
Shared Genetic Regulatory Networks Contribute to Neuropathic and Inflammatory Pain: Multi-Omics Systems Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Filtration of GWASs on NP and IP
2.2. Curation of Association between SNPs and Genes
2.3. Construction of Gene Co-Expression Modules
2.4. Identification of Associated Modules in NP and IP
2.5. Evaluation of Shared Genetic Mechanism between NP and IP
2.6. Identification and Consolidation of Key Drivers in NP and IP
2.7. Systematic Analyses of Conservative KDs
2.8. Changes in KD Expression in NP and IP Mouse Models
3. Results
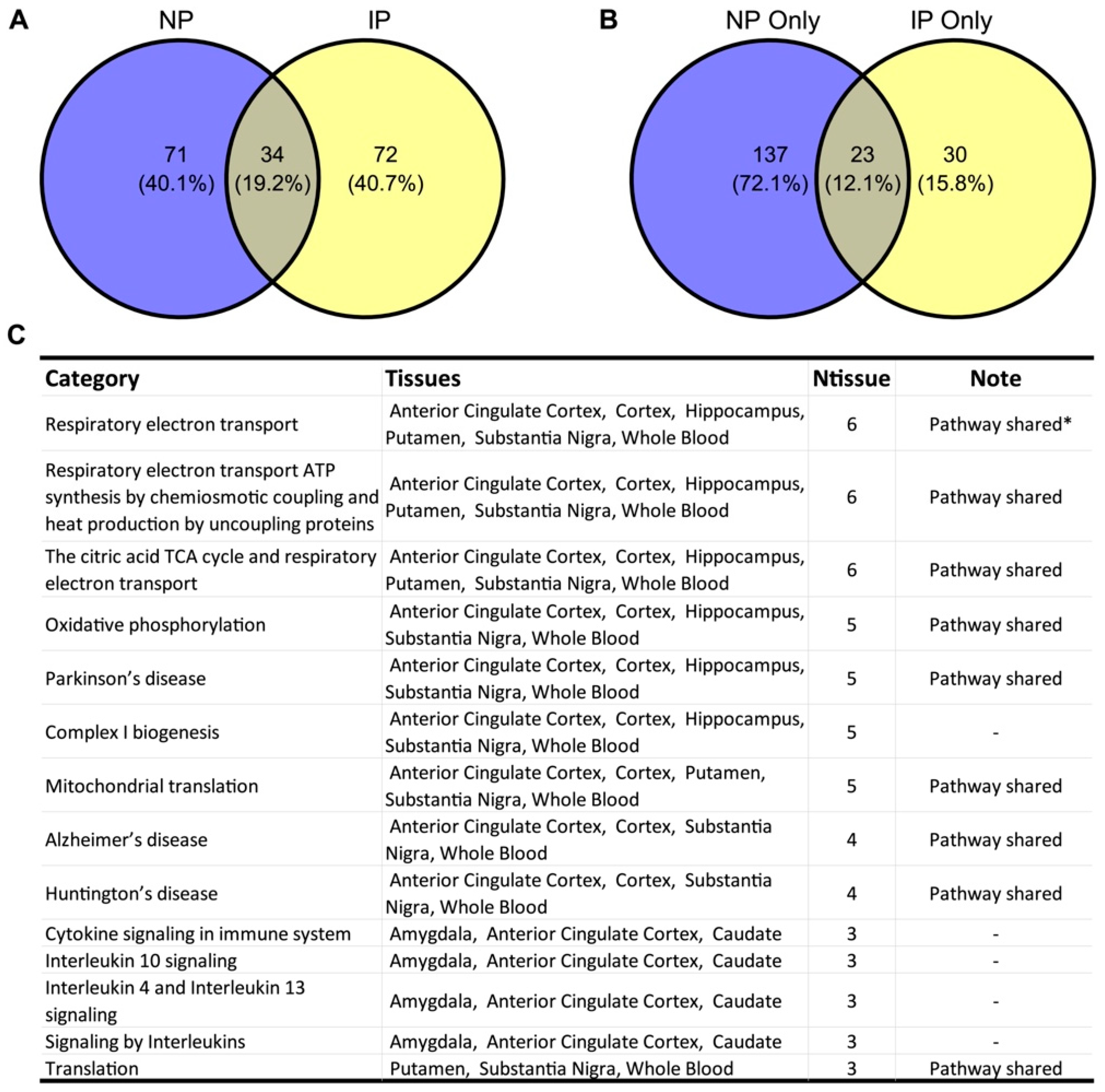
3.1. NP- and IP-Related Co-Expression Modules Are Significantly Overlapped
3.2. Shared Biological Pathways Contribute to NP and IP
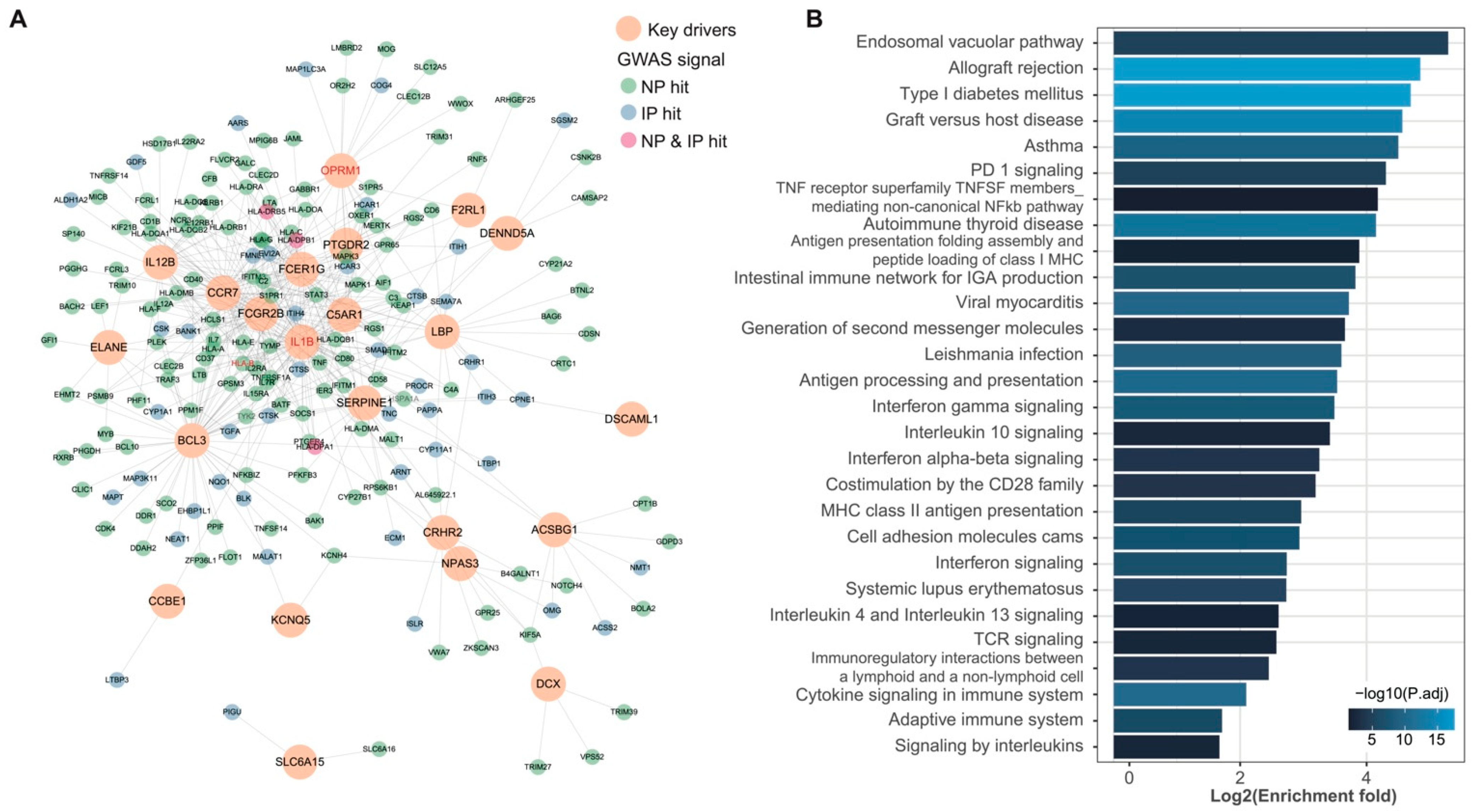
3.3. wKDA Identified 24 Conservative KDs Shared in NP and IP
3.4. Shared KDs Significantly Overlapped with Known Pain Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Souza, J.B.; Grossmann, E.; Perissinotti, D.M.N.; de Oliveira Junior, J.O.; da Fonseca, P.R.B.; Posso, I.P. Prevalence of Chronic Pain, Treatments, Perception, and Interference on Life Activities: Brazilian Population-Based Survey. Pain Res. Manag. 2017, 2017, 4643830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenewald, C.B.; Essner, B.S.; Wright, D.; Fesinmeyer, M.D.; Palermo, T.M. The Economic Costs of Chronic Pain among a Cohort of Treatment-Seeking Adolescents in the United States. J. Pain 2014, 15, 925–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeau, S.E.; Wu, J.K.; Lawhern, R.A. Opioids and Chronic Pain: An Analytic Review of the Clinical Evidence. Front. Pain Res. 2021, 2, 721357. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP Classification of Chronic Pain for ICD-11: Chronic Neuropathic Pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: From Mechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- Vasko, M.R. Inflammatory Pain. In Encyclopedia of Neuroscience; Binder, M.D., Hirokawa, N., Windhorst, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1952–1955. [Google Scholar]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic Pain. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kidd, B.L.; Urban, L.A. Mechanisms of Inflammatory Pain. Br. J. Anaesth. 2001, 87, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Yaksh, T.L. A Brief Comparison of the Pathophysiology of Inflammatory versus Neuropathic Pain. Curr. Opin. Anaesthesiol. 2011, 24, 400–407. [Google Scholar] [CrossRef] [Green Version]
- Diatchenko, L.; Slade, G.D.; Nackley, A.G.; Bhalang, K.; Sigurdsson, A.; Belfer, I.; Goldman, D.; Xu, K.; Shabalina, S.A.; Shagin, D.; et al. Genetic Basis for Individual Variations in Pain Perception and the Development of a Chronic Pain Condition. Hum. Mol. Genet. 2005, 14, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Hocking, L.J.; Morris, A.D.; Dominiczak, A.F.; Porteous, D.J.; Smith, B.H. Heritability of Chronic Pain in 2195 Extended Families. Eur. J. Pain 2012, 16, 1053–1063. [Google Scholar] [CrossRef]
- Nielsen, C.S.; Knudsen, G.P.; Steingrímsdóttir, Ó.A. Twin Studies of Pain. Clin. Genet. 2012, 82, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pain Research Forum. Available online: https://www.painresearchforum.org/ (accessed on 28 August 2022).
- Meloto, C.B.; Benavides, R.; Lichtenwalter, R.N.; Wen, X.; Tugarinov, N.; Zorina-Lichtenwalter, K.; Chabot-Doré, A.-J.; Piltonen, M.H.; Cattaneo, S.; Verma, V.; et al. Human Pain Genetics Database: A Resource Dedicated to Human Pain Genetics Research. Pain 2018, 159, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Mogil, J.S. Pain Genetics: Past, Present and Future. Trends Genet. 2012, 28, 258–266. [Google Scholar] [CrossRef]
- Kocot-Kępska, M.; Zajączkowska, R.; Mika, J.; Kopsky, D.J.; Wordliczek, J.; Dobrogowski, J.; Przeklasa-Muszyńska, A. Topical Treatments and Their Molecular/Cellular Mechanisms in Patients with Peripheral Neuropathic Pain-Narrative Review. Pharmaceutics 2021, 13, 450. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.M.; Day, R.O. Nonsteroidal Antiinflammatory Drugs—Differences and Similarities. N. Engl. J. Med. 1991, 324, 1716–1725. [Google Scholar] [PubMed]
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic Pain: An Update on Burden, Best Practices, and New Advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef]
- Lyon, M.S.; Andrews, S.J.; Elsworth, B.; Gaunt, T.R.; Hemani, G.; Marcora, E. The Variant Call Format Provides Efficient and Robust Storage of GWAS Summary Statistics. Genome Biol. 2021, 22, 32. [Google Scholar] [CrossRef]
- IEU OpenGWAS Project. Available online: https://gwas.mrcieu.ac.uk (accessed on 27 September 2022).
- Shu, L.; Zhao, Y.; Kurt, Z.; Byars, S.G.; Tukiainen, T.; Kettunen, J.; Orozco, L.D.; Pellegrini, M.; Lusis, A.J.; Ripatti, S.; et al. Mergeomics: Multidimensional Data Integration to Identify Pathogenic Perturbations to Biological Systems. BMC Genom. 2016, 17, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, s13742–015–0047–8. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An Integrated Map of Structural Variation in 2504 Human Genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef]
- GTEx. Consortium The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- GTEx Portal. Available online: https://www.gtexportal.org/home/datasets (accessed on 27 September 2022).
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of Functional Variation in Personal Genomes Using Regulome DB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 1–45. [Google Scholar] [CrossRef]
- GTEx Portal Frequently Asked Questions. Available online: https://www.gtexportal.org/home/faq#diseased (accessed on 26 September 2022).
- Carithers, L.J.; Ardlie, K.; Barcus, M.; Branton, P.A.; Britton, A.; Buia, S.A.; Compton, C.C.; DeLuca, D.S.; Peter-Demchok, J.; Gelfand, E.T.; et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv. Biobank. 2015, 13, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Shu, L.; Chan, K.H.K.; Zhang, G.; Huan, T.; Kurt, Z.; Zhao, Y.; Codoni, V.; Trégouët, D.-A.; Cardiogenics Consortium; Yang, J.; et al. Shared Genetic Regulatory Networks for Cardiovascular Disease and Type 2 Diabetes in Multiple Populations of Diverse Ethnicities in the United States. PLoS Genet. 2017, 13, e1007040. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.Y.; Papp, J.C.; Pellegrini, M.; Yu, H.; Sobel, E.M. Molecular Biology Networks and Key Gene Regulators for Inflammatory Biomarkers Shared by Breast Cancer Development: Multi-Omics Systems Analysis. Biomolecules 2021, 11, 1379. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding Multicellular Function and Disease with Human Tissue-Specific Networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.K.; Carlin, D.E.; Yu, M.K.; Zhang, W.; Kreisberg, J.F.; Tamayo, P.; Ideker, T. Systematic Evaluation of Molecular Networks for Discovery of Disease Genes. Cell Syst 2018, 6, 484–495.e5. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- International HapMap Consortium A Haplotype Map of the Human Genome. Nature 2005, 437, 1299–1320. [CrossRef] [PubMed] [Green Version]
- Gene Ontology. Consortium Gene Ontology Consortium: Going Forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Gene Ontology Resource. Available online: http://geneontology.org/ (accessed on 27 September 2022).
- Parisien, M.; Samoshkin, A.; Tansley, S.N.; Piltonen, M.H.; Martin, L.J.; El-Hachem, N.; Dagostino, C.; Allegri, M.; Mogil, J.S.; Khoutorsky, A.; et al. Genetic Pathway Analysis Reveals a Major Role for Extracellular Matrix Organization in Inflammatory and Neuropathic Pain. Pain 2019, 160, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International HapMap Consortium; Frazer, K.A.; Ballinger, D.G.; Cox, D.R.; Hinds, D.A.; Stuve, L.L.; Gibbs, R.A.; Belmont, J.W.; Boudreau, A.; Hardenbol, P.; et al. A Second Generation Human Haplotype Map of over 3.1 Million SNPs. Nature 2007, 449, 851–861. [Google Scholar]
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Khadijah Adam, S.; Abdul Manan, N.; Basir, R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int. J. Mol. Sci. 2018, 19, 2164. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Blencowe, M.; Nghiem, T.; Ha, S.-M.; Chen, Y.-W.; Li, G.; Yang, X. Mergeomics 2.0: A Web Server for Multi-Omics Data Integration to Elucidate Disease Networks and Predict Therapeutics. Nucleic Acids Res. 2021, 49, W375–W387. [Google Scholar] [CrossRef]
- Joseph, E.K.; Levine, J.D. Mitochondrial Electron Transport in Models of Neuropathic and Inflammatory Pain. Pain 2006, 121, 105–114. [Google Scholar] [CrossRef]
- Kato, Y.; Hiasa, M.; Ichikawa, R.; Hasuzawa, N.; Kadowaki, A.; Iwatsuki, K.; Shima, K.; Endo, Y.; Kitahara, Y.; Inoue, T.; et al. Identification of a Vesicular ATP Release Inhibitor for the Treatment of Neuropathic and Inflammatory Pain. Proc. Natl. Acad. Sci. USA 2017, 114, E6297–E6305. [Google Scholar] [CrossRef] [Green Version]
- Ford, B. Pain in Parkinson’s Disease. Mov. Disord. 2010, 25, S98–S103. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Sethi, K. Nonmotor Manifestations of Parkinson’s Disease. Ann. Neurol. 2008, 64, S65–S80. [Google Scholar] [CrossRef] [PubMed]
- Pautex, S.; Michon, A.; Guedira, M.; Emond, H.; Le Lous, P.; Samaras, D.; Michel, J.-P.; Herrmann, F.; Giannakopoulos, P.; Gold, G. Pain in Severe Dementia: Self-Assessment or Observational Scales? J. Am. Geriatr. Soc. 2006, 54, 1040–1045. [Google Scholar] [CrossRef]
- Mostofi, A.; Morgante, F.; Edwards, M.J.; Brown, P.; Pereira, E.A.C. Pain in Parkinson’s Disease and the Role of the Subthalamic Nucleus. Brain 2021, 144, 1342–1350. [Google Scholar] [CrossRef]
- Cao, S.; Fisher, D.W.; Yu, T.; Dong, H. The Link between Chronic Pain and Alzheimer’s Disease. J. Neuroinflammation 2019, 16, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.-S.; Chen, Q.-Y.; Chen, X.; Li, X.-H.; Zhou, Z.; Liu, Q.; Lin, Y.; Zhou, M.; Xu, P.-Y.; Zhuo, M. Cellular and Synaptic Mechanisms for Parkinson’s Disease-Related Chronic Pain. Mol. Pain 2021, 17, 1744806921999025. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the Expanded Endocannabinoid System in Neurological Disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Brainstorm Consortium; Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; et al. Analysis of Shared Heritability in Common Disorders of the Brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef] [Green Version]
- Bouza, A.A.; Isom, L.L. Voltage-Gated Sodium Channel β Subunits and Their Related Diseases. Handb. Exp. Pharmacol. 2018, 246, 423–450. [Google Scholar]
- Lin, C.-H.; Chaudhuri, K.R.; Fan, J.-Y.; Ko, C.-I.; Rizos, A.; Chang, C.-W.; Lin, H.-I.; Wu, Y.-R. Depression and Catechol-O-Methyltransferase (COMT) Genetic Variants Are Associated with Pain in Parkinson’s Disease. Sci. Rep. 2017, 7, 6306. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Du, B.; Wang, B.; Yin, R.; Lv, X.; Dai, Y.; Zhang, W.; Xia, K. Joint Analysis of Genome-Wide Association Data Reveals No Genetic Correlations Between Low Back Pain and Neurodegenerative Diseases. Front. Genet. 2021, 12, 744299. [Google Scholar] [CrossRef] [PubMed]
- Mocci, E.; Ward, K.; Dorsey, S.G.; Ament, S.A. GWAS Meta-Analysis Reveals Dual Neuronal and Immunological Etiology for Pain Susceptibility. medRxiv 2021, medRxiv:08.23.21262510. [Google Scholar]
- Zorina-Lichtenwalter, K.; Bango, C.I.; Van Oudenhove, L.; Čeko, M.; Lindquist, M.A.; Grotzinger, A.D.; Keller, M.C.; Friedman, N.P.; Wager, T.D. Identification and Characterization of Genetic Risk Shared across 24 Chronic Pain Conditions in the UK Biobank. medRxiv 2022, medRxiv:06.28.22277025. [Google Scholar]
- Nadeau, S.; Filali, M.; Zhang, J.; Kerr, B.J.; Rivest, S.; Soulet, D.; Iwakura, Y.; de Rivero Vaccari, J.P.; Keane, R.W.; Lacroix, S. Functional Recovery after Peripheral Nerve Injury Is Dependent on the pro-Inflammatory Cytokines IL-1β and TNF: Implications for Neuropathic Pain. J. Neurosci. 2011, 31, 12533–12542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; Gabay, E.; Tal, M.; Yirmiya, R.; Shavit, Y. Genetic Impairment of Interleukin-1 Signaling Attenuates Neuropathic Pain, Autotomy, and Spontaneous Ectopic Neuronal Activity, Following Nerve Injury in Mice. Pain 2006, 120, 315–324. [Google Scholar] [CrossRef]
- Bond, C.; LaForge, K.S.; Tian, M.; Melia, D.; Zhang, S.; Borg, L.; Gong, J.; Schluger, J.; Strong, J.A.; Leal, S.M.; et al. Single-Nucleotide Polymorphism in the Human Mu Opioid Receptor Gene Alters Beta-Endorphin Binding and Activity: Possible Implications for Opiate Addiction. Proc. Natl. Acad. Sci. USA 1998, 95, 9608–9613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaForge, K.S.; Yuferov, V.; Kreek, M.J. Opioid Receptor and Peptide Gene Polymorphisms: Potential Implications for Addictions. Eur. J. Pharmacol. 2000, 410, 249–268. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Johnson, A.D.; Papp, A.C.; Sadée, W. Allelic Expression Imbalance of Human Mu Opioid Receptor (OPRM1) Caused by Variant A118G. J. Biol. Chem. 2005, 280, 32618–32624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological Pain and the Neuroimmune Interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Sommer, C.; Leinders, M.; Üçeyler, N. Inflammation in the Pathophysiology of Neuropathic Pain. Pain 2018, 159, 595–602. [Google Scholar] [CrossRef]
- Moriconi, A.; Cunha, T.M.; Souza, G.R.; Lopes, A.H.; Cunha, F.Q.; Carneiro, V.L.; Pinto, L.G.; Brandolini, L.; Aramini, A.; Bizzarri, C.; et al. Targeting the Minor Pocket of C5aR for the Rational Design of an Oral Allosteric Inhibitor for Inflammatory and Neuropathic Pain Relief. Proc. Natl. Acad. Sci. USA 2014, 111, 16937–16942. [Google Scholar] [CrossRef] [Green Version]
- Doolen, S.; Cook, J.; Riedl, M.; Kitto, K.; Kohsaka, S.; Honda, C.N.; Fairbanks, C.A.; Taylor, B.K.; Vulchanova, L. Complement 3a Receptor in Dorsal Horn Microglia Mediates Pronociceptive Neuropeptide Signaling. Glia 2017, 65, 1976–1989. [Google Scholar] [CrossRef] [Green Version]
- Levin, M.E.; Jin, J.G.; Ji, R.-R.; Tong, J.; Pomonis, J.D.; Lavery, D.J.; Miller, S.W.; Chiang, L.W. Complement Activation in the Peripheral Nervous System Following the Spinal Nerve Ligation Model of Neuropathic Pain. Pain 2008, 137, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Warwick, C.A.; Shutov, L.P.; Shepherd, A.J.; Mohapatra, D.P.; Usachev, Y.M. Mechanisms Underlying Mechanical Sensitization Induced by Complement C5a: The Roles of Macrophages, TRPV1, and Calcitonin Gene-Related Peptide Receptors. Pain 2019, 160, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, C.; Zippoli, M.; Cocchiaro, P.; Castelli, V.; Varrassi, G.; Aramini, A.; Allegretti, M.; Brandolini, L.; Cesta, M.C. Emerging Role of C5 Complement Pathway in Peripheral Neuropathies: Current Treatments and Future Perspectives. Biomedicines 2021, 9, 399. [Google Scholar] [CrossRef]
- Warwick, C.A.; Keyes, A.L.; Woodruff, T.M.; Usachev, Y.M. The Complement Cascade in the Regulation of Neuroinflammation, Nociceptive Sensitization, and Pain. J. Biol. Chem. 2021, 297, 101085. [Google Scholar] [CrossRef] [PubMed]
- Party, H.; Dujarrier, C.; Hébert, M.; Lenoir, S.; Martinez de Lizarrondo, S.; Delépée, R.; Fauchon, C.; Bouton, M.-C.; Obiang, P.; Godefroy, O.; et al. Plasminogen Activator Inhibitor-1 (PAI-1) Deficiency Predisposes to Depression and Resistance to Treatments. Acta Neuropathol. Commun. 2019, 7, 153. [Google Scholar] [CrossRef]
- Angelucci, F.; Čechová, K.; Průša, R.; Hort, J. Amyloid Beta Soluble Forms and Plasminogen Activation System in Alzheimer’s Disease: Consequences on Extracellular Maturation of Brain-Derived Neurotrophic Factor and Therapeutic Implications. CNS Neurosci. Ther. 2019, 25, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Yepes, M. The Plasminogen Activating System in the Pathogenesis of Alzheimer’s Disease. Neural Regen. Res. 2021, 16, 1973–1977. [Google Scholar] [CrossRef]
- Jiang, H.; Li, X.; Chen, S.; Lu, N.; Yue, Y.; Liang, J.; Zhang, Z.; Yuan, Y. Plasminogen Activator Inhibitor-1 in Depression: Results from Animal and Clinical Studies. Sci. Rep. 2016, 6, 30464. [Google Scholar] [CrossRef] [Green Version]
- Kohli, M.A.; Lucae, S.; Saemann, P.G.; Schmidt, M.V.; Demirkan, A.; Hek, K.; Czamara, D.; Alexander, M.; Salyakina, D.; Ripke, S.; et al. The Neuronal Transporter Gene SLC6A15 Confers Risk to Major Depression. Neuron 2011, 70, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Santarellia, S.; Namendorf, C.; Anderzhanova, E.; Gerlach, T.; Bedenk, B.; Kaltwasser, S.; Wagner, K.; Labermaier, C.; Reichel, J.; Drgonova, J.; et al. The Amino Acid Transporter SLC6A15 Is a Regulator of Hippocampal Neurochemistry and Behavior. J. Psychiatr. Res. 2015, 68, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Schraut, K.; Kalnytska, O.; Lamp, D.; Jastroch, M.; Eder, M.; Hausch, F.; Gassen, N.C.; Moore, S.; Nagaraj, N.; Lopez, J.P.; et al. Loss of the Psychiatric Risk Factor SLC6A15 Is Associated with Increased Metabolic Functions in Primary Hippocampal Neurons. Eur. J. Neurosci. 2021, 53, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.W. KCNQs: Ligand- and Voltage-Gated Potassium Channels. Front. Physiol. 2020, 11, 583. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Trussell, L.O. KCNQ5 Channels Control Resting Properties and Release Probability of a Synapse. Nat. Neurosci. 2011, 14, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manville, R.W.; Abbott, G.W. Gabapentin Is a Potent Activator of KCNQ3 and KCNQ5 Potassium Channels. Mol. Pharmacol. 2018, 94, 1155–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Iwata, M.; Tsuchimori, N.; Matsumoto, T. Activation of Peripheral KCNQ Channels Attenuates Inflammatory Pain. Mol. Pain 2014, 10, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.-H.; Estacion, M.; Mis, M.A.; Tanaka, B.S.; Schulman, B.R.; Chen, L.; Liu, S.; Dib-Hajj, F.B.; Dib-Hajj, S.D.; Waxman, S.G. KCNQ Variants and Pain Modulation: A Missense Variant in Kv7.3 Contributes to Pain Resilience. Brain Commun. 2021, 3, fcab212. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Tissue | Ntissue | If Known |
|---|---|---|---|
| SERPINE1 | amygdala, blood, caudate nucleus, caudate putamen, cerebellum, frontal lobe, hippocampus, spinal cord, substantia nigra | 9 | no |
| IL1B | blood, caudate nucleus, caudate putamen, cerebellum, frontal lobe, hippocampus, spinal cord, substantia nigra | 8 | yes |
| C5AR1 | amygdala, caudate nucleus, hippocampus, substantia nigra | 4 | no |
| CCR7 | amygdala, cerebellar cortex, frontal lobe, hippocampus | 4 | no |
| ACSBG1 | amygdala, caudate nucleus, substantia nigra | 3 | no |
| BCL3 | blood, frontal lobe, nucleus accumbens | 3 | no |
| DCX | caudate nucleus, frontal lobe, substantia nigra | 3 | no |
| ELANE | caudate putamen, hippocampus, nucleus accumbens | 3 | no |
| FCER1G | caudate nucleus, hypothalamus, spinal cord | 3 | no |
| IL12B | caudate putamen, frontal lobe, hypothalamus | 3 | no |
| OPRM1 | caudate putamen, hypothalamus, nucleus accumbens | 3 | yes |
| SLC6A15 | caudate putamen, hypothalamus, nucleus accumbens | 3 | no |
| CCBE1 | amygdala, caudate nucleus | 2 | no |
| CRHR2 | hypothalamus, nucleus accumbens | 2 | no |
| DENND5A | amygdala, spinal cord | 2 | no |
| DSCAML1 | cerebellar cortex, substantia nigra | 2 | no |
| F2RL1 | blood, caudate nucleus | 2 | no |
| FCGR2B | amygdala, cerebellum | 2 | no |
| KCNQ5 | amygdala, caudate putamen | 2 | no |
| LBP | cerebellum, hippocampus | 2 | no |
| NPAS3 | frontal lobe, substantia nigra | 2 | no |
| PTGDR2 | caudate putamen, hypothalamus | 2 | no |
| TMPRSS3 | caudate putamen, substantia nigra | 2 | no |
| TRPC7 | nucleus accumbens, substantia nigra | 2 | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, F.; Du, L.; Huang, W.; Wang, S. Shared Genetic Regulatory Networks Contribute to Neuropathic and Inflammatory Pain: Multi-Omics Systems Analysis. Biomolecules 2022, 12, 1454. https://doi.org/10.3390/biom12101454
Ye F, Du L, Huang W, Wang S. Shared Genetic Regulatory Networks Contribute to Neuropathic and Inflammatory Pain: Multi-Omics Systems Analysis. Biomolecules. 2022; 12(10):1454. https://doi.org/10.3390/biom12101454
Chicago/Turabian StyleYe, Fang, Li Du, Wenqi Huang, and Sheng Wang. 2022. "Shared Genetic Regulatory Networks Contribute to Neuropathic and Inflammatory Pain: Multi-Omics Systems Analysis" Biomolecules 12, no. 10: 1454. https://doi.org/10.3390/biom12101454
APA StyleYe, F., Du, L., Huang, W., & Wang, S. (2022). Shared Genetic Regulatory Networks Contribute to Neuropathic and Inflammatory Pain: Multi-Omics Systems Analysis. Biomolecules, 12(10), 1454. https://doi.org/10.3390/biom12101454