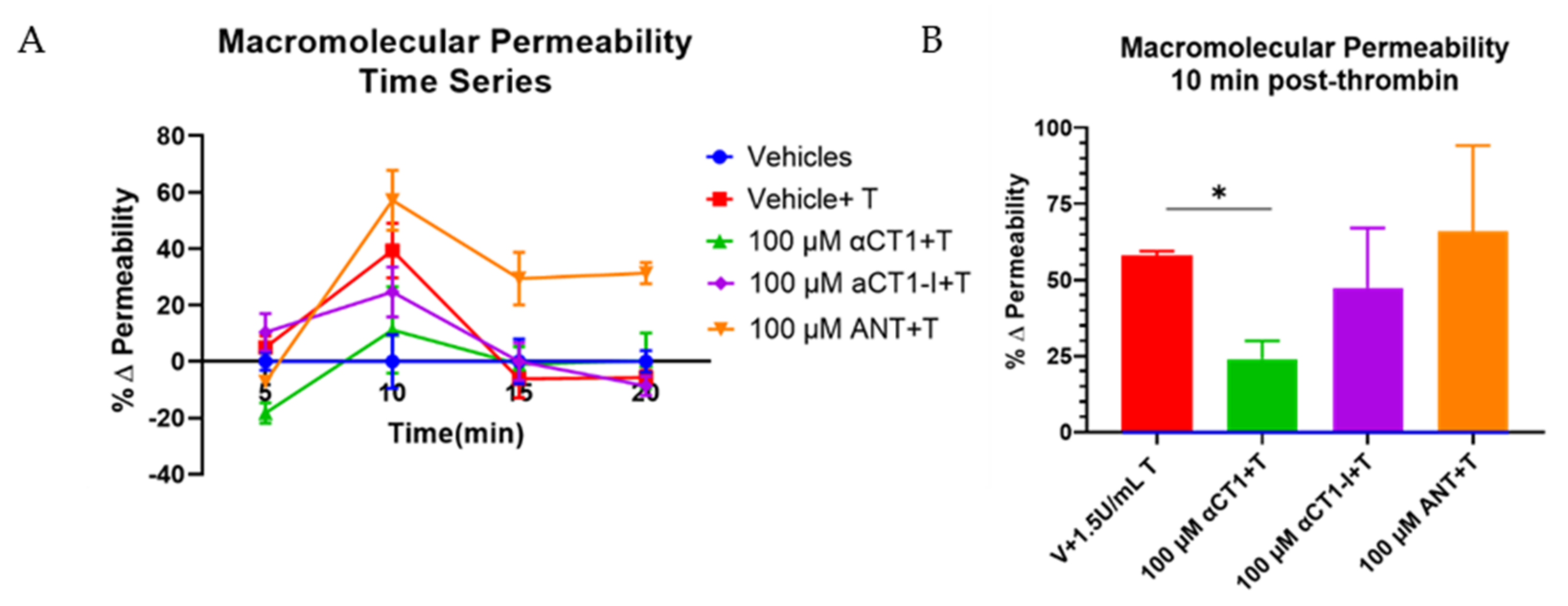
2.4. Macromolecular Permeability (MP)
2.5. Proximity Ligation Assay
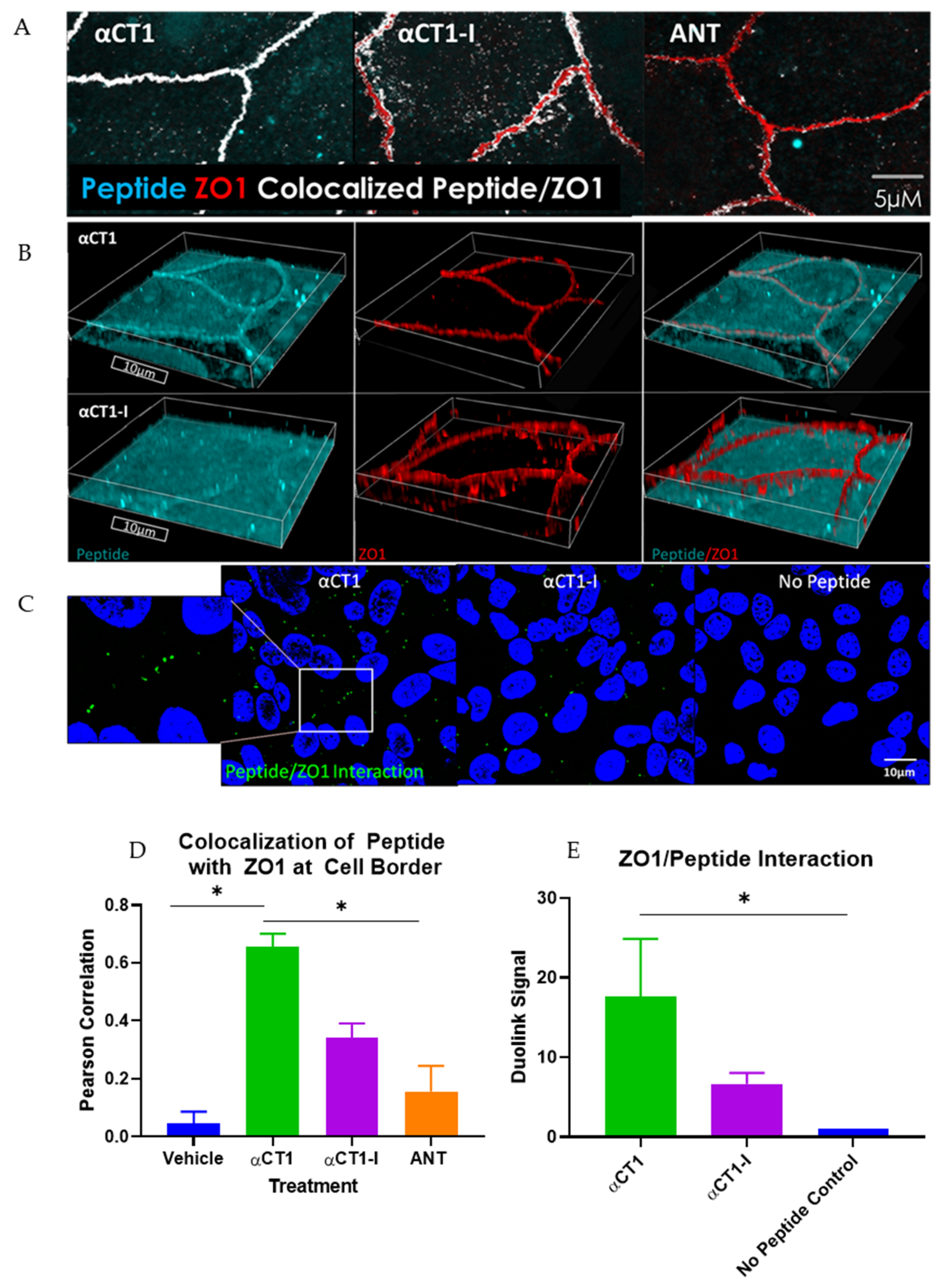
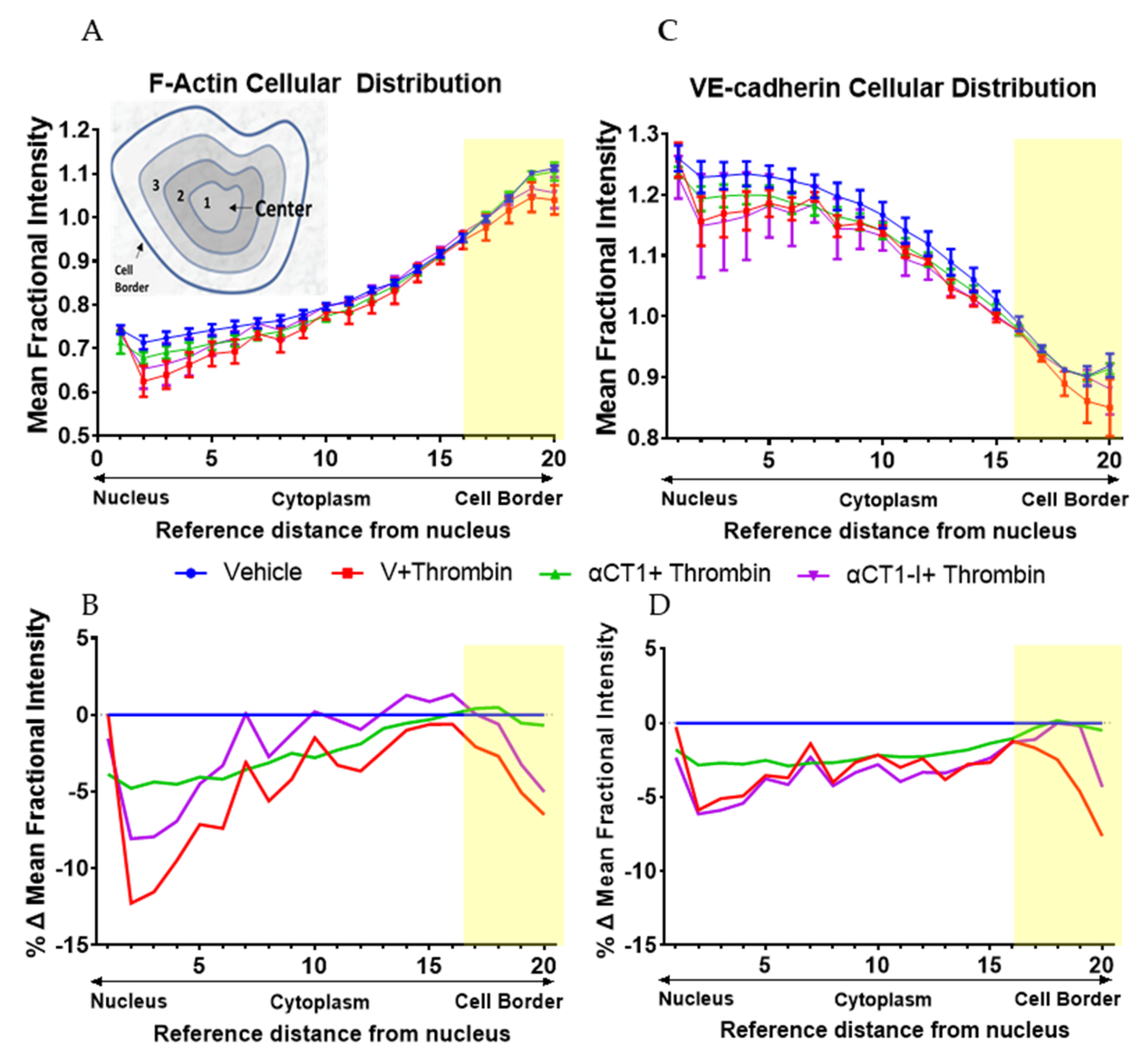
2.6. Immunostaining and Quantitative Image Analysis
2.7. Statistics
3. Results
3.1. αCT1 Requires a CT Isoleucine to Associate with ZO1 at Borders between MDCK Cells
3.2. αCT1 Inhibits Thrombin-Induced Disruption of Endothelial Barrier Function in a ZO1 Binding-Competent Manner
3.3. αCT1 Prevents Thrombin-Induced Changes in Endothelial F-Actin and VE-Cadherin Distribution in a ZO1 Binding-Competent Manner
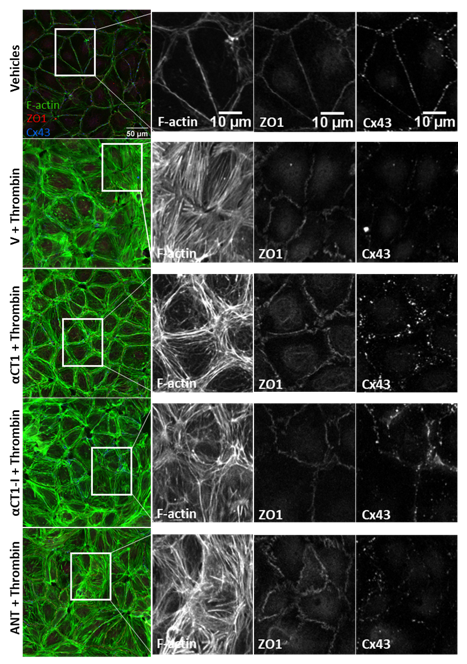
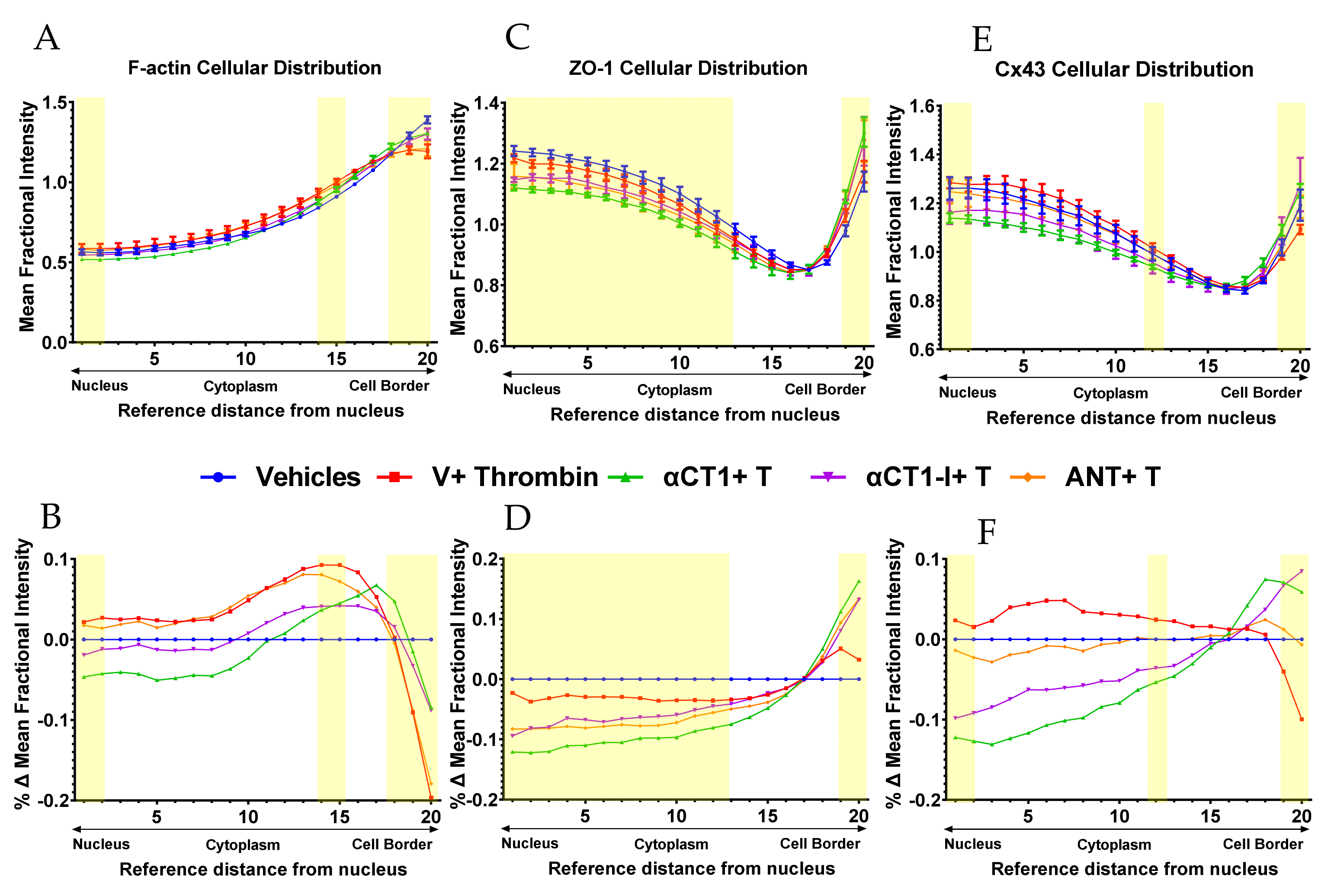
3.4. αCT1 Requires ZO1 Binding Competency to Modulate the Distribution of F-Actin, ZO1 and Cx43 in Cultured Endothelial Cells
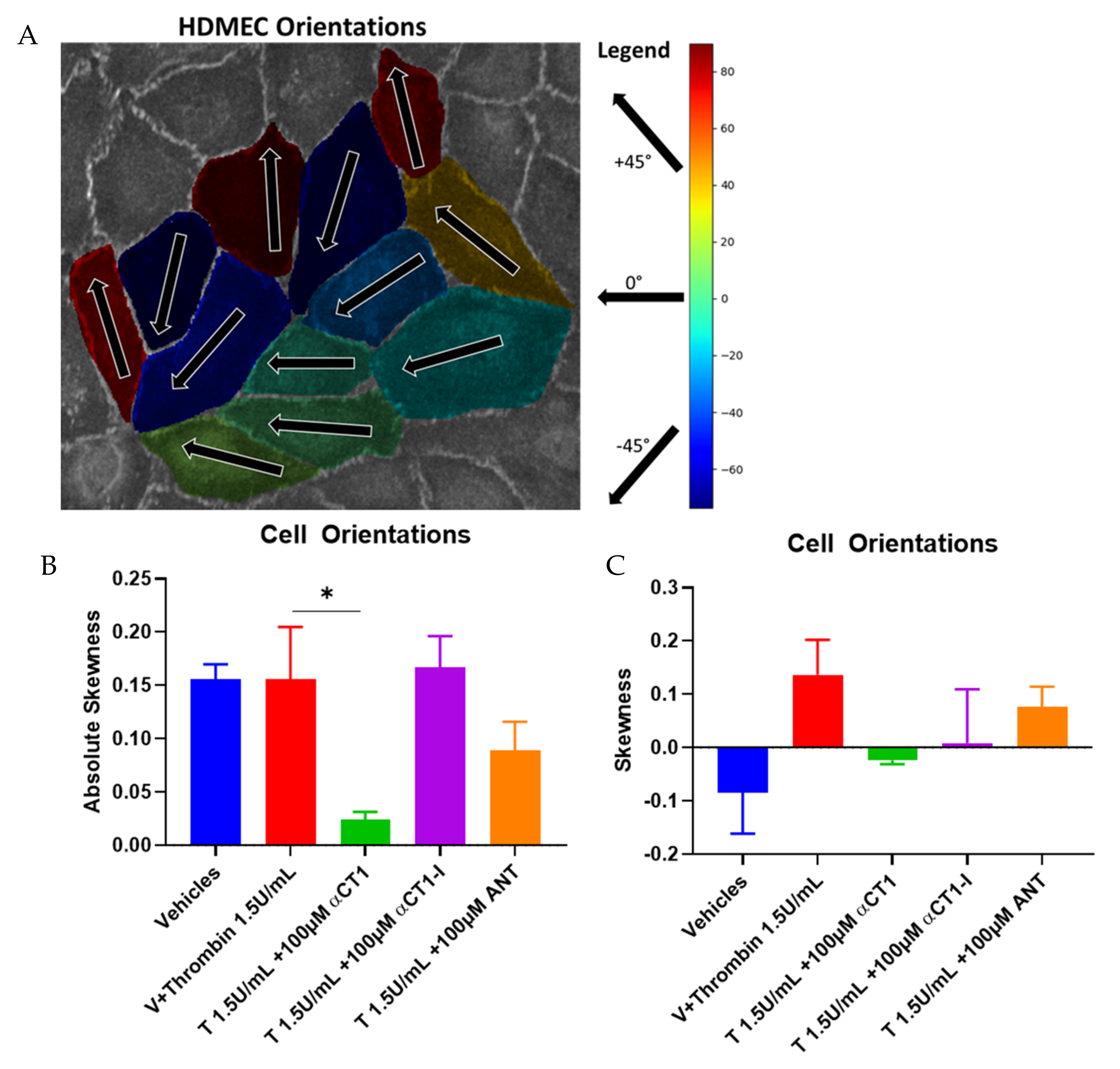
3.5. αCT1 Requires ZO1 Binding Competency to Exert Changes in Distribution of Cell Orientations
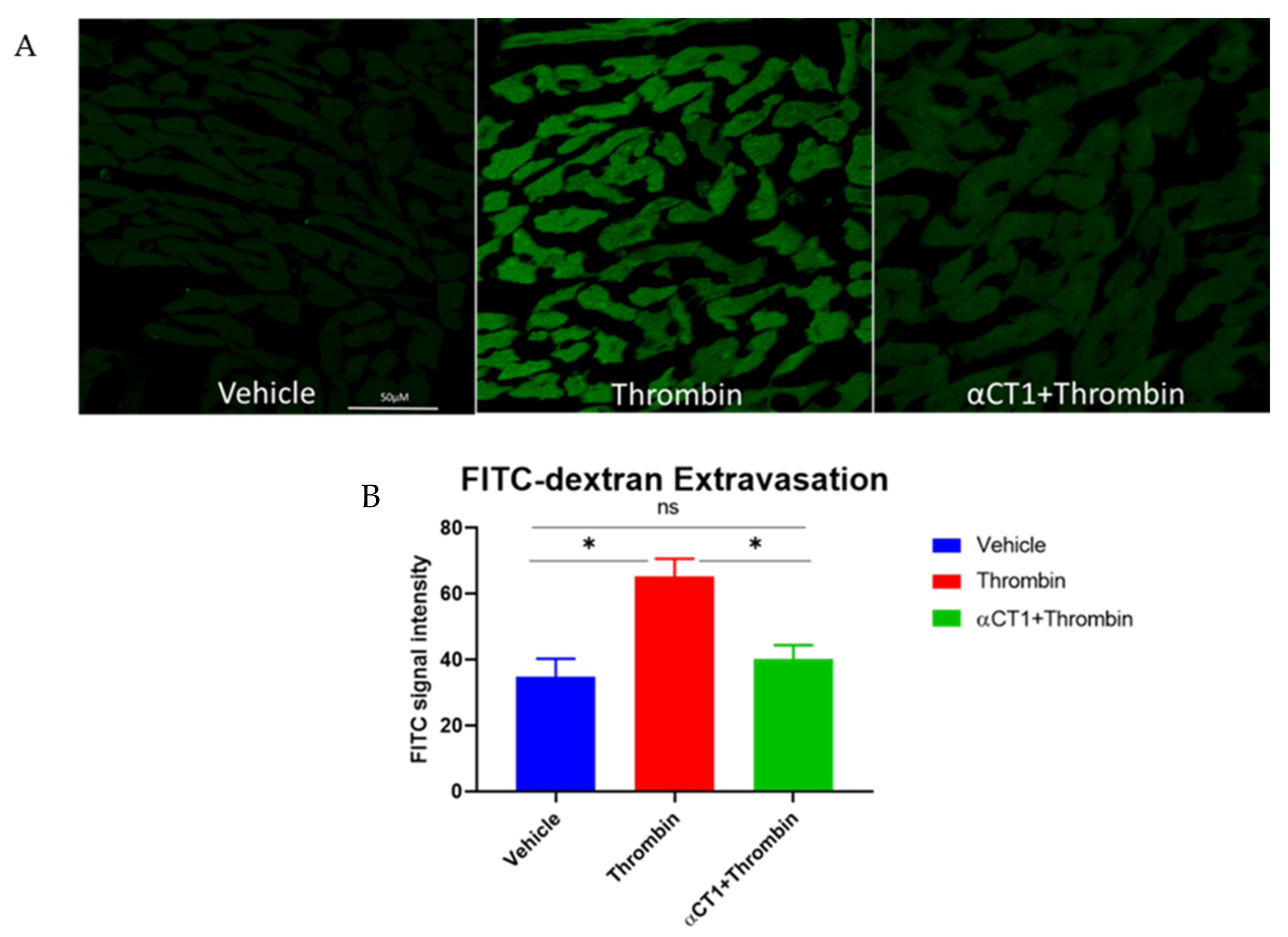
3.6. αCT1 Reduces Vascular Leak in Langendorff-Perfused Mouse Hearts
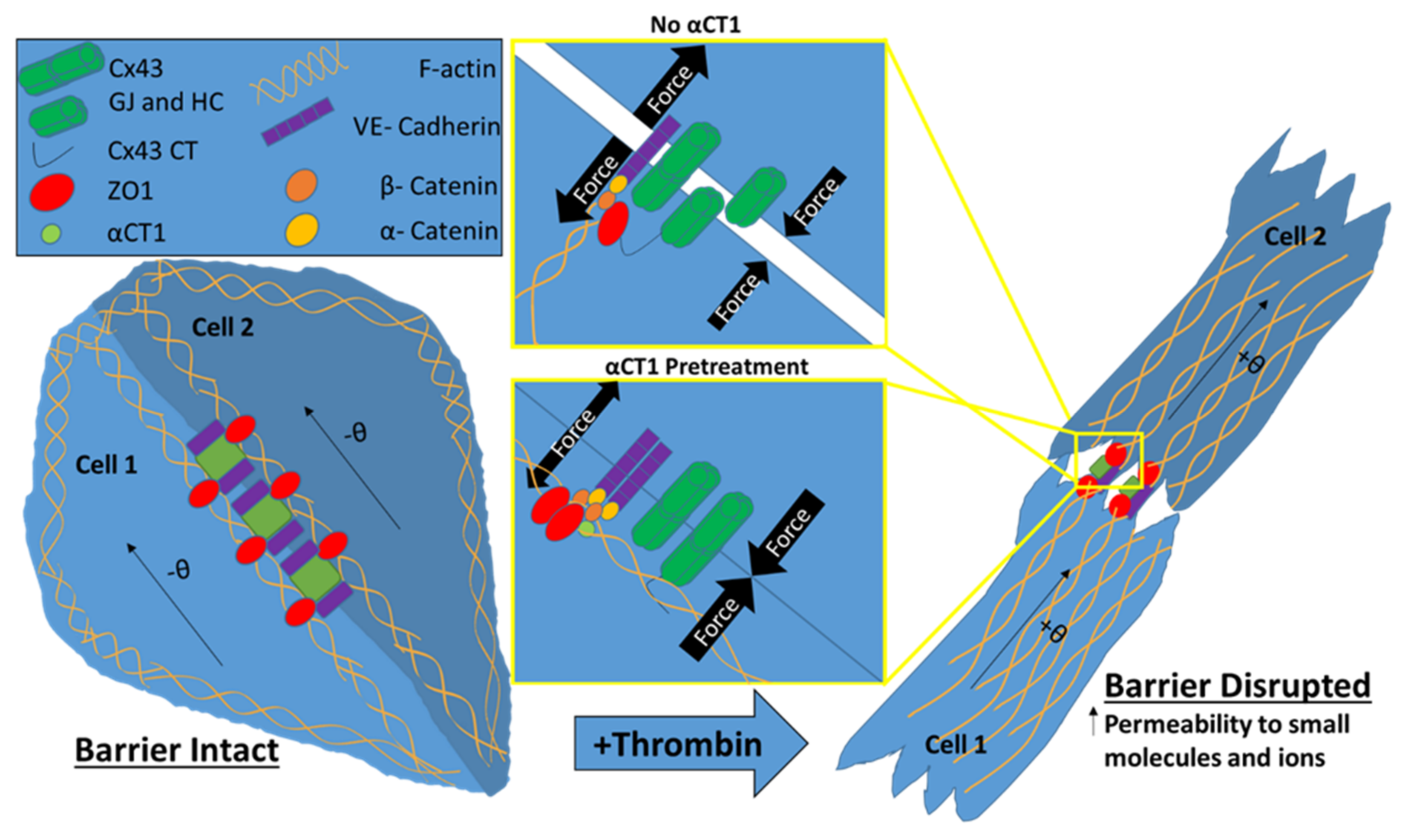
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zihni, C.; Mills, C.; Matter, K.; Balda, M. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Higashi, T.; Miller, A.L. Tricellular junctions: How to build junctions at the TRICkiest points of epithelial cells. Mol. Biol. Cell 2017, 28, 2023–2034. [Google Scholar] [CrossRef]
- Escribano, J.; Chen, M.B.; Moeendarbary, E.; Cao, X.; Shenoy, V.; Garcia-Aznar, J.M.; Kamm, R.D.; Spill, F. Balance of me-chanical forces drives endothelial gap formation and may facilitate cancer and immune-cell extravasation. PLoS Comput. Biol. 2019, 15, e1006395. [Google Scholar] [CrossRef] [Green Version]
- Herrero, R.; Sanchez, G.; Lorente, J.A. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann. Transl. Med. 2018, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. The Coronary Circulation as a Target of Cardioprotection. Circ. Res. 2016, 118, 1643–1658. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Protection of the human coronary circulation by remote ischemic conditioning. Int. J. Cardiol. 2018, 252, 35–36. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.; Erfinanda, L.; Bartz, C.; Kuebler, W.M. Novel mechanisms regulating endothelial barrier function in the pulmonary microcirculation. J. Physiol. 2018, 597, 997–1021. [Google Scholar] [CrossRef] [PubMed]
- Soon, A.S.C.; Chua, J.W.; Becker, D.L. Connexins in endothelial barrier function—Novel therapeutic targets countering vascular hyperpermeability. Thromb. Haemost. 2016, 116, 852–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghajanian, A.; Wittchen, E.S.; Allingham, M.J.; Garrett, T.A.; Burridge, K. Endothelial cell junctions and the regulation of vascular permeability and leukocyte transmigration. J. Thromb. Haemost. 2008, 6, 1453–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komarova, Y.; Malik, A.B. Regulation of Endothelial Permeability via Paracellular and Transcellular Transport Pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef]
- Belvitch, P.; Htwe, Y.M.; Brown, M.E.; Dudek, S. Cortical Actin Dynamics in Endothelial Permeability. Basement Membr. 2018, 82, 141–195. [Google Scholar] [CrossRef]
- Giepmans, B. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Derangeon, M.; Spray, D.; Bourmeyster, N.; Sarrouilhe, D.; Hervé, J.-C. Reciprocal influence of connexins and apical junction proteins on their expressions and functions. BBA Biomembr. 2009, 1788, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radeva, M.Y.; Waschke, J.; Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2018, 222, e12860. [Google Scholar] [CrossRef]
- Hervé, J.-C.; Bourmeyster, N.; Sarrouilhe, D.; Duffy, H.S. Gap junctional complexes: From partners to functions. Prog. Biophys. Mol. Biol. 2007, 94, 29–65. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G. Concepts in microvascular endothelial barrier regulation in health and disease. Microvasc. Res. 2009, 77, 1–3. [Google Scholar] [CrossRef]
- Strauss, R.E.; Gourdie, R.G. Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation. Biomolecules 2020, 10, 1656. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.; Xu, X.; Park, H.; Wei, C.-J.; Chang, S.; Chatterjee, B.; Lo, C. Connexin43 Modulates Cell Polarity and Directional Cell Migration by Regulating Microtubule Dynamics. PLoS ONE 2011, 6, e26379. [Google Scholar] [CrossRef] [Green Version]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. BBA Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Kameritsch, P.; Pogoda, K.; Pohl, U. Channel-independent influence of connexin 43 on cell migration. BBA Biomembr. 2012, 1818, 1993–2001. [Google Scholar] [CrossRef] [Green Version]
- Olk, S.; Zoidl, G.; Dermietzel, R. Connexins, cell motility, and the cytoskeleton. Cell Motil Cytoskeleton. 2009, 66, 1000–1016. [Google Scholar] [CrossRef]
- Matsuuchi, L.; Naus, C.C. Gap junction proteins on the move: Connexins, the cytoskeleton and migration. BBA Biomembr. 2013, 1828, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein–Protein Interactions with Connexin 43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef] [Green Version]
- Sorgen, P.L.; Duffy, H.S.; Sahoo, P.; Coombs, W.; Delmar, M.; Spray, D.C. Structural Changes in the Carboxyl Terminus of the Gap Junction Protein Connexin43 Indicates Signaling between Binding Domains for c-Src and Zonula Occludens-1. J. Biol. Chem. 2004, 279, 54695–54701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giepmans, B.; Moolenaar, W.H. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr. Biol. 1998, 8, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar]
- Giepmans, B.N.G.; Moolenaar, W.H. Pitfalls in assessing microvascular endothelial barrier function: Impedance-based devices versus the classic macromolecular tracer assay. Sci. Rep. 2016, 6, 23671. [Google Scholar]
- Gullberg, M.; Andersson, A.-C. Visualization and quantification of protein-protein interactions in cells and tissues. Nat. Methods 2010, 7, v–vi. [Google Scholar] [CrossRef]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. Core R: A Language and Environment for Statistical Computing; Version 2.6.2 (2008-02-08); R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Obert, E.; Strauss, R.; Brandon, C.; Grek, C.; Ghatnekar, G.; Gourdie, R.; Rohrer, B. Targeting the tight junction protein, zonula occludens-1, with the connexin43 mimetic peptide, αCT1, reduces VEGF-dependent RPE pathophysiology. J. Mol. Med. 2017, 95, 535–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula Occludens-1 Alters Connexin43 Gap Junction Size and Organization by Influencing Channel Accretion. Mol. Bio. Cell 2005, 16, 5686–5698. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Hoagland, D.; Palatinus, J.A.; He, H.; Iyyathurai, J.; Jourdan, L.J.; Bultynck, G.; Wang, Z.; Zhang, Z.; Schey, K.; et al. Interaction of α Carboxyl Terminus 1 Peptide With the Connexin 43 Carboxyl Terminus Preserves Left Ventricular Function After Ischemia-Reperfusion Injury. J. Am. Hear. Assoc. 2019, 8, e012385. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting co-agulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslin, J.W.; Zhang, X.E.; Worthylake, R.A.; Souza-Smith, F.M. Involvement of Local Lamellipodia in Endothelial Barrier Function. PLoS ONE 2015, 10, e0117970. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Tanislav, C.; Troidl, C.; Schulz, R.; Hamm, C.; Gündüz, D. cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol. Rep. 2014, 2, e12175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Cell Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [Green Version]
- Rabiet, M.-J.; Plantier, J.-L.; Rival, Y.; Genoux, Y.; Lampugnani, M.-G.; Dejana, E. Thrombin-Induced Increase in Endo-thelial Permeability Is Associated With Changes in Cell-to-Cell Junction Organization. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 488–496. [Google Scholar] [CrossRef]
- Doggett, T.M.; Breslin, J.W. Study of the Actin Cytoskeleton in Live Endothelial Cells Expressing GFP-Actin. J. Vis. Exp. 2011, e3187. [Google Scholar] [CrossRef] [Green Version]
- Elias, L.A.B.; Wang, D.D.; Kriegstein, A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nat. Cell Biol. 2007, 448, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef]
- Fan, J.; Ray, P.; Lu, Y.W.; Kaur, G.; Schwarz, J.J.; Wan, L.Q. Cell chirality regulates intercellular junctions and endothelial permeability. Sci. Adv. 2018, 4, eaat2111. [Google Scholar] [CrossRef] [Green Version]
- Maass, K.; Ghanem, A.; Kim, J.-S.; Saathoff, M.; Urschel, S.; Kirfel, G.; Grümmer, R.; Kretz, M.; Lewalter, T.; Tiemann, K.; et al. Defective Epidermal Barrier in Neonatal Mice Lacking the C-Terminal Region of Connexin43. Mol. Biol. Cell 2004, 15, 4597–4608. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.C.; Tiruppathi, R.; Sandoval, R.D.; Minshall, M.; Holinstat, M.; Malik, A.B. Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J. Physiol. 2002, 539, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Wittchen, E.S. The tension mounts: Stress fibers as force-generating mechanotransducers. J. Cell Biol. 2013, 200, 9–19. [Google Scholar] [CrossRef]
- Marsh, S.R.; Williams, Z.J.; Pridham, K.J.; Gourdie, R.G. Peptidic Connexin43 Therapeutics in Cardiac Reparative Medi-cine. J. Cardiovasc. Dev. Dis. 2021, 8, 52. [Google Scholar] [CrossRef]
- Schulz, R.; Görge, P.M.; Görbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vouret-Craviari, V.; Boquet, P.; Pouysségur, J.; Van Obberghen-Schilling, E. Regulation of the Actin Cytoskeleton by Thrombin in Human Endothelial Cells: Role of Rho Proteins in Endothelial Barrier Function. Mol. Biol. Cell 1998, 9, 2639–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiers, J.L.; Peng, X.; Fanning, A.S.; DeMali, K.A. ZO-1 recruitment to α-catenin: A novel mechanism for coupling the assembly of tight junctions to adherens junctions. J. Cell Sci. 2013, 126, 3904–3915. [Google Scholar] [CrossRef] [Green Version]
- Bogatcheva, N.V.; Garcia, J.G.; Verin, A.D. Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry 2002, 67, 75–84. [Google Scholar]
- Riesen, F.K.; Rothen-Rutishauser, B.; Wunderli-Allenspach, H. A ZO1-GFP fusion protein to study the dynamics of tight junctions in living cells. Histochem. Cell Biol. 2002, 117, 307–315. [Google Scholar] [CrossRef]
- Cotrina, M.L.; Lin, J.H.-C.; Nedergaard, M. Adhesive properties of connexin hemichannels. Glia 2008, 56, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.-C.; Takano, T.; Cotrina, M.L.; Arcuino, G.; Kang, J.; Liu, S.; Gao, Q.; Jiang, L.; Li, F.; Lichtenberg-Frate, H.; et al. Connexin 43 Enhances the Adhesivity and Mediates the Invasion of Malignant Glioma Cells. J. Neurosci. 2002, 22, 4302–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Francis, R.; Wei, C.J.; Linask, K.L.; Lo, C.W. Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development 2006, 133, 3629–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameritsch, P.; Kiemer, F.; Beck, H.; Pohl, U.; Pogoda, K. Cx43 increases serum induced filopodia formation via activation of p21-activated protein kinase 1. BBA Bioenerg. 2015, 1853, 2907–2917. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Bleher, R.; Brown, M.E.; Garcia, J.G.N.; Dudek, S.M.; Shekhawat, G.S.; Dravid, V.P. Nano-Biomechanical Study of Spatio-Temporal Cytoskeleton Rearrangements that Determine Subcellular Mechanical Properties and Endothelial Permeability. Sci. Rep. 2015, 5, 11097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-F.; Zeng, Q.; Ozaki, H.; Wang, L.; Hand, A.R.; Hla, T.; Wang, E.; Lee, M.-J. Dual Roles of Tight Junction-associated Protein, Zonula Occludens-1, in Sphingosine 1-Phosphate-mediated Endothelial Chemotaxis and Barrier Integrity. J. Biol. Chem. 2006, 281, 29190–29200. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Montgomery, J.; Sharma, M.; Ravi, A.; Rajkumar, J.; Moyer, K.E.; Gourdie, R.G.; Ghatnekar, G.S. A Multicenter Randomized Controlled Trial Evaluating a Cx43-Mimetic Peptide in Cutaneous Scarring. J. Investig. Dermatol. 2017, 137, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Prasad, G.; Viswanathan, V.; Armstrong, D.G.; Gourdie, R.G.; Dvm, G.S.G. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial. Wound Repair Regen. 2015, 23, 203–212. [Google Scholar] [CrossRef]
- Ghatnekar, G.S.; Grek, C.L.; Armstrong, D.G.; Desai, S.C.; Gourdie, R.G. The Effect of a Connexin43-Based Peptide on the Healing of Chronic Venous Leg Ulcers: A Multicenter, Randomized Trial. J. Investig. Dermatol. 2015, 135, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin 43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Endothelial Barrier and Its Abnormalities in Cardiovascular Disease. Front. Physiol. 2015, 6, 365. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Hoeker, G.S.; Alvarez-Laviada, A.; Hoagland, D.; Wan, X.; King, D.R.; Sanchez-Alonso, J.; Chen, C.; Jourdan, J.; Isom, L.L.; et al. The adhesion function of the sodium channel beta subunit (β1) contributes to cardiac action potential propagation. Elife 2018, 7, e37610. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Lin, J.; Hoeker, G.S.; Keener, J.P.; Gourdie, R.G.; Poelzing, S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: An experimental and modeling study. Pflügers Arch. Eur. J. Physiol. 2015, 467, 2093–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strauss, R.E.; Mezache, L.; Veeraraghavan, R.; Gourdie, R.G. The Cx43 Carboxyl-Terminal Mimetic Peptide αCT1 Protects Endothelial Barrier Function in a ZO1 Binding-Competent Manner. Biomolecules 2021, 11, 1192. https://doi.org/10.3390/biom11081192
Strauss RE, Mezache L, Veeraraghavan R, Gourdie RG. The Cx43 Carboxyl-Terminal Mimetic Peptide αCT1 Protects Endothelial Barrier Function in a ZO1 Binding-Competent Manner. Biomolecules. 2021; 11(8):1192. https://doi.org/10.3390/biom11081192
Chicago/Turabian StyleStrauss, Randy E., Louisa Mezache, Rengasayee Veeraraghavan, and Robert G. Gourdie. 2021. "The Cx43 Carboxyl-Terminal Mimetic Peptide αCT1 Protects Endothelial Barrier Function in a ZO1 Binding-Competent Manner" Biomolecules 11, no. 8: 1192. https://doi.org/10.3390/biom11081192