
STAT3 Inhibitors: A Novel Insight for Anticancer Therapy of Pancreatic Cancer

Abstract
:1. Introduction
2. STAT3 and Its Pathways
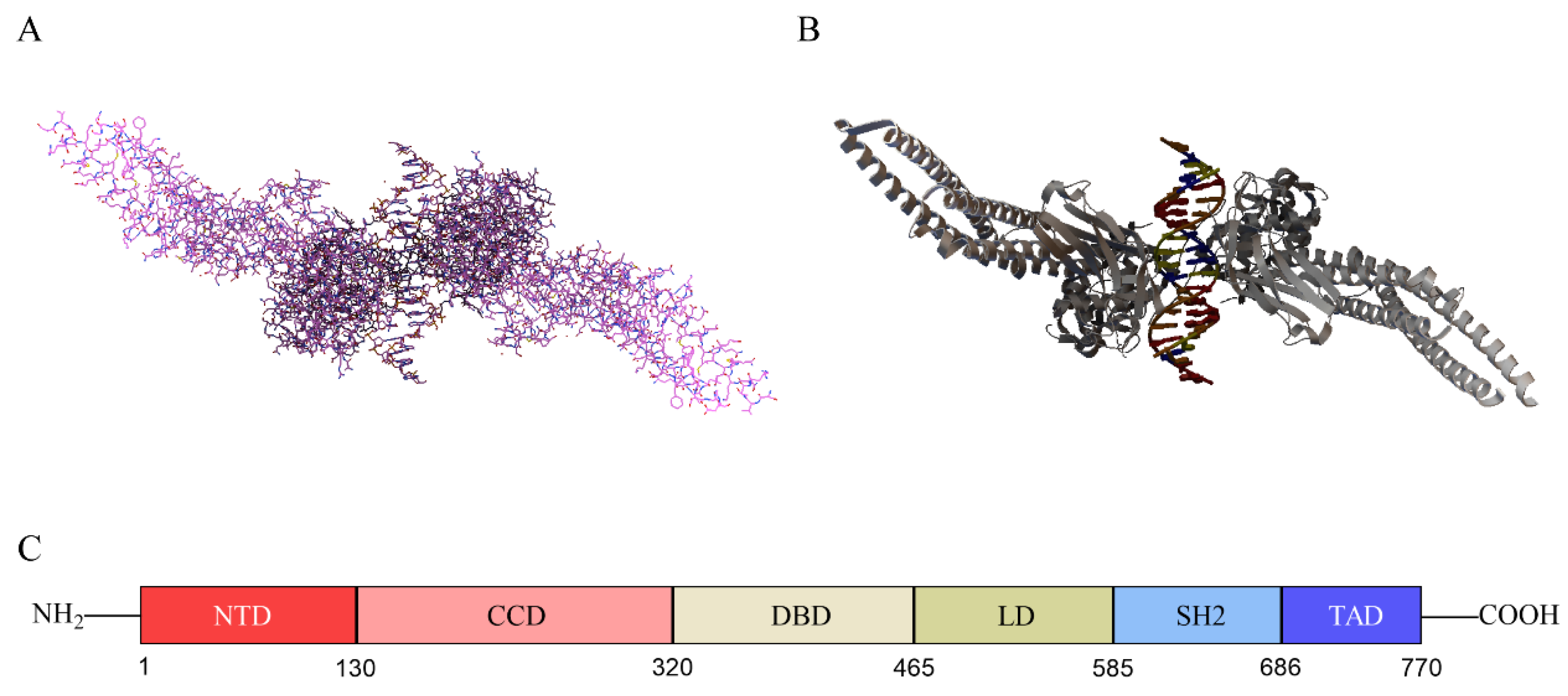
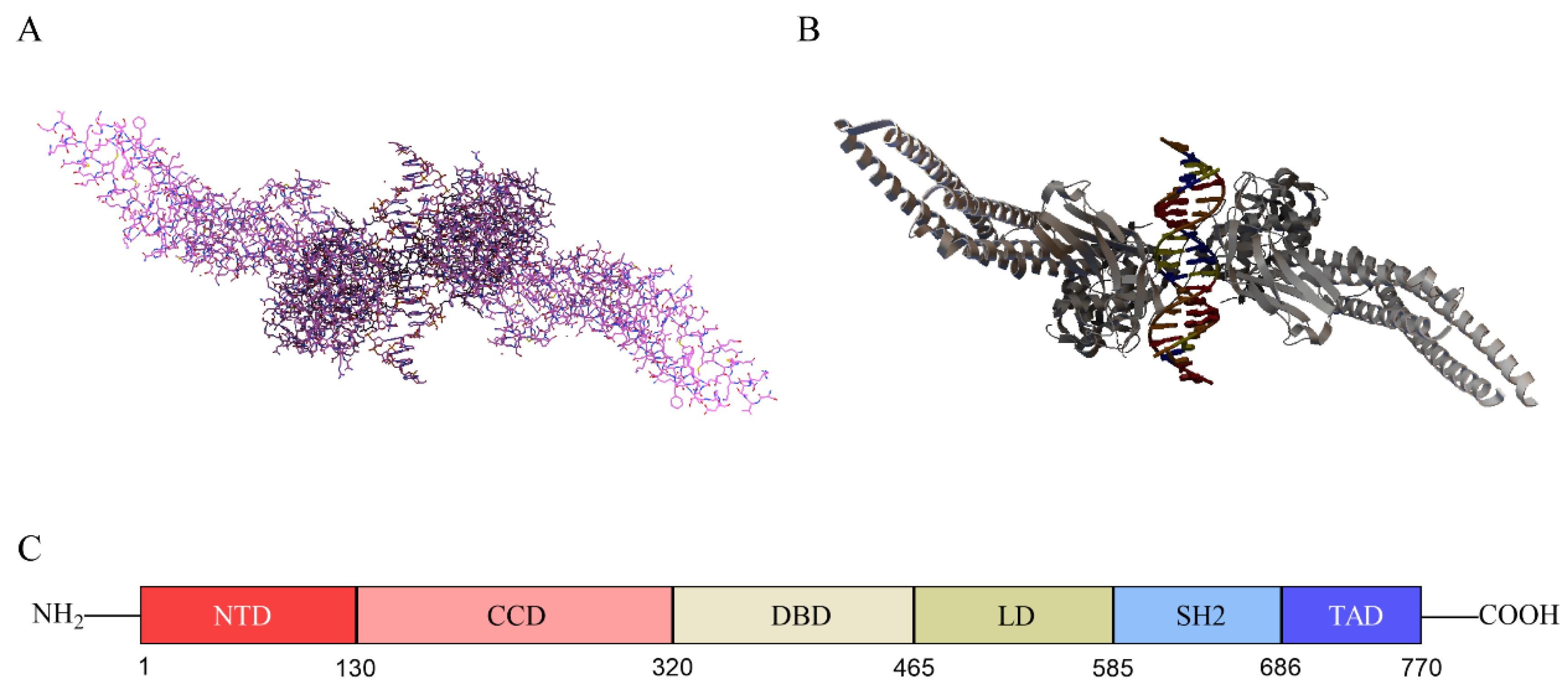
2.1. Structure of STAT3
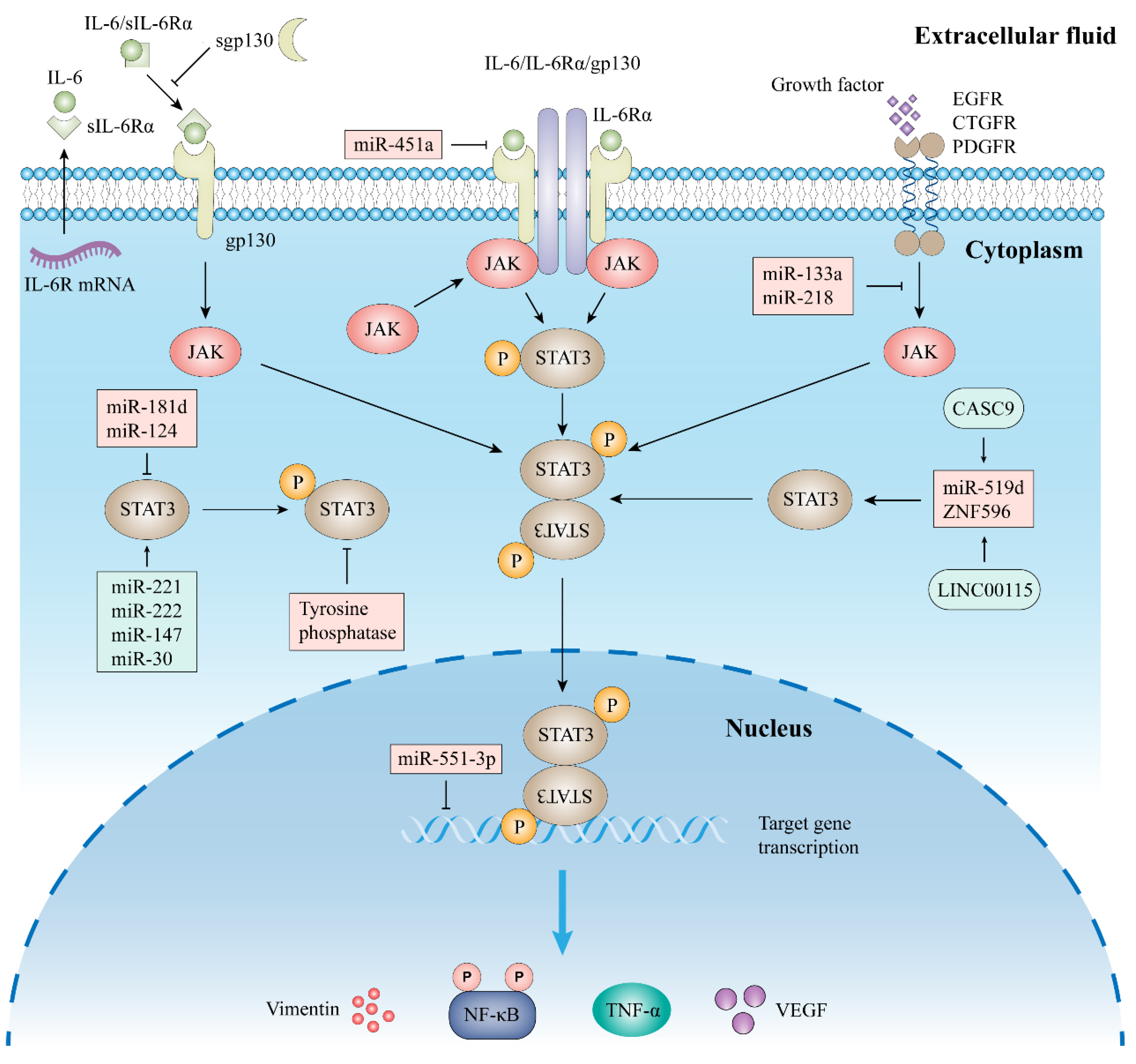
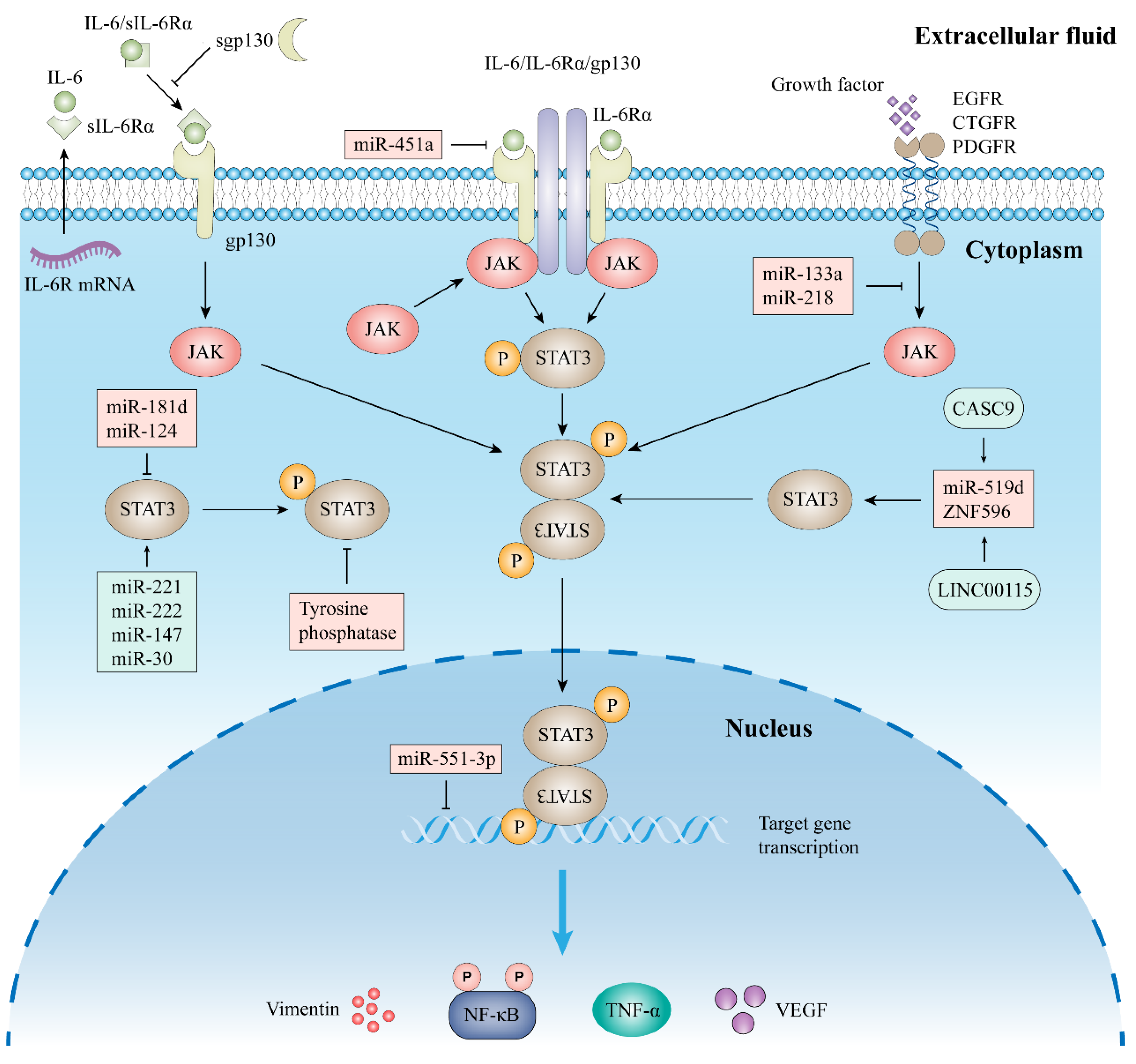
2.2. Signal Pathway of STAT3
2.2.1. Classic Signaling
2.2.2. Trans-Signaling
2.2.3. Other Signal Pathway
2.3. STAT3 in Immune Cells, Stromal Cells, and Epithelial Cells
3. STAT3 in Pancreatic Cancer
3.1. Tumor Progression and STAT3
3.2. Metastasis and STAT3
3.3. Immunosuppression and STAT3
3.4. STAT3 and Drug Resistance
4. STAT3 Inhibitors and Cancer
4.1. Direct Targeting of STAT3 Inhibitors
4.2. Indirect Targeting of STAT3 Inhibitors
4.2.1. Inhibitors of IL-6 and IL-6R
4.2.2. Inhibitors of JAKs
4.2.3. Other Indirect Inhibitors

{kind=link}
{kind=link}
| Targets | Inhibitors | Effects | Ref. |
|---|---|---|---|
| IL-6/IL-6R | Tocilizumab | Inhibition of stromal cell function and inhibition of EMT | [97,98,99] |
| Siltuximab | Inhibition of cancer cell growth | [104] | |
| Bazedoxifene | Inhibition of cancer cell growth | [106,107,108] | |
| JAK | Baricitinib | Inhibition of chemokines | [112] |
| Ruxolitinib | Enhancing anti-tumor immunity and inhibiting inflammation | [114,117] | |
| EGFR | Erlotinib | Inhibition of cancer cell growth | [126] |
| STAT3 | C188-9 | Reversing the hypermethylation status of tumor suppressor genes | [79] |
| Shikonin | Promoting apoptosis of cancer cells | [83] | |
| Triptomycin A | Inhibiting the proliferation, migration and invasion of cancer cells | [84] |
4.3. Combined STAT3 Inhibitor and Immune Checkpoint Inhibitor (ICI)
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- El-Tanani, M.; Al Khatib, A.O.; Aladwan, S.M.; Abuelhana, A.; McCarron, P.A.; Tambuwala, M.M. Importance of STAT3 signalling in cancer, metastasis and therapeutic inter-ventions. Cell. Signal. 2022, 92, 110275. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenviron-ment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- GBD 2017 Pancreatic Cancer Collaborators. The global, regional, and national burden of pancreatic cancer and its attributa-ble risk factors in 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Caldenhoven, E.; van Dijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.; Lammers, J.W.J.; Koenderman, L.; de Groot, R.P. STAT3β, a splice variant of transcription factor STAT3, is a dominant nega-tive regulator of transcription. J. Biol. Chem. 1996, 271, 13221–13227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, T.S.; Sanders, L.K.; Nathans, D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc. Natl. Acad. Sci. USA 1995, 92, 9097–9101. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kee, W.H.; Seow, K.T.; Fung, W.; Cao, X. The Coiled-Coil Domain of Stat3 Is Essential for Its SH2 Domain-Mediated Receptor Binding and Subsequent Activation Induced by Epidermal Growth Factor and Interleukin-6. Mol. Cell. Biol. 2000, 20, 7132–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.; Wen, Z.; Haspel, R.L.; Zhang, J.J.; Darnell, J.E. The Linker Domain of Stat1 Is Required for Gamma Interferon-Driven Transcription. Mol. Cell. Biol. 1999, 19, 5106–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgrignani, J.; Garofalo, M.; Matkovic, M.; Merulla, J.; Catapano, C.V.; Cavalli, A. Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development. Int. J. Mol. Sci. 2018, 19, 1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, C.M. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 2000, 25, 496–502. [Google Scholar] [CrossRef]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.-I.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lym-phocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Blocking only the bad side of IL-6 in inflammation and cancer. Cytokine 2021, 148, 155690. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cai, J.; Chen, K.; Zhu, M.; Li, Z.; Liu, H.; Liu, T.; Mao, J.; Ding, Q.; Zhu, Y.Z. STAT3-NAV2 axis as a new therapeutic target for rheumatoid arthritis via activating SSH1L/Cofilin-1 signaling pathway. Signal Transduct. Target. Ther. 2022, 7, 209. [Google Scholar] [CrossRef]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Casteele, N.V. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef]
- Shawky, A.M.; Almalki, F.A.; Abdalla, A.N.; Abdelazeem, A.H.; Gouda, A.M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 2022, 14, 1001. [Google Scholar] [CrossRef]
- Witthuhn, B.A.; Silvennoinen, O.; Miura, O.; Lai, K.S.; Cwik, C.; Liu, E.T.; Ihle, J.N. Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature 1994, 370, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of neoplastic and inflammatory disorders. Pharmacol. Res. 2022, 183, 106362. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, C.; Deng, J.; Zhou, J. JAK inhibition as a new treatment strategy for patients with COVID-19. Biochem. Pharmacol. 2022, 181, 467–475. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 al-pha-receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.-W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front. Oncol. 2022, 12, 866014. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, M.; Krawczyńska, A.; Antushevich, H.; Herman, A.P. Post-Receptor Inhibitors of the GHR-JAK2-STAT Pathway in the Growth Hormone Signal Transduction. Int. J. Mol. Sci. 2018, 19, 1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mülberg, J.; Schooltink, H.; Stoyan, T.; Günther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Riethmueller, S.; Ehlers, J.C.; Lokau, J.; Düsterhöft, S.; Knittler, K.; Dombrowsky, G.; Grötzinger, J.; Rabe, B.; Rose-John, S.; Garbers, C. Cleavage Site Localization Differentially Controls Interleukin-6 Receptor Proteol-ysis by ADAM10 and ADAM17. Sci. Rep. 2016, 6, 25550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostock, T.; Müllberg, J.; Özbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001, 268, 160–167. [Google Scholar] [CrossRef]
- Wei, J.; Wang, F.; Kong, L.-Y.; Xu, S.; Doucette, T.; Ferguson, S.D.; Yang, Y.; McEnery, K.; Jethwa, K.; Gjyshi, O.; et al. miR-124 Inhibits STAT3 Signaling to Enhance T Cell–Mediated Immune Clearance of Glioma. Cancer Res. 2013, 73, 3913–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.W.; Lee, P.M.; Bamodu, O.A.; Su, Y.K.; Fong, I.H.; Yeh, C.T.; Chien, M.-H.; Kan, I.-H.; Lin, C.-M. Enhanced Hsa-miR-181d/p-STAT3 and Hsa-miR-181d/p-STAT5A Ratios Mediate the Anticancer Effect of Garcinol in STAT3/5A-Addicted Glioblastoma. Cancers 2019, 11, 1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Chen, F.Z.; Jia, Q.B.; Hu, D.F. Upregulation of microRNA-133a and downregulation of connective tissue growth factor suppress cell proliferation, migration, and invasion in human glioma through the JAK/STAT signaling pathway. IUBMB Life 2019, 71, 1857–1875. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Xu, Z.; Jin, X.; He, Y.; Zhang, J.; Jiang, T.; Chen, J. miR-451a suppression of IL-6R can inhibit proliferation and increase apoptosis through the JAK2/STAT3 pathway in multiple myeloma. Oncol. Lett. 2020, 20, 339. [Google Scholar] [CrossRef] [PubMed]
- Mathew, L.K.; Huangyang, P.; Mucaj, V.; Lee, S.S.; Skuli, N.; Eisinger-Mathason, T.K.; Biju, K.; Li, B.; Venneti, S.; Lal, P. Feedback circuitry between miR-218 repression and RTK activation in glioblastoma. Sci. Signal. 2015, 8, ra42. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.H.; Liu, Y.; Xiao, L.M.; Chen, L.K.; Zheng, S.Y.; Zeng, E.M.; Li, D.-H.; Li, Y.-P. Silencing microRNA-221/222 cluster suppresses glioblastoma angiogenesis by suppressor of cytokine signaling-3-dependent JAK/STAT pathway. J. Cell. Physiol. 2019, 234, 22272–22284. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Ge, X.; Shi, Z.; Yu, C.; Lu, C.; Wei, Y.; Zeng, A.; Wang, X.; Yan, W.; Zhang, J.; et al. Extracellular vesicles derived from hypoxic glioma stem-like cells confer temozolomide resistance on glioblastoma by delivering miR-30b-3p. Theranostics 2021, 11, 1763–1779. [Google Scholar] [CrossRef]
- Sajjadi-Dokht, M.; Mohamad, T.A.M.; Rahman, H.S.; Maashi, M.S.; Danshina, S.; Shomali, N.; Solali, S.; Marofi, F.; Zeinalzadeh, E.; Akbari, M.; et al. MicroRNAs and JAK/STAT3 signaling: A new promising therapeutic axis in blood cancers. Genes Dis. 2021, 9, 849–867. [Google Scholar] [CrossRef]
- Tang, J.; Yu, B.; Li, Y.; Zhang, W.; Alvarez, A.A.; Hu, B.; Cheng, S.Y.; Feng, H. TGF-β-activated lncRNA LINC00115 is a critical regulator of glioma stem-like cell tumorigenicity. EMBO Rep. 2019, 20, e48170. [Google Scholar] [CrossRef]
- Yuan, B.; Sun, R.; Du, Y.; Jia, Z.; Yao, W.; Yang, J. STAT3-Induced Upregulation of lncRNA CASC9 Promotes the Progression of Bladder Cancer by Interacting with EZH2 and Affecting the Expression of PTEN. OncoTargets Ther. 2020, 13, 9147–9157. [Google Scholar] [CrossRef]
- Giordano, M.; Decio, A.; Battistini, C.; Baronio, M.; Bianchi, F.; Villa, A.; Bertalot, G.; Freddi, S.; Lupia, M.; Jodice, M.G.; et al. L1CAM promotes ovarian cancer stemness and tumor initiation via FGFR1/SRC/STAT3 signaling. J. Exp. Clin. Cancer Res. 2021, 40, 319. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ke, S.; Wang, C.; Xu, Y.; Li, Z.; Song, K.; Bai, M.; Zhou, M.; Yu, H.; Yin, B.; et al. NNMT promotes the progression of intrahepatic cholangiocarcinoma by regulating aerobic glycolysis via the EGFR-STAT3 axis. Oncogenesis 2022, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ji, Y.R.; Lee, Y.M. Crosstalk between angiogenesis and immune regulation in the tumor microenvironment. Arch. Pharmacal Res. 2022, 45, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Mirlekar, B.; Michaud, D.; Lee, S.J.; Kren, N.P.; Harris, C.; Greene, K.; Goldman, E.C.; Gupta, G.P.; Fields, R.C.; Hawkins, W.G.; et al. B cell-Derived IL35 Drives STAT3-Dependent CD8+ T-cell Exclusion in Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 292–308. [Google Scholar] [CrossRef] [Green Version]
- Kujawski, M.; Zhang, C.; Herrmann, A.; Reckamp, K.; Scuto, A.; Jensen, M.; Deng, J.; Forman, S.; Figlin, R.; Yu, H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010, 70, 9599–9610. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Zhang, D.; Gong, B.; Wang, P.; Liu, F. CD163 as a novel target gene of STAT3 is a potential therapeutic target for gastric cancer. Oncotarget 2017, 8, 87244–87262. [Google Scholar] [CrossRef] [Green Version]
- Solís-Martínez, R.; Cancino-Marentes, M.; Hernández-Flores, G.; Ortiz-Lazareno, P.; Mandujano-Álvarez, G.; Cruz-Gálvez, C.; Sierra-Díaz, E.; Rodríguez-Padilla, C.; Jave-Suárez, L.F.; Aguilar-Lemarroy, A.; et al. Regulation of immunophenotype modulation of mono-cytes-macrophages from M1 into M2 by prostate cancer cell-culture supernatant via transcription factor STAT3. Immunol. Lett. 2018, 196, 140–148. [Google Scholar] [CrossRef]
- Damiani, V.; Cufaro, M.C.; Fucito, M.; Dufrusine, B.; Rossi, C.; Del Boccio, P.; Federici, L.; Turco, M.C.; Sallese, M.; Pieragostino, D.; et al. Proteomics Approach Highlights Early Changes in Human Fibroblasts-Pancreatic Ductal Adenocarcinoma Cells Crosstalk. Cells 2022, 11, 1160. [Google Scholar] [CrossRef]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef]
- Dawson, R.E.; Deswaerte, V.; West, A.C.; Tang, K.; West, A.J.; Balic, J.J.; Gearing, L.J.; Saad, M.I.; Yu, L.; Wu, Y.; et al. STAT3-mediated upregulation of the AIM2 DNA sensor links innate immunity with cell migration to promote epithelial tumourigenesis. Gut 2022, 71, 1515–1531. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Block, K.M.; Hanke, N.T.; Maine, E.A.; Baker, A.F. IL-6 Stimulates STAT3 and Pim-1 Kinase in Pancreatic Cancer Cell Lines. Pancreas 2012, 41, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Y.; Wang, Y.; Li, W.; Wang, X.; Liu, X.; Chen, Y.; Ouyang, C.; Wang, J. Quantification of STAT3 and VEGF expression for molecular diagnosis of lymph node metastasis in breast cancer. Medicine 2017, 96, e8488. [Google Scholar] [CrossRef]
- Huang, C.; Huang, R.; Chang, W.; Jiang, T.; Huang, K.; Cao, J.; Sun, X.; Qiu, Z. The Expression and Clinical Significance of pSTAT3, VEGF and VEGF-C in Pancreatic Adenocarcinoma. Neoplasma 2012, 59, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Liu, C.; Guan, X.; Wang, B.; Yao, Y.; Su, D.; Ma, Y.; Fang, L.; Zhang, Y. Ubiquitin specific peptidase 5 enhances STAT3 signaling and promotes migration and invasion in Pancreatic Cancer. J. Cancer 2020, 11, 6802–6811. [Google Scholar] [CrossRef]
- He, Z.; Wang, J.; Zhu, C.; Xu, J.; Chen, P.; Jiang, X.; Chen, Y.; Jiang, J.; Sun, C. Exosome-derived FGD5-AS1 promotes tumor-associated macrophage M2 polarization-mediated pancreatic cancer cell proliferation and metastasis. Cancer Lett. 2022, 548, 215751. [Google Scholar] [CrossRef]
- Ganesh, K.; Massagué, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef]
- Shukla, S.K.; Markov, S.D.; Attri, K.S.; Vernucci, E.; King, R.J.; Dasgupta, A.; Grandgenett, P.M.; Hollingsworth, M.A.; Singh, P.K.; Yu, F.; et al. Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia. Cancer Lett. 2020, 484, 29–39. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, Q.; Chen, W.; Wu, T.; Liu, S.; Li, X.; Luo, B.; Zhang, T.; Yan, G.; Lu, H.; et al. MicroRNA-301a promotes pancreatic cancer invasion and metastasis through the JAK/STAT3 signaling pathway by targeting SOCS5. Carcinogenesis 2020, 41, 502–514. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Hu, S.; Liu, K.; Sun, G.; Zhang, Y. The Role of MicroRNA in the Regulation of Tumor Epithelial–Mesenchymal Transition. Cells 2022, 11, 1981. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial–mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, N.; Hirano, K.; Shichi, Y.; Gomi, F.; Yoshimura, H.; Matsushita, A.; Toyoda, M.; Ishiwata, T. Gp130-Mediated STAT3 Activation Contributes to the Aggressiveness of Pancreatic Cancer through H19 Long Non-Coding RNA Expression. Cancers 2022, 14, 2055. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ren, T.; Huang, C.; Li, Y.; Yang, P.; Che, G.; Luo, L.; Chen, Y.; Peng, S.; Lin, Y.; et al. S100A16 induces epithelial-mesenchymal transition in human PDAC cells and is a new therapeutic target for pancreatic cancer treatment that synergizes with gemcitabine. Biochem. Pharmacol. 2021, 189, 114396. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, Z.; Han, Q.; Zhao, H.; Pan, Z.; Lei, Z.; Zhang, J. Targeted inhibition of STAT3 induces immunogenic cell death of hepatocellular carcinoma cells via glycolysis. Mol. Oncol. 2022, 16, 2861–2880. [Google Scholar] [CrossRef]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, K.; Xiong, H.; Huang, Y.; Chen, X.; Zhou, Y.; Qin, W.; Su, J.; Chen, R.; Qiu, H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Provides a New Combination Therapy in Pancreatic Cancer. Front. Immunol. 2021, 12, 762989. [Google Scholar] [CrossRef]
- Koltai, T.; Reshkin, S.J.; Carvalho, T.; Di Molfetta, D.; Greco, M.R.; Alfarouk, K.O.; Cardone, R.A. Resistance to Gemcitabine in Pancreatic Ductal Adenocarcinoma: A Physiopathologic and Pharmacologic Review. Cancers 2022, 14, 2486. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, D.; Wu, X.; Lin, X.; Ye, L.; Lin, C.; Wu, S.; Zhu, J.; Peng, X.; Song, L. miR-1266 Contributes to Pancreatic Cancer Progression and Chemoresistance by the STAT3 and NF-κB Signaling Pathways. Mol. Ther. Nucleic Acids 2018, 11, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L.; et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Iizuka, A.; Maeda, C.; Tanaka, E.; Kondou, R.; Miyata, H.; Sugino, T.; Kawata, T.; Deguchi, S.; Mitsuya, K.; et al. Impact of combination therapy with anti-PD-1 blockade and a STAT3 inhibitor on the tumor-infiltrating lymphocyte status. Immunol. Lett. 2019, 216, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.-F.; Zuo, F.-F.; Yin, B.-C.; Ye, B.-C. Delivery of siRNA based on engineered exosomes for glioblastoma therapy by targeting STAT3. Biomater. Sci. 2022, 10, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Shakeran, Z.; Varshosaz, J.; Keyhanfar, M.; Mohammad-Beigi, H.; Rahimi, K.; Sutherland, D.S. Co-delivery of STAT3 siRNA and methotrexate in breast cancer cells. Artif. Cells Nanomed. Biotechnol. 2022, 50, 29–39. [Google Scholar] [CrossRef]
- Luo, K.; Gao, Y.; Yin, S.; Yao, Y.; Yu, H.; Wang, G.; Li, J. Co-delivery of paclitaxel and STAT3 siRNA by a multifunctional nanocomplex for targeted treatment of metastatic breast cancer. Acta Biomater. 2021, 134, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; Nott, L.; Yoshino, T.; Gill, S.; Shapiro, J.; Ohtsu, A.; Zalcberg, J.; Vickers, M.M.; Wei, A.C.; Gao, Y.; et al. Napabucasin versus placebo in refractory advanced colorectal cancer: A randomised phase 3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 263–270. [Google Scholar] [CrossRef]
- Chen, H.; Bian, A.; Yang, L.-F.; Yin, X.; Wang, J.; Ti, C.; Miao, Y.; Peng, S.; Xu, S.; Liu, M.; et al. Targeting STAT3 by a small molecule suppresses pancreatic cancer progression. Oncogene 2021, 40, 1440–1457. [Google Scholar] [CrossRef]
- Redell, M.S.; Ruiz, M.J.; Alonzo, T.A.; Gerbing, R.B.; Tweardy, D.J. Stat3 signaling in acute myeloid leukemia: Ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood 2011, 117, 5701–5709. [Google Scholar] [CrossRef]
- Kong, R.; Sun, G.; Li, X.; Wu, L.; Li, L.; Li, Y.; Wang, F.; Xuan, P.; Yang, S.; Sun, B.; et al. Small Molecule Inhibitor C188-9 Synergistically Enhances the Demethylated Activity of Low-Dose 5-Aza-2′-Deoxycytidine Against Pancreatic Cancer. Front. Oncol. 2020, 10, 612. [Google Scholar] [CrossRef]
- Bai, E.; Yang, L.; Xiang, Y.; Hu, W.; Li, C.; Lin, J.; Dai, X.; Liang, G.; Jin, R.; Zhao, C. L61H46 shows potent efficacy against human pancreatic cancer through inhibiting STAT3 pathway. Cancer Manag. Res. 2018, 10, 565–581. [Google Scholar] [CrossRef]
- Jiang, W.; Li, X.; Dong, S.; Zhou, W. Betulinic acid in the treatment of tumour diseases: Application and research progress. Biomed. Pharmacother. 2021, 142, 111990. [Google Scholar] [CrossRef]
- Shi, S.; Cao, H. Shikonin promotes autophagy in BXPC-3 human pancreatic cancer cells through the PI3K/Akt signaling pathway. Oncol. Lett. 2014, 8, 1087–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Z.; Liang, M.; Shang, L.; Lai, M.; Deng, X.; Su, X. Shikonin-mediated PD-L1 degradation suppresses immune evasion in pancreatic cancer by inhibiting NF-κB/STAT3 and NF-κB/CSN5 signaling pathways. Pancreatology 2021, 21, 630–641. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Tang, J.; Liu, Y.; Chen, Z.; Liu, Y.; Chen, H.; Li, D.; Yi, Z.; Gao, J. The natural product trienomycin A is a STAT3 pathway inhibitor that exhibits potent in vitro and in vivo efficacy against pancreatic cancer. Br. J. Pharmacol. 2021, 178, 2496–2515. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, S.; Kang, Y.; Xiang, Y.; Xu, L.; Li, J.; Dai, X.; Liang, G.; Huang, X.; Zhao, C. Rhein sensitizes human pancreatic cancer cells to EGFR inhibitors by inhibiting STAT3 path-way. J. Exp. Clin. Cancer Res. 2019, 38, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Yang, L.; Kang, Y.; Chen, M.; Lin, S.; Xiang, Y.; Li, C.; Dai, X.; Huang, X.; Liang, G.; et al. Alantolactone sensitizes human pancreatic cancer cells to EGFR inhibitors through the inhibition of STAT3 signaling. Mol. Carcinog. 2019, 58, 565–576. [Google Scholar] [CrossRef]
- Hu, B.; Cai, H.; Yang, S.; Tu, J.; Huang, X.; Chen, G. Berbamine Enhances the Efficacy of Gefitinib by Suppressing STAT3 Signaling in Pancreatic Cancer Cells. OncoTargets Ther. 2019, 12, 11437–11451. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [Green Version]
- Odate, S.; Veschi, V.; Yan, S.; Lam, N.; Woessner, R.; Thiele, C.J. Inhibition of STAT3 with the Generation 2.5 Antisense Oligonucleotide, AZD9150, Decreases Neuroblastoma Tumorigenicity and Increases Chemosensitivity. Clin. Cancer Res. 2017, 23, 1771–1784. [Google Scholar] [CrossRef] [Green Version]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pre-treated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhou, J.; Wu, Y.; Zhao, L. The emerging role of Interleukin 37 in bone homeostasis and inflammatory bone diseases. Int. Immunopharmacol. 2021, 98, 107803. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.E.; Fisher, J.L.; Flick, H.L.; Wang, C.; Sun, L.; Ernstoff, M.S.; Alvarez, J.C.; Losey, H.C. ALKS 4230: A novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000673. [Google Scholar] [CrossRef]
- Abidi, E.; El Nekidy, W.S.; Alefishat, E.; Rahman, N.; Petroianu, G.A.; El-Lababidi, R.; Mallat, J. Tocilizumab and COVID-19: Timing of Administration and Efficacy. Front. Pharmacol. 2022, 13, 825749. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Terao, K.; Mima, T.; Nakahara, H.; Takagi, N.; Kakehi, T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008, 112, 3959–3964. [Google Scholar] [CrossRef]
- Cabezas, L.; Jouve, T.; Malvezzi, P.; Janbon, B.; Giovannini, D.; Rostaing, L.; Noble, J. Tocilizumab and Active Antibody-Mediated Rejection in Kidney Transplantation: A Literature Review. Front. Immunol. 2022, 14, 1689. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Santegoets SJ, A.M.; Reyners AK, L.; Goedemans, R.; Wouters MC, A.; Kenter, G.G.; van Erkel, A.R.; van Poelgeest, M.I.E.; Nijman, H.W.; van der Hoeven, J.J.M.; et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an an-ti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2015, 26, 2141–2149. [Google Scholar] [CrossRef]
- Antoon, R.; Wang, X.-H.; Saleh, A.H.; Warrington, J.; Hedley, D.W.; Keating, A. Pancreatic cancer growth promoted by bone marrow mesenchymal stromal cell–derived IL-6 is reversed predominantly by IL-6 blockade. Cytotherapy 2022, 24, 699–710. [Google Scholar] [CrossRef]
- Otto, L.; Rahn, S.; Daunke, T.; Walter, F.; Winter, E.; Möller, J.; Rose-John, S.; Wesch, D.; Schäfer, H.; Sebens, S. Initiation of Pancreatic Cancer: The Interplay of Hyperglycemia and Macrophages Promotes the Acquisition of Malignancy-Associated Properties in Pancreatic Ductal Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 5086. [Google Scholar] [CrossRef]
- Magni, L.; Bouazzi, R.; Olmedilla, H.H.; Petersen, P.; Tozzi, M.; Novak, I. The P2X7 Receptor Stimulates IL-6 Release from Pancreatic Stellate Cells and Tocilizumab Prevents Activation of STAT3 in Pancreatic Cancer Cells. Cells 2021, 10, 1928. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Sande, B.; Brodkin, S.; van Rhee, F. Idiopathic multicentric Castleman disease treated with siltuximab for 15 years: A case report. Ther. Adv. Hematol. 2022, 13, 20406207221082552. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Smith, M.A.; Doshi, P.; Sasser, K.; Fulp, W.; Altiok, S.; Haura, E.B. Antitumor Efficacy of the Anti-Interleukin-6 (IL-6) Antibody Siltuximab in Mouse Xenograft Models of Lung Cancer. J. Thorac. Oncol. 2014, 9, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen ML, T.; Bui, K.C.; Scholta, T.; Xing, J.; Bhuria, V.; Sipos, B.; Sipos, B.; Wilkens, L.; Linh, T.N.; Velavan, T.P.; et al. Targeting interleukin 6 signaling by monoclonal antibody siltuximab on cholangiocarcinoma. J. Gastroenterol. Hepatol. 2021, 36, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-Y.; Lin, H.-C.; Tsai, F.-C.; Ko, J.-Y.; Kok, S.-H.; Cheng, S.-J.; Lee, J.-J.; Chia, J.-S. Effects of Interleukin-6 on STAT3-regulated signaling in oral cancer and as a prognosticator of patient survival. Oral Oncol. 2022, 124, 105665. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; Van Laethem, J.L.; Ottensmeier, C.; Lopez-Martin, J.A.; Clive, S.; Joly, F.; et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavropoulou, M.P.; Makras, P.; Anastasilakis, A.D. Bazedoxifene for the treatment of osteoporosis. Expert Opin. Pharmacother. 2019, 20, 1201–1210. [Google Scholar] [CrossRef]
- Chen, X.; Tian, J.; Su, G.H.; Lin, J. Blocking IL-6/GP130 Signaling Inhibits Cell Viability/Proliferation, Glycolysis, and Colony Forming Activity in Human Pancreatic Cancer Cells. Curr. Cancer Drug Targets 2019, 19, 417–427. [Google Scholar] [CrossRef]
- Fu, S.; Lin, J. Blocking Interleukin-6 and Interleukin-8 Signaling Inhibits Cell Viability, Colony-forming Activity, and Cell Migration in Human Triple-negative Breast Cancer and Pancreatic Cancer Cells. Anticancer Res. 2018, 38, 6271–6279. [Google Scholar] [CrossRef]
- Wu, X.; Cao, Y.; Xiao, H.; Li, C.; Lin, J. Bazedoxifene as a Novel GP130 Inhibitor for Pancreatic Cancer Therapy. Mol. Cancer Ther. 2016, 15, 2609–2619. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, S.J.; Bingham, C.O.; van Vollenhoven, R.; Murray, C.; Gruben, D.; Gold, D.A.; Cella, D. The impact of tofacitinib on fatigue, sleep, and health-related quality of life in patients with rheumatoid arthritis: A post hoc analysis of data from Phase 3 trials. Arthritis Res. Ther. 2022, 24, 83. [Google Scholar] [CrossRef]
- Assadiasl, S.; Fatahi, Y.; Mosharmovahed, B.; Mohebbi, B.; Nicknam, M.H. Baricitinib: From Rheumatoid Arthritis to COVID-19. J. Clin. Pharmacol. 2021, 61, 1274–1285. [Google Scholar] [CrossRef]
- Okita, R.; Shimizu, K.; Nojima, Y.; Saisho, S.; Nakata, M. Tofacitinib overcomes an IFNγ-induced decrease in NK cell-mediated cytotoxicity via the regulation of immune-related molecules in LC-2/ad. Thorac. Cancer. 2021, 12, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.N.; Pino, M.; Boddapati, A.K.; Viox, E.G.; Starke, C.E.; Upadhyay, A.A.; Gumber, S.; Nekorchuk, M.; Nekorchuk, M.; Busman-Sahay, K.; et al. Baricitinib treatment resolves lower-airway macrophage inflammation and neutrophil recruitment in SARS-CoV-2-infected rhesus macaques. Cell 2021, 184, 460–475.e21. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Talukder, A.; Savage, N.M.; Singh, N.; Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. OncoImmunology 2017, 6, e1291106. [Google Scholar] [CrossRef] [Green Version]
- Felt, S.A.; Droby, G.N.; Grdzelishvili, V.Z. Ruxolitinib and Polycation Combination Treatment Overcomes Multiple Mechanisms of Resistance of Pancreatic Cancer Cells to Oncolytic Vesicular Stomatitis Virus. J. Virol. 2017, 91, e00461-17. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, H.; Van Cutsem, E.; Bendell, J.; Hidalgo, M.; Li, C.-P.; Salvo, M.G.; Macarulla, T.; Sahai, V.; Sama, A.; Greeno, E.; et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Investig. New Drugs 2018, 36, 683–695. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Uppal, N.; Wagner, S.A.; Bendell, J.C.; Beck, J.T.; Wade, S.M., III; Nemunaitis, J.J.; Stella, P.J.; Pipas, J.M.; Wainberg, Z.A.; et al. Randomized, Double-Blind, Phase II Study of Ruxolitinib or Placebo in Combination With Capecitabine in Patients With Metastatic Pancreatic Cancer for Whom Therapy With Gemcitabine Has Failed. J. Clin. Oncol. 2015, 33, 4039–4047. [Google Scholar] [CrossRef] [Green Version]
- Plimack, E.R.; LoRusso, P.M.; McCoon, P.; Tang, W.; Krebs, A.D.; Curt, G.; Eckhardt, S.G. AZD1480: A Phase I Study of a Novel JAK2 Inhibitor in Solid Tumors. Oncologist 2013, 18, 819–820. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Herrmann, A.; Reckamp, K.; Zhang, W.; Pal, S.; Hedvat, M.; Zhang, C.; Liang, W.; Scuto, A.; Weng, S.; et al. Antiangiogenic and Antimetastatic Activity of JAK Inhibitor AZD1480. Cancer Res. 2011, 71, 6601–6610. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Hong, C.C.; Jark, P.C.; Assumpção AL, F.V.; Bollig, N.; Kong, G.; Pan, X. JAK1/2 Inhibitors AZD1480 and CYT387 Inhibit Canine B-Cell Lymphoma Growth by In-creasing Apoptosis and Disrupting Cell Proliferation. J. Vet. Intern. Med. 2017, 31, 1804–1815. [Google Scholar] [CrossRef]
- Murakami, T.; Takigawa, N.; Ninomiya, T.; Ochi, N.; Yasugi, M.; Honda, Y.; Kubo, T.; Ichihara, E.; Hotta, K.; Tanimoto, M.; et al. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer 2014, 83, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryani, S.; Bracken, L.S.; Harvey, R.C.; Sia, K.C.; Carol, H.; Chen, I.-M.; Evans, K.; Dietrich, P.A.; Roberts, K.G.; Kurmasheva, R.T.; et al. Evaluation of the In Vitro and In Vivo Efficacy of the JAK Inhibitor AZD1480 against JAK-Mutated Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2015, 14, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peisl, S.; Mellenthin, C.; Vignot, L.; Gonelle-Gispert, C.; Bühler, L.; Egger, B. Therapeutic targeting of STAT3 pathways in pancreatic adenocarcinoma: A systematic review of clinical and preclinical literature. PLoS ONE 2021, 16, e0252397. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Hendifar, A.; Starodub, A.; Chaves, J.; Yang, Y.; Koh, B.; Barbie, D.; Hahn, W.C.; Fuchs, C.S. Phase 1 dose-escalation study of momelotinib, a Janus kinase 1/2 inhibitor, combined with gemcitabine and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2019, 37, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Kenchappa, R.S.; Dovas, A.; Argenziano, M.G.; Meyer, C.T.; Stopfer, L.E.; Banu, M.A.; Pereira, B.; Griffith, J.; Mohammad, A.; Talele, S.; et al. Activation of STAT3 through combined SRC and EGFR signaling drives resistance to a mitotic kinesin inhibitor in glioblastoma. Cell Rep. 2022, 39, 110991. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belli, S.; Esposito, D.; Servetto, A.; Pesapane, A.; Formisano, L.; Bianco, R. c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve? Cancers 2020, 12, 1489. [Google Scholar] [CrossRef]
- Yang, Z.; Xie, J.; Fang, J.; Lv, M.; Yang, M.; Deng, Z.; Xie, Y.; Cai, L. Nigericin exerts anticancer effects through inhibition of the SRC/STAT3/BCL-2 in osteosarcoma. Biochem. Pharmacol. 2022, 198, 114938. [Google Scholar] [CrossRef]
- Liu, Y.-X.; Xu, B.-W.; Niu, X.-D.; Chen, Y.-J.; Fu, X.-Q.; Wang, X.-Q.; Yin, C.-L.; Chou, J.-Y.; Li, J.-K.; Wu, J.-Y.; et al. Inhibition of Src/STAT3 signaling-mediated angiogenesis is involved in the anti-melanoma effects of dioscin. Pharmacol. Res. 2022, 175, 105983. [Google Scholar] [CrossRef]
- An, E.-J.; Kim, Y.; Lee, S.-H.; Ko, H.M.; Chung, W.-S.; Jang, H.-J. Anti-Cancer Potential of Oxialis obtriangulata in Pancreatic Cancer Cell through Regulation of the ERK/Src/STAT3-Mediated Pathway. Molecules 2020, 25, 2301. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Eckhardt, S.G.; De Porre, P.; Smith, D.; Maurel, J.; Steward, W.P.; Bouche, O.; van de Velde, H.; Michiels, B.; Bugat, R. Patient-reported outcomes as a component of the primary endpoint in a double-blind, placebo-controlled trial in advanced pancreatic cancer. J. Pain Symptom Manag. 2009, 37, 135–143. [Google Scholar] [CrossRef]
| STAT3 Inhibitors | Cancer Types | Targets | Phase | NCT Number |
|---|---|---|---|---|
| IMX-110 | Solid Tumor * | STAT3/NF-kB | I/II | NCT03382340 |
| AZD9150 | PC, NSCLC, CRC | STAT3 | II | NCT02983578 |
| Napabucasin | PC | STAT3 | III | NCT02993731 |
| Bazedoxifene | PC | IL-6 and gp130 | NA | NCT04812808 |
| Siltuximab | PC | IL-6 | I/II | NCT04191421 |
| CNTO 328 | Solid Tumor | IL-6 | II | NCT00841191 |
| Ruxolitinib | PC | JAK1 and JAK2 | I | NCT05440942 |
| PC | JAK1 and JAK2 | II | NCT01423604 | |
| PC, CRC | JAK1 and JAK2 | I | NCT04303403 | |
| Itacitinib | Solid Tumor * | JAK1 | I | NCT02646748 |
| Ponatinib | CML | FGFR | II | NCT04043676 |
| Sunitinib | RCCC | VEGFR, PDGFR | II | NCT03066427 |
| pNET | VEGFR, PDGFR | II | NCT02713763 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Jiang, W.; Dong, S.; Li, W.; Zhu, W.; Zhou, W. STAT3 Inhibitors: A Novel Insight for Anticancer Therapy of Pancreatic Cancer. Biomolecules 2022, 12, 1450. https://doi.org/10.3390/biom12101450
Li X, Jiang W, Dong S, Li W, Zhu W, Zhou W. STAT3 Inhibitors: A Novel Insight for Anticancer Therapy of Pancreatic Cancer. Biomolecules. 2022; 12(10):1450. https://doi.org/10.3390/biom12101450
Chicago/Turabian StyleLi, Xin, Wenkai Jiang, Shi Dong, Wancheng Li, Weixiong Zhu, and Wence Zhou. 2022. "STAT3 Inhibitors: A Novel Insight for Anticancer Therapy of Pancreatic Cancer" Biomolecules 12, no. 10: 1450. https://doi.org/10.3390/biom12101450
APA StyleLi, X., Jiang, W., Dong, S., Li, W., Zhu, W., & Zhou, W. (2022). STAT3 Inhibitors: A Novel Insight for Anticancer Therapy of Pancreatic Cancer. Biomolecules, 12(10), 1450. https://doi.org/10.3390/biom12101450