



Deciphering the Alphabet of Disorder—Glu and Asp Act Differently on Local but Not Global Properties

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Techniques
2.2. Protein Purification
2.3. NMR Assignment
2.4. Secondary Chemical Shift (SCS) Analysis
2.5. Ubiquitin Binding
2.6. Small-Angle X-ray Scattering (SAXS)
2.7. Diffusion-Ordered NMR Spectroscopy
2.8. Dss1 Peptide Assignment
2.9. Molecular Dynamics Simulations
3. Results
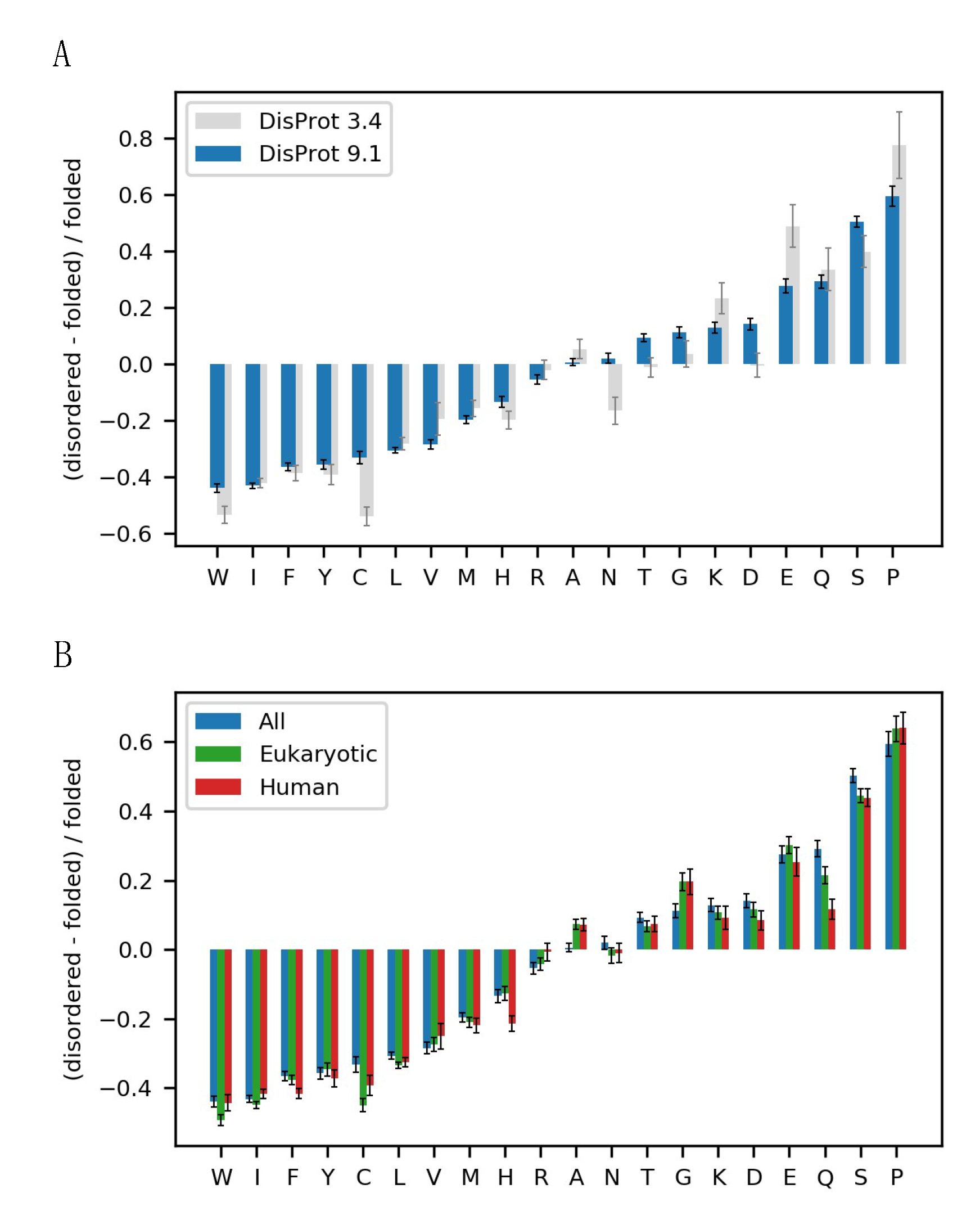
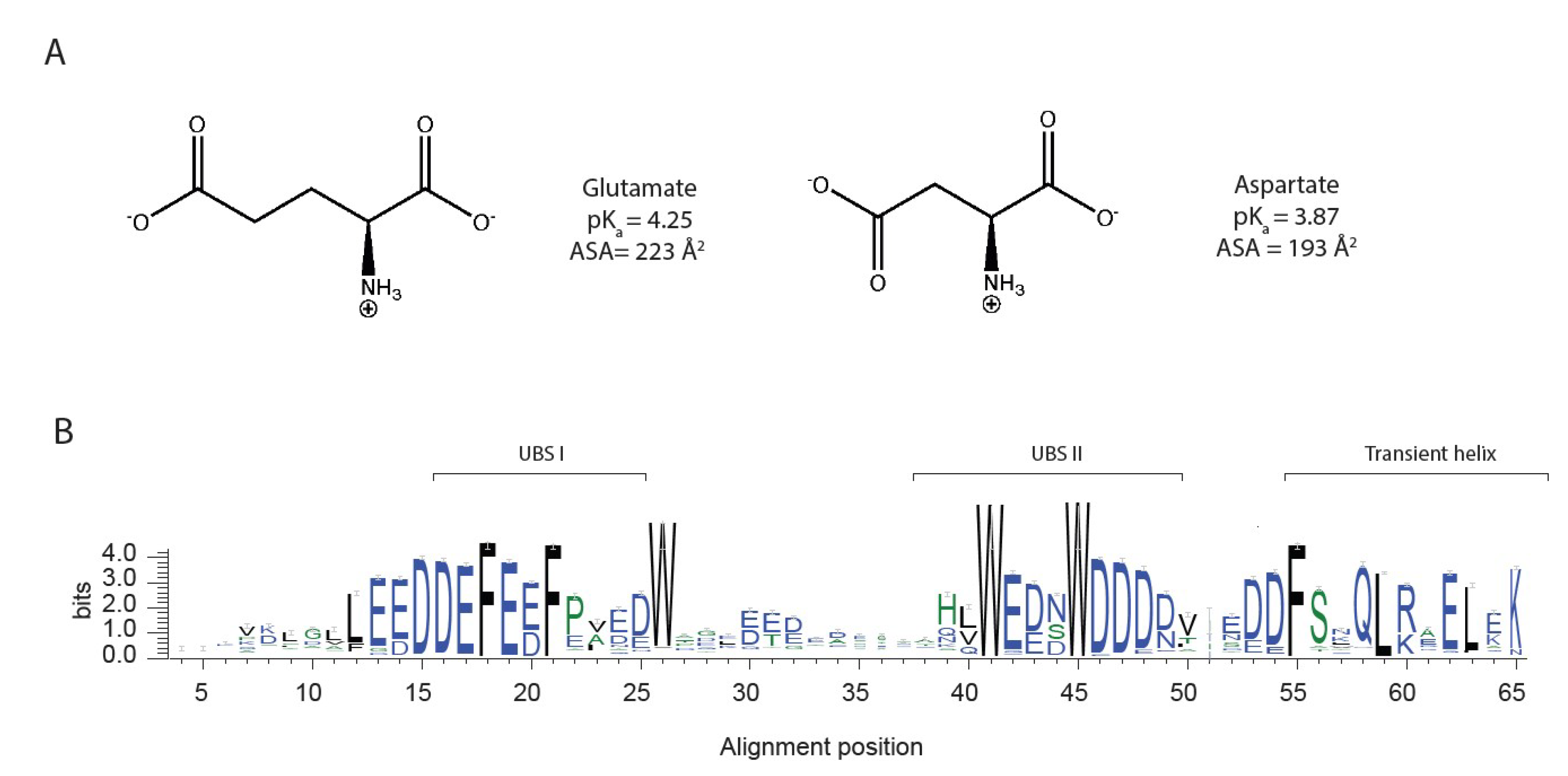
3.1. The Alphabet of Disorder
3.2. Functional Effect of Aspartate and Glutamate in Dss1
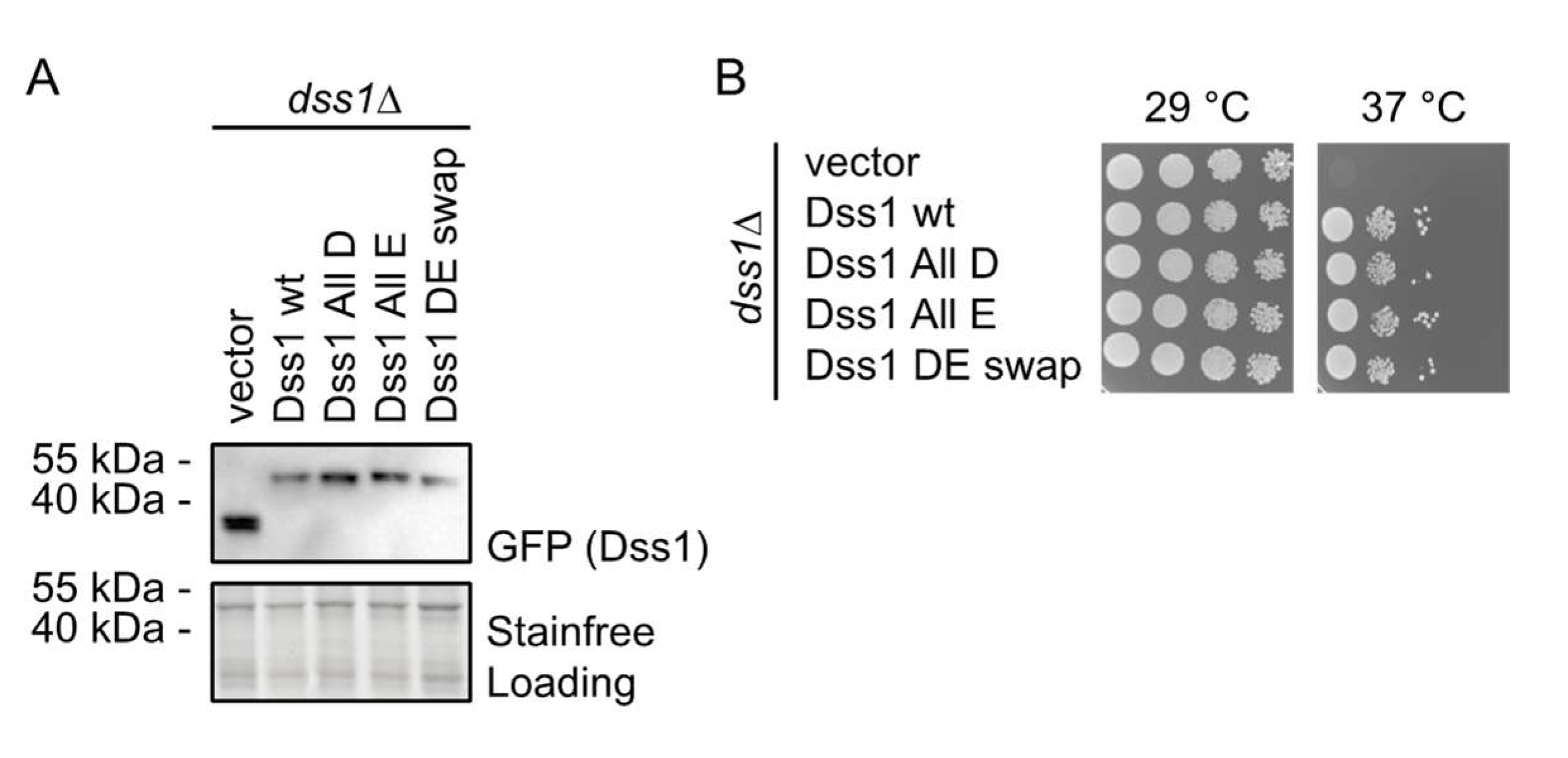
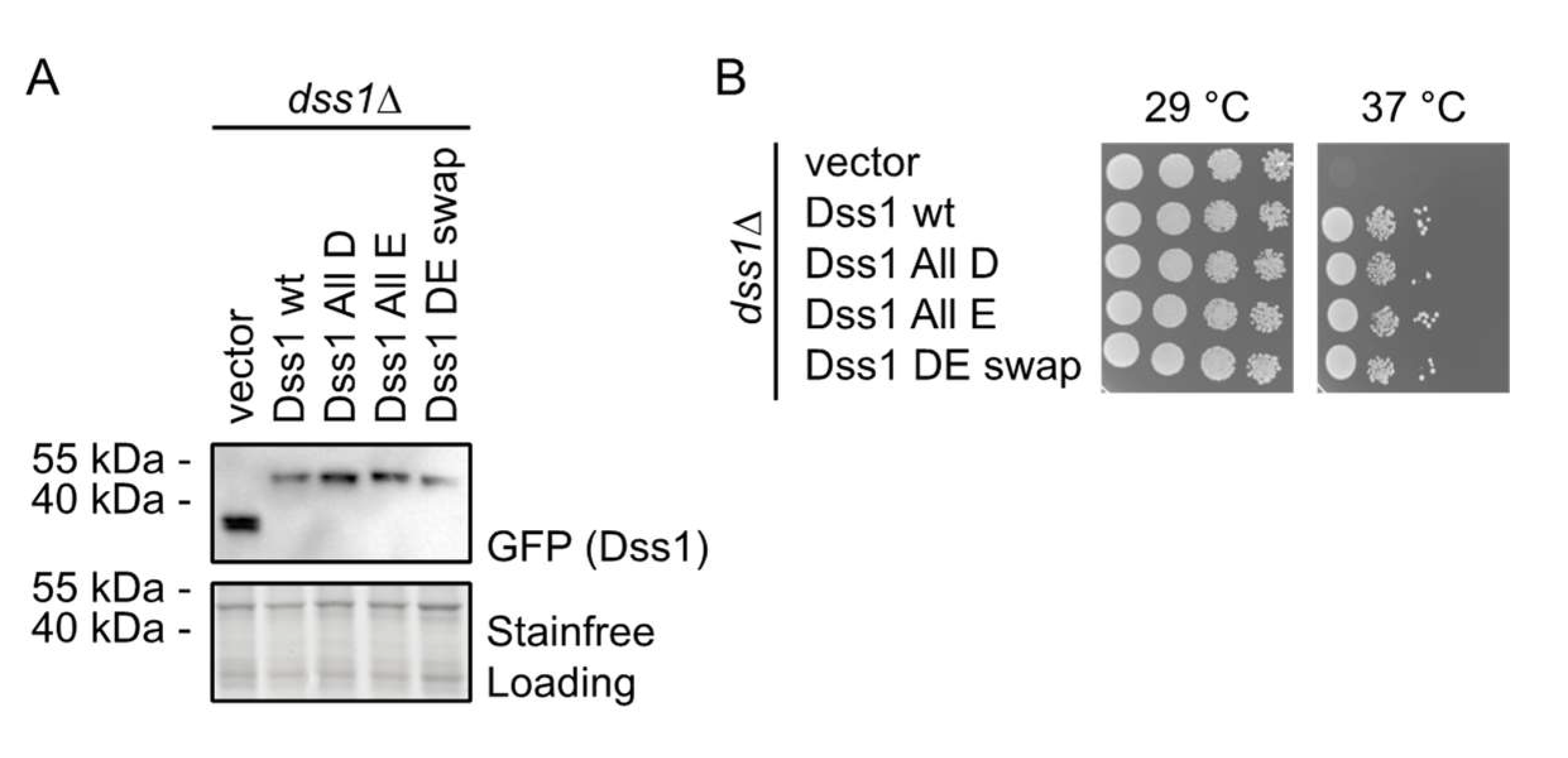
3.2.1. The Glu/Asp Variants Are Functional in Vivo
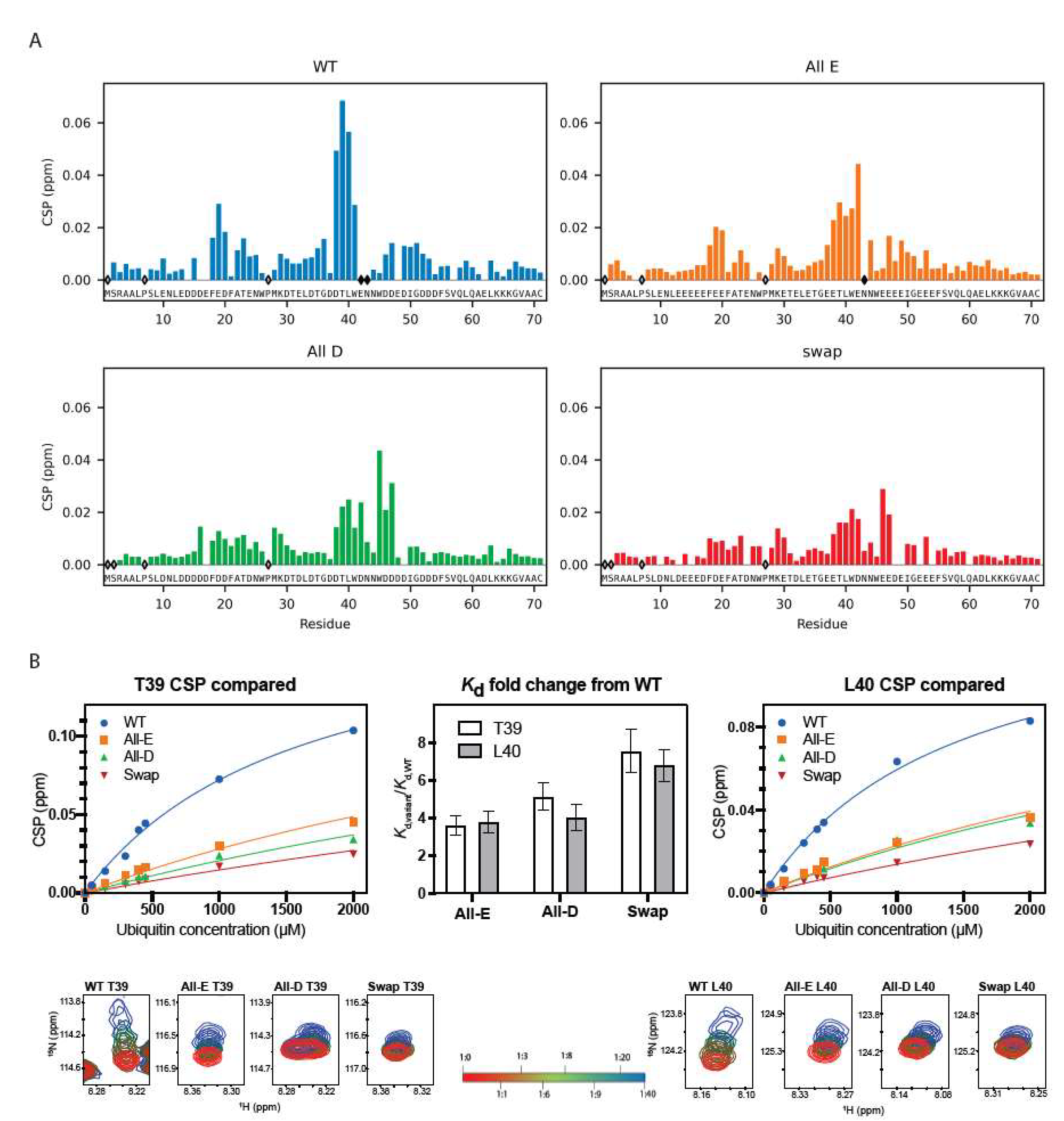
3.2.2. Ubiquitin Binding Affinity, but Not Binding Ability, Depends on Glutamate
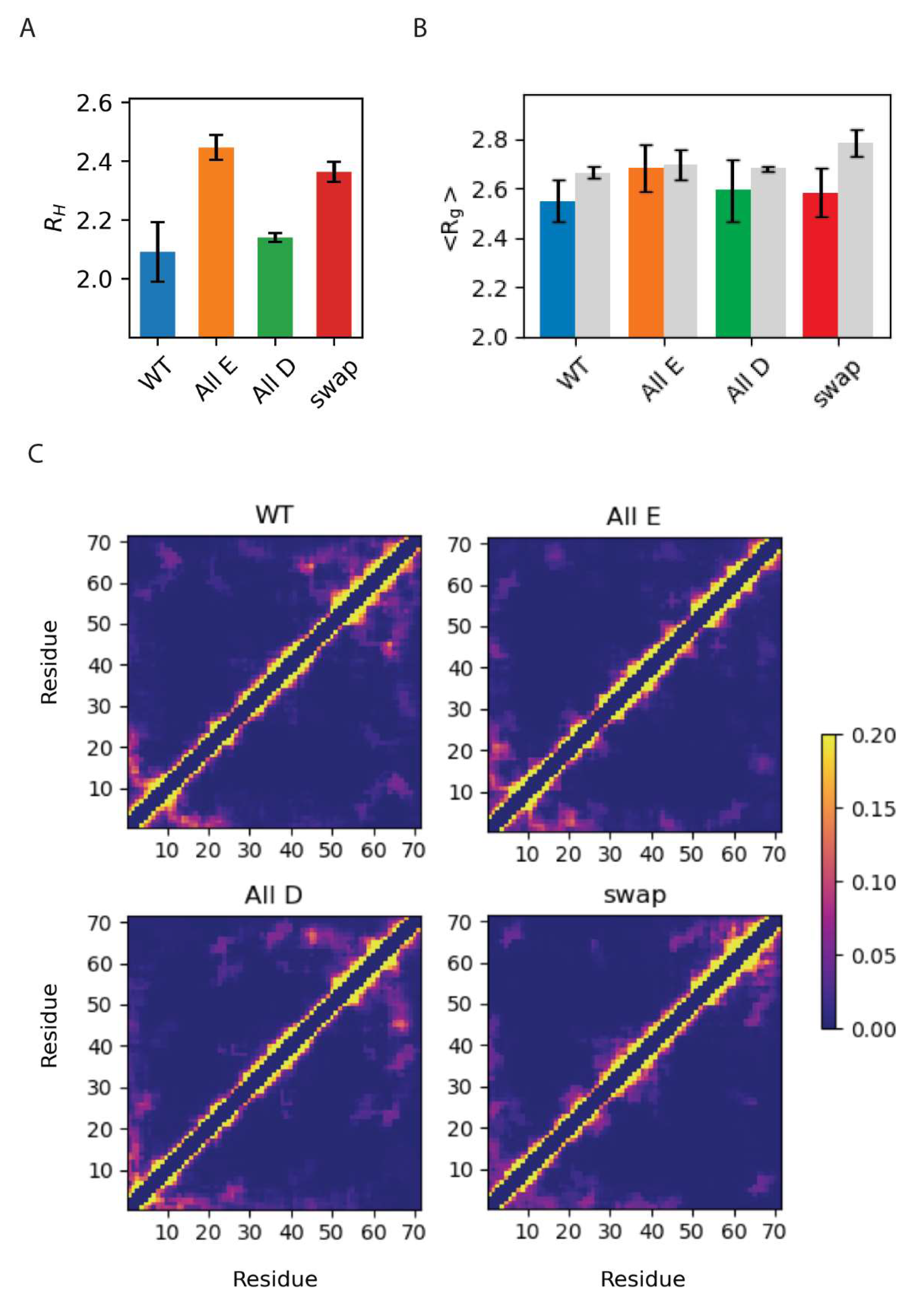
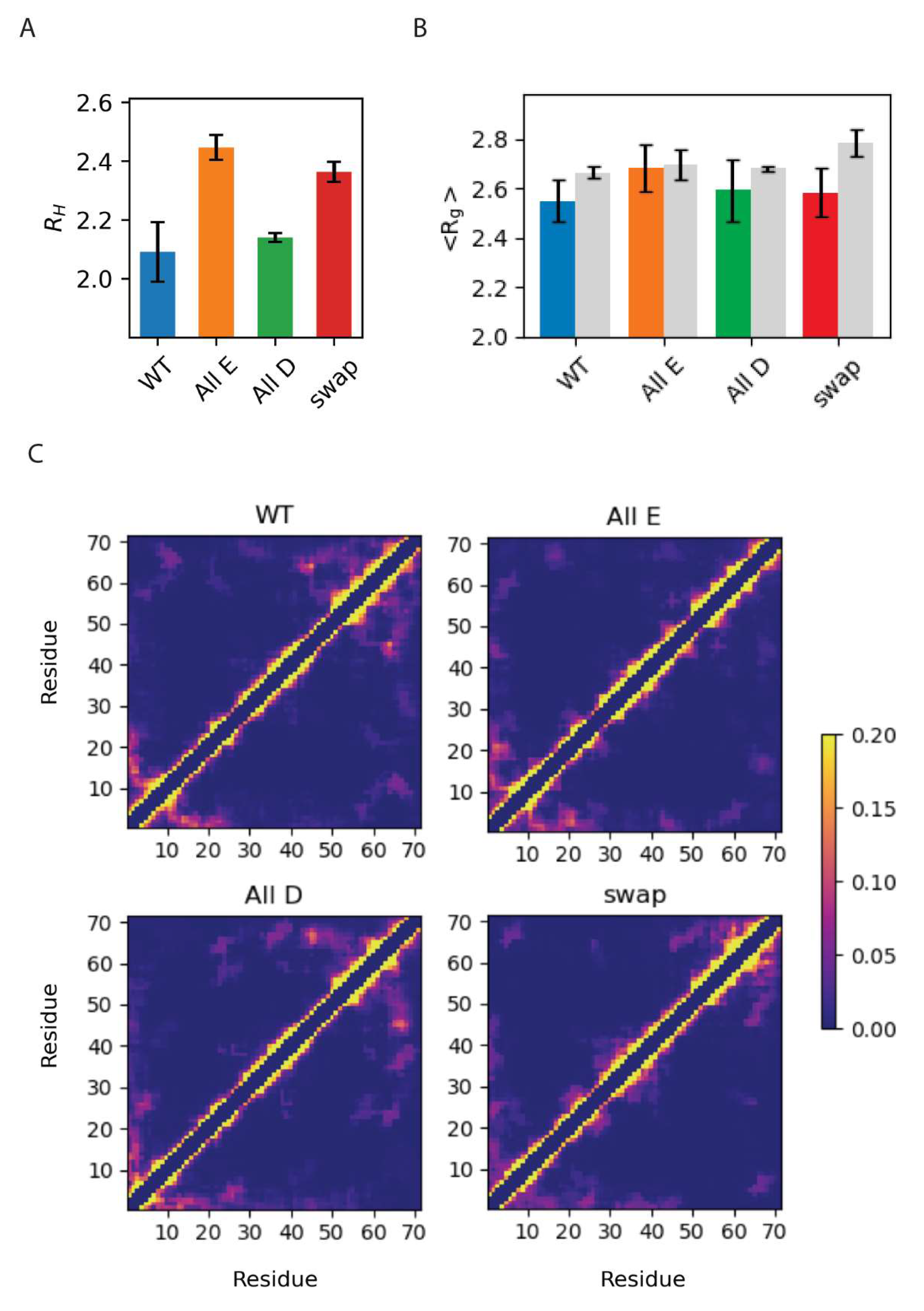
3.3. Global Compaction Does Depend on Glu vs. Asp Ratio
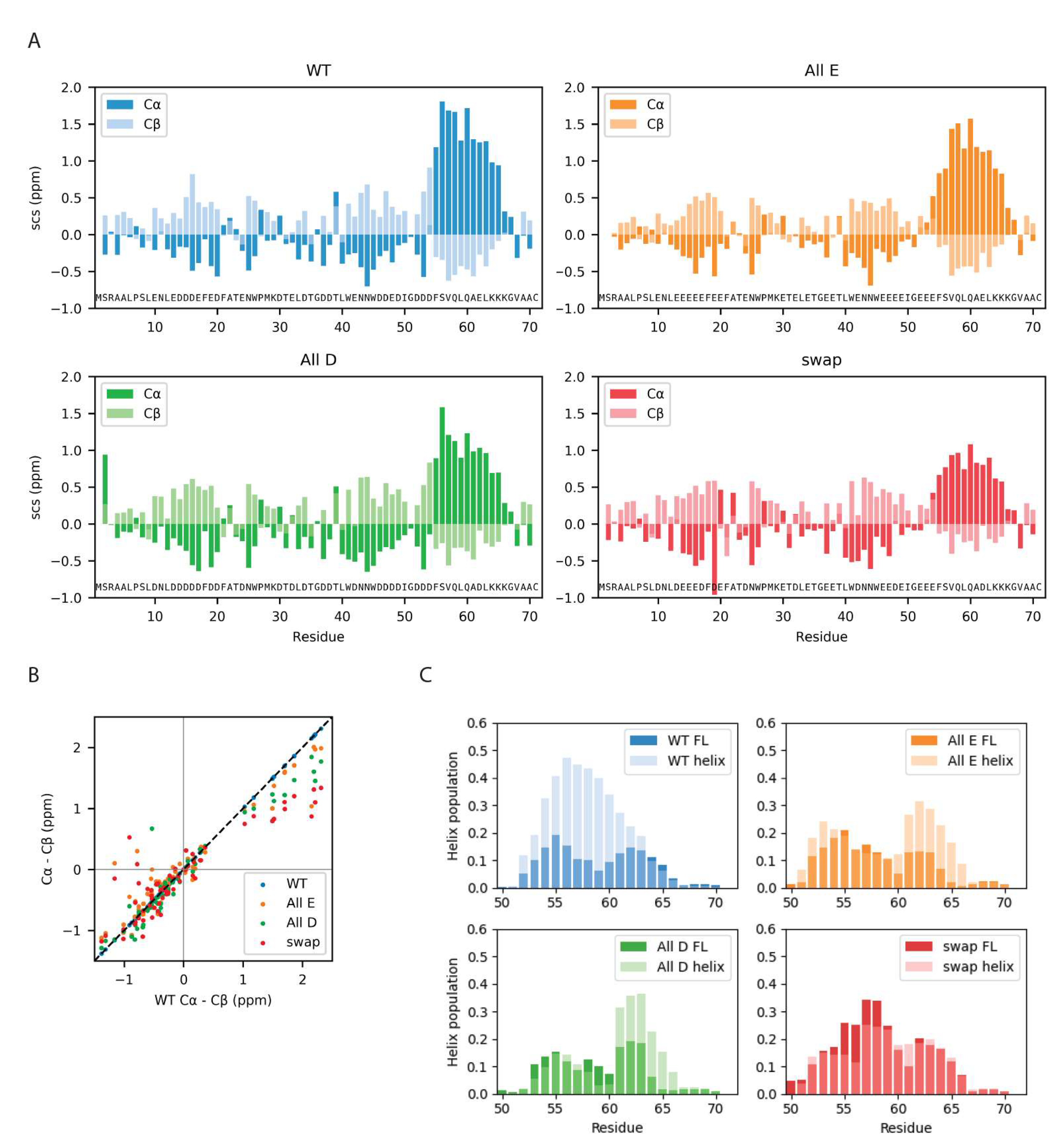
3.4. Local Structural Changes in Dss1 Depending on Glu/Asp Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.H.S.; Lopes, F.C.; John, E.B.O.; Carlini, C.R.; Ligabue-Braun, R. Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. Int. J. Mol. Sci. 2019, 20, 1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsic Disorder, Protein-Protein Interactions, and Disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph Model 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: Amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 2001, 2000, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. The alphabet of intrinsic disorder: II. Various roles of glutamic acid in ordered and intrinsically disordered proteins. IDP 2013, 1, e24684. [Google Scholar] [CrossRef] [Green Version]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Nagano, K. Logical analysis of the mechanism of protein folding. I. Predictions of helices, loops and beta-structures from primary structure. J. Mol. Biol. 1973, 75, 401–420. [Google Scholar] [CrossRef]
- Pace, C.N.; Scholtz, J.M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef]
- Daughdrill, G.W. Disorder for Dummies: Functional Mutagenesis of Transient Helical Segments in Disordered Proteins. Methods. Mol. Biol. 2020, 2141, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Iesmantavicius, V.; Dogan, J.; Jemth, P.; Teilum, K.; Kjaergaard, M. Helical propensity in an intrinsically disordered protein accelerates ligand binding. Angew. Chem. Int. Ed. Engl. 2014, 53, 1548–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaglia, F.; Meszaros, B.; Salladini, E.; Hatos, A.; Pancsa, R.; Chemes, L.B.; Pajkos, M.; Lazar, T.; Pena-Diaz, S.; Santos, J.; et al. DisProt in 2022: Improved quality and accessibility of protein intrinsic disorder annotation. Nucleic Acids Res. 2022, 50, D480–D487. [Google Scholar] [CrossRef] [PubMed]
- Hatos, A.; Hajdu-Soltesz, B.; Monzon, A.M.; Palopoli, N.; Alvarez, L.; Aykac-Fas, B.; Bassot, C.; Benitez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, D.; Tabaro, F.; Micetic, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidovic, R.; Dosztanyi, Z.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017, 45, D219–D227. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome. Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Schenstrom, S.M.; Rebula, C.A.; Tatham, M.H.; Hendus-Altenburger, R.; Jourdain, I.; Hay, R.T.; Kragelund, B.B.; Hartmann-Petersen, R. Expanded Interactome of the Intrinsically Disordered Protein Dss1. Cell. Rep. 2018, 25, 862–870. [Google Scholar] [CrossRef]
- Ellisdon, A.M.; Dimitrova, L.; Hurt, E.; Stewart, M. Structural basis for the assembly and nucleic acid binding of the TREX-2 transcription-export complex. Nat. Struct. Mol. Biol. 2012, 19, 328–336. [Google Scholar] [CrossRef]
- Sone, T.; Saeki, Y.; Toh-e, A.; Yokosawa, H. Sem1p is a novel subunit of the 26 S proteasome from Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 28807–28816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kragelund, B.B.; Schenstrom, S.M.; Rebula, C.A.; Panse, V.G.; Hartmann-Petersen, R. DSS1/Sem1, a Multifunctional and Intrinsically Disordered Protein. Trends Biochem. Sci. 2016, 41, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulos, K.; Kriegenburg, F.; Tatham, M.H.; Rosner, H.I.; Medina, B.; Larsen, I.B.; Brandstrup, R.; Hardwick, K.G.; Hay, R.T.; Kragelund, B.B.; et al. Dss1 is a 26S proteasome ubiquitin receptor. Mol. Cell. 2014, 56, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, J.E.; Prestel, A.; Martins, J.M.; Brondum, S.S.; Nielsen, O.; Garbers, A.E.; Suga, H.; Boomsma, W.; Rogers, J.M.; Hartmann-Petersen, R.; et al. A context-dependent and disordered ubiquitin-binding motif. Cell. Mol. Life Sci. 2022, 79, 484. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Matsuyama, A.; Shirai, A.; Yashiroda, Y.; Kamata, A.; Horinouchi, S.; Yoshida, M. pDUAL, a multipurpose, multicopy vector capable of chromosomal integration in fission yeast. Yeast 2004, 21, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Suga, M.; Hatakeyama, T. A rapid and simple procedure for high-efficiency lithium acetate transformation of cryopreserved Schizosaccharomyces pombe cells. Yeast 2005, 22, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.P.; Seiffert, P.; Brakti, I.; Bugge, K. Production of Intrinsically Disordered Proteins for Biophysical Studies: Tips and Tricks. Methods Mol. Biol. 2020, 2141, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, E.A.; Fernandes, C.B.; Lundsgaard, J.E.; Brakti, I.; Lindorff-Larsen, K.; Langkilde, A.E.; Skriver, K.; Kragelund, B.B. Insight into Calcium-Binding Motifs of Intrinsically Disordered Proteins. Biomolecules 2021, 11, 1173. [Google Scholar] [CrossRef] [PubMed]
- Ruidiaz, S.F.; Dreier, J.E.; Hartmann-Petersen, R.; Kragelund, B.B. The disordered PCI-binding human proteins CSNAP and DSS1 have diverged in structure and function. Protein Sci. 2021, 30, 2069–2082. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, M.; Poulsen, F.M. Sequence correction of random coil chemical shifts: Correlation between neighbor correction factors and changes in the Ramachandran distribution. J. Biomol. NMR 2011, 50, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, M.; Brander, S.; Poulsen, F.M. Random coil chemical shift for intrinsically disordered proteins: Effects of temperature and pH. J. Biomol. NMR 2011, 49, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Prestel, A.; Bugge, K.; Staby, L.; Hendus-Altenburger, R.; Kragelund, B.B. Characterization of Dynamic IDP Complexes by NMR Spectroscopy. Methods Enzymol. 2018, 611, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Teilum, K.; Kunze, M.B.; Erlendsson, S.; Kragelund, B.B. (S)Pinning down protein interactions by NMR. Protein Sci. 2017, 26, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Borgia, A.; Zheng, W.; Buholzer, K.; Borgia, M.B.; Schuler, A.; Hofmann, H.; Soranno, A.; Nettels, D.; Gast, K.; Grishaev, A.; et al. Consistent View of Polypeptide Chain Expansion in Chemical Denaturants from Multiple Experimental Methods. J. Am. Chem. Soc. 2016, 138, 11714–11726. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.H.; Chen, A.D.; Johnson, C.S. Johnson. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv. Protein Chem. 2002, 62, 311–340. [Google Scholar] [CrossRef]
- Mielke, S.P.; Krishnan, V.V. Characterization of protein secondary structure from NMR chemical shifts. Prog. Nucl. Magn. Reson. Spectrosc 2009, 54, 141–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spera, S.; Ikura, M.; Bax, A. Measurement of the exchange rates of rapidly exchanging amide protons: Application to the study of calmodulin and its complex with a myosin light chain kinase fragment. J. Biomol. NMR 1991, 1, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory. Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, E.; Abraham, M.J.; Hess, B.; van der Spoel, D. GROMACS 2019.6 Source Code. Available online: https://zenodo.org/record/3685922#.YdzzHfnYsdU(accessed on 16 August 2022).
- Consortium, P. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaendtner, J.; Bonomi, M. Efficient Sampling of High-Dimensional Free-Energy Landscapes with Parallel Bias Metadynamics. J. Chem. Theory Comput. 2015, 11, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [Green Version]
- Raiteri, P.; Laio, A.; Gervasio, F.L.; Micheletti, C.; Parrinello, M. Efficient reconstruction of complex free energy landscapes by multiple walkers metadynamics. J. Chem. Phys. B 2006, 110, 3533–3539. [Google Scholar] [CrossRef] [PubMed]
- Branduardi, D.; Bussi, G.; Parrinello, M. Metadynamics with Adaptive Gaussians. J. Chem. Theory. Comput. 2012, 8, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.; Fraaije, J. LINCS: A Linear Constraint Solver for molecular simulations. J. Chem. Phys. 1998, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comp. Phy. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Flyvbjerg, H. Error estimates on averages of correlated data. In Advances in Computer Simulation; Springer: Berlin/Heidelberg, Germany, 1998; pp. 88–103. [Google Scholar]
- Bussi, G.; Tribello, G.A. Analyzing and Biasing Simulations with PLUMED. Methods Mol. Biol. 2019, 2022, 529–578. [Google Scholar] [CrossRef] [Green Version]
- Grudinin, S.; Garkavenko, M.; Kazennov, A. Pepsi-SAXS: An adaptive method for rapid and accurate computation of small-angle X-ray scattering profiles. Acta Crystallogr. D Struct. Biol. 2017, 73, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.H.; Pedersen, M.C. Experimental noise in small-angle scattering can be assessed using the Bayesian indirect Fourier transformation. J. Appl. Crystallogr. 2021, 54, 1281–1289. [Google Scholar] [CrossRef]
- Hansen, S. BayesApp: A web site for indirect transformation of small-angle scattering data. J. Appl. Crystallogr. 2012, 45, 566–567. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernandez, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomko, R.J., Jr.; Hochstrasser, M. The intrinsically disordered Sem1 protein functions as a molecular tether during proteasome lid biogenesis. Mol. Cell. 2014, 53, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Funakoshi, M.; Li, X.; Velichutina, I.; Hochstrasser, M.; Kobayashi, H. Sem1, the yeast ortholog of a human BRCA2-binding protein, is a component of the proteasome regulatory particle that enhances proteasome stability. J. Cell. Sci. 2004, 117, 6447–6454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josse, L.; Harley, M.E.; Pires, I.M.; Hughes, D.A. Fission yeast Dss1 associates with the proteasome and is required for efficient ubiquitin-dependent proteolysis. Biochem. J. 2006, 393, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhang, W.P.; Xing, Q.; Ren, X.; Liu, M.; Tang, C. Noncovalent dimerization of ubiquitin. Angew. Chem. Int. Ed. Engl. 2012, 51, 469–472. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Ruff, K.M.; Pappu, R.V. Competing interactions give rise to two-state behavior and switch-like transitions in charge-rich intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2022, 119, e2200559119. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.; Farag, M.; Borcherds, W.M.; Peran, I.; Martin, E.W.; Pappu, R.V.; Mittag, T. Deciphering how naturally occurring sequence features impact the phase behaviours of disordered prion-like domains. Nat. Chem. 2022, 14, 196–207. [Google Scholar] [CrossRef]
- Crabtree, M.D.; Borcherds, W.; Poosapati, A.; Shammas, S.L.; Daughdrill, G.W.; Clarke, J. Conserved Helix-Flanking Prolines Modulate Intrinsically Disordered Protein:Target Affinity by Altering the Lifetime of the Bound Complex. Biochemistry 2017, 56, 2379–2384. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Baldwin, R.L. N- and C-capping preferences for all 20 amino acids in alpha-helical peptides. Protein Sci. 1995, 4, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.R.; Communie, G.; Ribeiro, E.A., Jr.; Martinez, N.; Desfosses, A.; Salmon, L.; Mollica, L.; Gabel, F.; Jamin, M.; Longhi, S.; et al. Intrinsic disorder in measles virus nucleocapsids. Proc. Natl. Acad. Sci. USA 2011, 108, 9839–9844. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roesgaard, M.A.; Lundsgaard, J.E.; Newcombe, E.A.; Jacobsen, N.L.; Pesce, F.; Tranchant, E.E.; Lindemose, S.; Prestel, A.; Hartmann-Petersen, R.; Lindorff-Larsen, K.; et al. Deciphering the Alphabet of Disorder—Glu and Asp Act Differently on Local but Not Global Properties. Biomolecules 2022, 12, 1426. https://doi.org/10.3390/biom12101426
Roesgaard MA, Lundsgaard JE, Newcombe EA, Jacobsen NL, Pesce F, Tranchant EE, Lindemose S, Prestel A, Hartmann-Petersen R, Lindorff-Larsen K, et al. Deciphering the Alphabet of Disorder—Glu and Asp Act Differently on Local but Not Global Properties. Biomolecules. 2022; 12(10):1426. https://doi.org/10.3390/biom12101426
Chicago/Turabian StyleRoesgaard, Mette Ahrensback, Jeppe E. Lundsgaard, Estella A. Newcombe, Nina L. Jacobsen, Francesco Pesce, Emil E. Tranchant, Søren Lindemose, Andreas Prestel, Rasmus Hartmann-Petersen, Kresten Lindorff-Larsen, and et al. 2022. "Deciphering the Alphabet of Disorder—Glu and Asp Act Differently on Local but Not Global Properties" Biomolecules 12, no. 10: 1426. https://doi.org/10.3390/biom12101426
APA StyleRoesgaard, M. A., Lundsgaard, J. E., Newcombe, E. A., Jacobsen, N. L., Pesce, F., Tranchant, E. E., Lindemose, S., Prestel, A., Hartmann-Petersen, R., Lindorff-Larsen, K., & Kragelund, B. B. (2022). Deciphering the Alphabet of Disorder—Glu and Asp Act Differently on Local but Not Global Properties. Biomolecules, 12(10), 1426. https://doi.org/10.3390/biom12101426