
Electrophilic Aldehyde 4-Hydroxy-2-Nonenal Mediated Signaling and Mitochondrial Dysfunction
 , , , and
, , , and 
Abstract
:1. Introduction
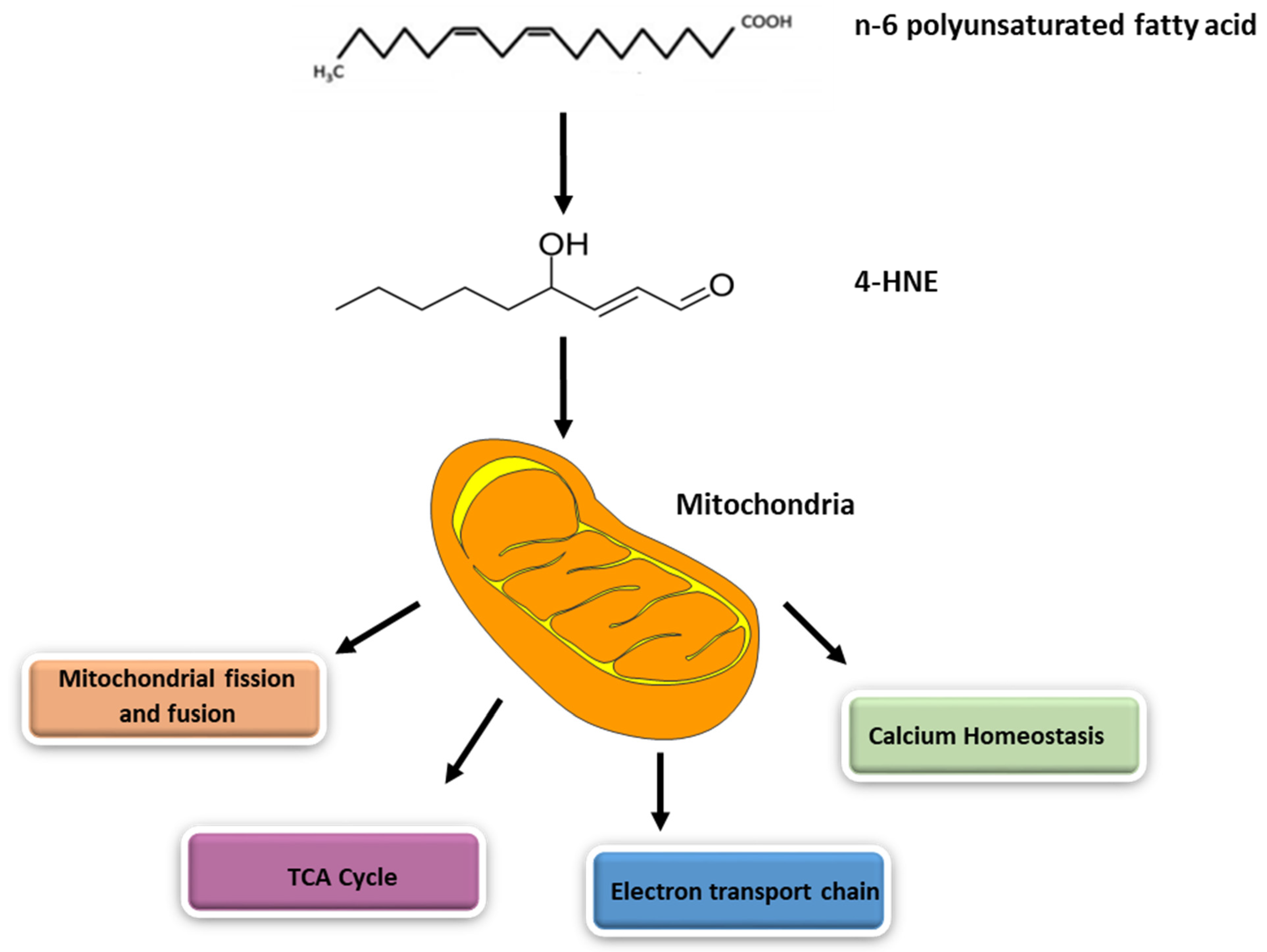
2. Generation of 4-HNE inside Mitochondria
3. Metabolism of 4-HNE
4. Mitochondrial Dysfunction Mediated by 4-HNE
4.1. Exogenous 4-HNE and Mitochondrial Dysfunction
4.2. Role of 4-HNE Causing Mitochondrial Dysfunction in Various Diseases
5. Signaling and Cytotoxic Functions of 4-HNE
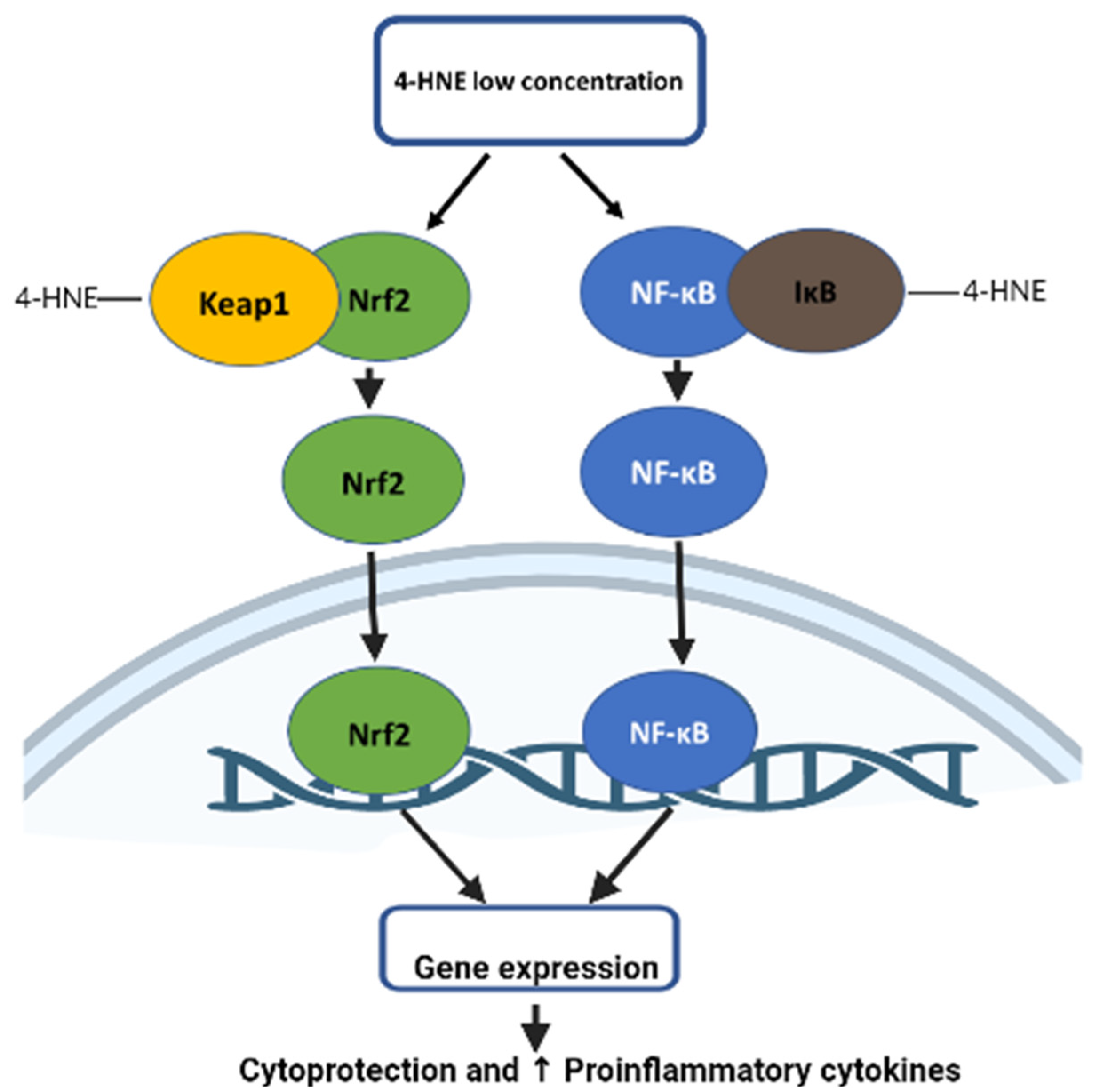
5.1. Role of 4-HNE in Nrf2 Signaling
5.2. Role of 4-HNE in NFkB Signaling
6. Role of 4-HNE in Aging
7. Role of 4-HNE in Stroke
8. Role of 4-HNE in Reproductive Physiology
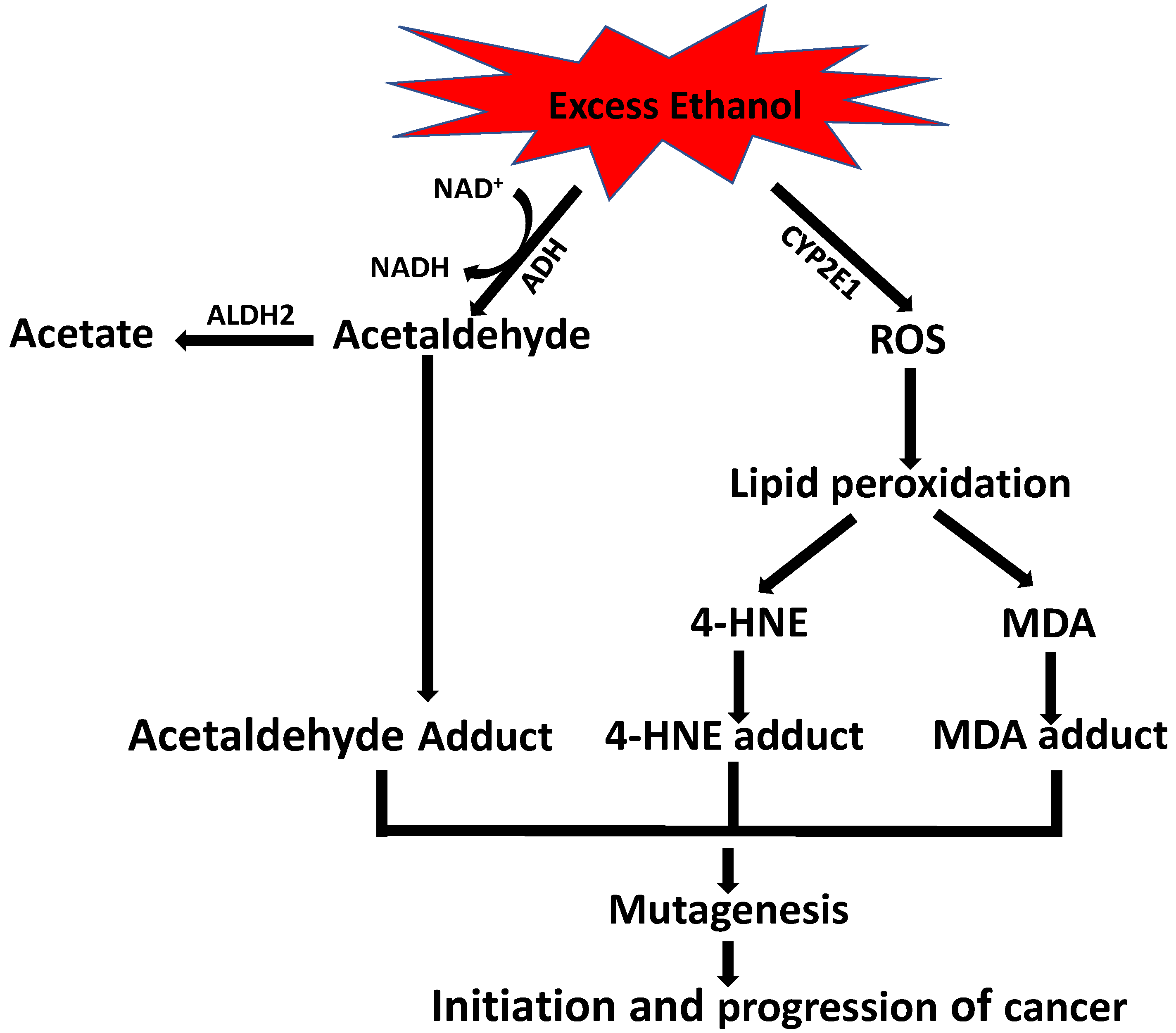
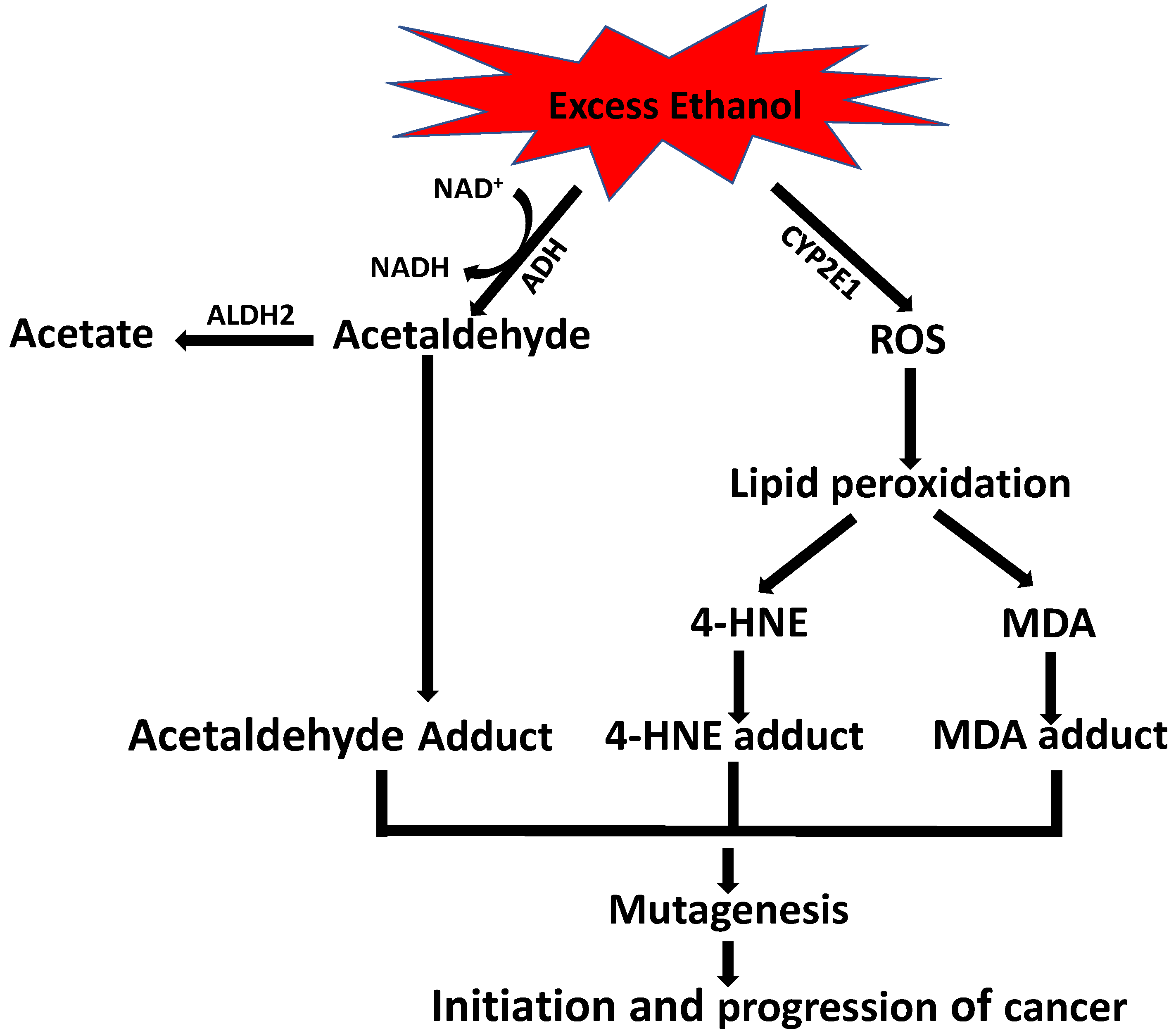
9. Role of Alcohol in 4-HNE-Induced Carcinogenesis
10. Potential Therapeutics Targeting 4-HNE
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar] [PubMed]
- Mustafa, A.G.; Alfaqih, M.A.; Al-Shboul, O. The 4-hydroxynonenal mediated oxidative damage of blood proteins and lipids involves secondary lipid peroxidation reactions. Exp. Ther. Med. 2018, 16, 2132–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cafe, S.L.; Nixon, B.; Dun, M.D.; Roman, S.D.; Bernstein, I.R.; Bromfield, E.G. Oxidative stress dysregulates protein homeostasis within the male germ line. Antioxid. Redox Signal. 2019, 32, 487–503. [Google Scholar] [CrossRef]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Walters, J.L.H.; De Iuliis, G.N.; Dun, M.D.; Aitken, R.J.; McLaughlin, E.A.; Nixon, B.; Bromfield, E.G. Pharmacological inhibition of arachidonate 15-lipoxygenase protects human spermatozoa against oxidative stress. Biol. Reprod. 2018, 98, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Miriyala, S.; Thippakorn, C.; Chaiswing, L.; Xu, Y.; Noel, T.; Tovmasyan, A.; Batinic-Haberle, I.; Vander Kooi, C.W.; Chi, W.; Latif, A.A.; et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic. Biol. Med. 2016, 91, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxid. Med. Cell Longev. 2016, 2016, 1656450. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- D’Souza, A.D.; Parish, I.A.; Krause, D.S.; Kaech, S.M.; Shadel, G.S. Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia-telangiectasia. Mol. Ther. 2013, 21, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.J.; Garg, N.J. Manganese superoxide dismutase deficiency exacerbates the mitochondrial ROS production and oxidative damage in Chagas disease. PLoS Negl. Trop. Dis. 2018, 12, e0006687. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalleau, S.; Baradat, M.; Gueraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Diffe.r 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Daum, G. Lipids of mitochondria. Biochim. Biophys. Acta 1985, 822, 1–42. [Google Scholar] [CrossRef]
- Martensson, C.U.; Doan, K.N.; Becker, T. Effects of lipids on mitochondrial functions. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 102–113. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bakovic, M. Formation and regulation of mitochondrial membranes. Int. J. Cell Biol. 2014, 2014, 709828. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; John, A. 4-hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicol. Appl. Pharmacol. 2006, 216, 309–318. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Reiter, R.J.; Ruggiero, F.M. Melatonin, cardiolipin and mitochondrial bioenergetics in health and disease. J. Pineal. Res. 2010, 48, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Serena, D.; Ruggiero, F.M. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic. Biol. Med. 1999, 27, 42–50. [Google Scholar] [CrossRef]
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial dysfunction in rat with nonalcoholic fatty liverInvolvement of complex I, reactive oxygen species and cardiolipin. Biochim. Et Biophys. Acta Bioenerg. 2007, 1767, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gredilla, R.; Lopez Torres, M.; Portero-Otin, M.; Pamplona, R.; Barja, G. Influence of hyper- and hypothyroidism on lipid peroxidation, unsaturation of phospholipids, glutathione system and oxidative damage to nuclear and mitochondrial DNA in mice skeletal muscle. Mol. Cell Biochem. 2001, 221, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Tyurin, V.A.; Tyurina, Y.Y.; Viner, R.; Ritov, V.; Amoscato, A.A.; Zhao, Q.; Zhang, X.J.; Janesko-Feldman, K.L.; Alexander, H.; et al. Selective early cardiolipin peroxidation after traumatic brain injury: An oxidative lipidomics analysis. Ann. Neurol. 2007, 62, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Lu, J.; Xia, L.; Zhu, M.; Yin, H. Formation of electrophilic oxidation products from mitochondrial cardiolipin in vitro and in vivo in the context of apoptosis and atherosclerosis. Redox Biol. 2014, 2, 878–883. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Porter, N.A.; Schneider, C.; Brash, A.R.; Yin, H. Formation of 4-hydroxynonenal from cardiolipin oxidation: Intramolecular peroxyl radical addition and decomposition. Free Radic. Biol. Med. 2011, 50, 166–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siems, W.; Grune, T. Intracellular metabolism of 4-hydroxynonenal. Mol. Asp. Med. 2003, 24, 167–175. [Google Scholar] [CrossRef]
- Zheng, R.; Dragomir, A.C.; Mishin, V.; Richardson, J.R.; Heck, D.E.; Laskin, D.L.; Laskin, J.D. Differential metabolism of 4-hydroxynonenal in liver, lung and brain of mice and rats. Toxicol. Appl. Pharm. 2014, 279, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, V.R.; Palaniyandi, S.S. Regulation and therapeutic strategies of 4-hydroxy-2-nonenal metabolism in heart disease. Free Radic. Res. 2014, 48, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, Y.C.; Ansari, G.A.; Awasthi, S. Regulation of 4-hydroxynonenal mediated signaling by glutathione S-transferases. Methods Enzym. 2005, 401, 379–407. [Google Scholar] [CrossRef]
- Vasiliou, V.; Pappa, A.; Estey, T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug Metab. Rev. 2004, 36, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Csala, M.; Kardon, T.; Legeza, B.; Lizak, B.; Mandl, J.; Margittai, E.; Puskas, F.; Szaraz, P.; Szelenyi, P.; Banhegyi, G. On the role of 4-hydroxynonenal in health and disease. Biochim. Biophys. Acta 2015, 1852, 826–838. [Google Scholar] [CrossRef] [Green Version]
- Mol, M.; Regazzoni, L.; Altomare, A.; Degani, G.; Carini, M.; Vistoli, G.; Aldini, G. Enzymatic and non-enzymatic detoxification of 4-hydroxynonenal: Methodological aspects and biological consequences. Free Radic. Biol. Med. 2017, 111, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Roede, J.R.; Jones, D.P. Reactive species and mitochondrial dysfunction: Mechanistic significance of 4-hydroxynonenal. Env. Mol. Mutagen. 2010, 51, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ren, J. ALDH2 in alcoholic heart diseases: Molecular mechanism and clinical implications. Pharmacol. Ther. 2011, 132, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.J.; Akbar, M.; Abdelmegeed, M.A.; Byun, K.; Lee, B.; Yoon, S.K.; Hardwick, J.P. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014, 3, 109–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, M.; Ito, Y. N-acetylcysteine and neurodegenerative diseases: Basic and clinical pharmacology. Cerebellum 2007, 6, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011, 32, 1025–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.M.; Liu, A.J.; Zang, P.; Dong, W.Z.; Ying, L.; Wang, W.; Xu, P.; Song, X.R.; Cai, J.; Zhang, S.Q.; et al. ALDH2 protects against stroke by clearing 4-HNE. Cell Res. 2013, 23, 915–930. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.S.; Hernandez-Cuervo, H.; Fukumoto, J.; Narala, V.R.; Saji, S.; Borra, M.; Alleyn, M.; Lin, M.; Soundararajan, R.; Lockey, R.; et al. Alda-1 attenuates hyperoxia-induced mitochondrial dysfunction in lung vascular endothelial cells. Aging 2019, 11, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, B.; Zhang, J.; He, D.; Zhang, Q.; Pan, C.; Yuan, Q.; Shi, Y.; Tang, H.; Xu, F.; et al. ALDH2 (Aldehyde Dehydrogenase 2) Protects Against Hypoxia-Induced Pulmonary Hypertension. Arter. Thromb. Vasc. Biol. 2019, 39, 2303–2319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, Q.; Ye, F.; Huang, C.Y.; Chen, W.M.; Huang, W.Q. Alda-1, an ALDH2 activator, protects against hepatic ischemia/reperfusion injury in rats via inhibition of oxidative stress. Free Radic. Res. 2018, 52, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Wang, Y.B.; Han, B.; Yang, B.; Qiang, Y.W.; Zhang, Y.; Wang, Z.; Huang, X.; Liu, J.; Chen, Y.D.; et al. Activation of aldehyde dehydrogenase 2 slows down the progression of atherosclerosis via attenuation of ER stress and apoptosis in smooth muscle cells. Acta Pharmacol. Sin. 2018, 39, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Keith, R.J.; Haberzettl, P.; Vladykovskaya, E.; Hill, B.G.; Kaiserova, K.; Srivastava, S.; Barski, O.; Bhatnagar, A. Aldose reductase decreases endoplasmic reticulum stress in ischemic hearts. Chem. Biol. Interact. 2009, 178, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Conklin, D.; Prough, R.; Bhatanagar, A. Aldehyde metabolism in the cardiovascular system. Mol. Biosyst. 2007, 3, 136–150. [Google Scholar] [CrossRef]
- Srivastava, S.; Chandra, A.; Bhatnagar, A.; Srivastava, S.K.; Ansari, N.H. Lipid peroxidation product, 4-hydroxynonenal and its conjugate with GSH are excellent substrates of bovine lens aldose reductase. Biochem. Biophys. Res. Commun. 1995, 217, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awasthi, S. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol. Appl. Pharmacol. 2015, 289, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.M.; Atkins, W.M. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab. Rev. 2011, 43, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesfaigzi, Y. Roles of apoptosis in airway epithelia. Am. J. Respir. Cell Mol. Biol. 2006, 34, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, G.F.; Manzo, N.D.; Cotich, K.L.; Shone, R.K.; Waxman, A.B. DNA damage induced by hyperoxia: Quantitation and correlation with lung injury. Am. J. Respir. Cell Mol. Biol. 2006, 35, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolliputi, N.; Waxman, A.B. IL-6 Cytoprotection in Hyperoxic Acute Lung Injury Occurs via Suppressor of Cytokine Signaling-1–Induced Apoptosis Signal–Regulating Kinase-1 Degradation. Am. J. Respir. Cell Mol. Biol. 2009, 40, 314–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galam, L.; Failla, A.; Soundararajan, R.; Lockey, R.F.; Kolliputi, N. 4-hydroxynonenal regulates mitochondrial function in human small airway epithelial cells. Oncotarget 2015, 6, 41508–41521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiryu-Seo, S.; Tamada, H.; Kato, Y.; Yasuda, K.; Ishihara, N.; Nomura, M.; Mihara, K.; Kiyama, H. Mitochondrial fission is an acute and adaptive response in injured motor neurons. Sci. Rep. 2016, 6, 28331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; Wani, W.Y.; Redmann, M.; Benavides, G.A.; Johnson, M.S.; Ouyang, X.; Cofield, S.S.; Mitra, K.; Darley-Usmar, V.; Zhang, J. Regulation of autophagy, mitochondrial dynamics, and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy 2017, 13, 1828–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, H.; John, A.; Brown, E.M.; Benedict, S.; Kambal, A. Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol. Appl. Pharmacol. 2008, 226, 161–168. [Google Scholar] [CrossRef]
- Al-Menhali, A.S.; Banu, S.; Angelova, P.R.; Barcaru, A.; Horvatovich, P.; Abramov, A.Y.; Jaganjac, M. Lipid peroxidation is involved in calcium dependent upregulation of mitochondrial metabolism in skeletal muscle. Gen. Subj. 2020, 1864, 129487. [Google Scholar] [CrossRef]
- Ugarte, N.; Petropoulos, I.; Friguet, B. Oxidized mitochondrial protein degradation and repair in aging and oxidative stress. Antioxid. Redox Signal. 2010, 13, 539–549. [Google Scholar] [CrossRef]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Administration of the Nrf2–ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free Radic. Biol. Medicine. 2013, 1, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y. Oxidative stress and cardiac repair/remodeling following infarction. Am. J. Med. Sci. 2007, 334, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Gomes, K.M.; Bechara, L.R.; Lima, V.M.; Ribeiro, M.A.; Campos, J.C.; Dourado, P.M.; Kowaltowski, A.J.; Mochly-Rosen, D.; Ferreira, J.C. Aldehydic load and aldehyde dehydrogenase 2 profile during the progression of post-myocardial infarction cardiomyopathy: Benefits of Alda-1. Int. J. Cardiol. 2015, 179, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Hao, Y.; Chen, H.; He, Q.; Yuan, Z.; Cheng, J. Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 2015, 6, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santin, Y.; Fazal, L.; Sainte-Marie, Y.; Sicard, P.; Maggiorani, D.; Tortosa, F.; Yücel, Y.Y.; Teyssedre, L.; Rouquette, J.; Marcellin, M. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa 2+ overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020, 27, 1907–1923. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.V.; Sandeep, N.; Paige, S.L.; Ranjbarvaziri, S.; Hu, D.Q.; Zhao, M.; Lan, I.S.; Coronado, M.; Kooiker, K.B.; Wu, S.M. 4HNE Impairs Myocardial Bioenergetics in Congenital Heart Disease-Induced Right Ventricular Failure. Circulation. Circulation. 2020, 142, 1667–1683. [Google Scholar] [CrossRef]
- Lashin, O.M.; Szweda, P.A.; Szweda, L.I.; Romani, A.M. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic. Biol. Med. 2006, 40, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Spencer, C.H.; Young, T.A.; Lively, M.O.; Cunningham, C.C. Effects of 4-hydroxynonenal on mitochondrial 3-hydroxy-3-methylglutaryl (HMG-CoA) synthase. Free Radic. Biol. Med. 2007, 43, 1499–1507. [Google Scholar] [CrossRef] [Green Version]
- Lord, T.; Martin, J.H.; Aitken, R.J. Accumulation of electrophilic aldehydes during postovulatory aging of mouse oocytes causes reduced fertility, oxidative stress, and apoptosis. Biol. Reprod. 2015, 92, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Miriyala, S.; Miao, L.; Mitov, M.; Schnell, D.; Dhar, S.K.; Cai, J.; Klein, J.B.; Sultana, R.; Butterfield, D.A.; et al. Redox proteomic identification of HNE-bound mitochondrial proteins in cardiac tissues reveals a systemic effect on energy metabolism after doxorubicin treatment. Free Radic. Biol. Med. 2014, 72, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Bhattarai, S.; Ara, H.; Sun, G.; St. Clair, D.K.; Bhuiyan, M.S.; Kevil, C.; Watts, M.N.; Dominic, P.; Shimizu, T.; et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol. 2020, 37, 101740. [Google Scholar] [CrossRef]
- Chandra, M.; Escalante-Alcalde, D.; Bhuiyan, M.S.; Orr, A.W.; Kevil, C.; Morris, A.J.; Nam, H.; Dominic, P.; McCarthy, K.J.; Miriyala, S.; et al. Cardiac-specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox Biol. 2018, 14, 261–271. [Google Scholar] [CrossRef]
- Bonora, E.; Muggeo, M. Postprandial blood glucose as a risk factor for cardiovascular disease in Type II diabetes: The epidemiological evidence. Diabetologia 2001, 44, 2107–2114. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.M.; Maddox, T.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 2015, 6, 1246–1258. [Google Scholar] [CrossRef]
- Cai, L.; Kang, Y.J. Oxidative stress and diabetic cardiomyopathy: A brief review. Cardiovasc. Toxicol. 2001, 1, 181–193. [Google Scholar] [CrossRef]
- Ansley, D.M.; Wang, B. Oxidative stress and myocardial injury in the diabetic heart. J. Pathol. 2013, 229, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, M.; Mukae, S.; Geshi, E.; Umetsu, K.; Nakatani, M.; Katagiri, T. Mitochondrial respiratory impairment in streptozotocin-induced diabetic rat heart. Jpn Circ. J. 1996, 60, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Cederbaum, A.I. Oxidative stress and alcoholic liver disease. Semin. Liver Dis. 2009, 29, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef]
- Andringa, K.K.; Udoh, U.S.; Landar, A.; Bailey, S.M. Proteomic analysis of 4-hydroxynonenal (4-HNE) modified proteins in liver mitochondria from chronic ethanol-fed rats. Redox Biol. 2014, 2, 1038–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihalas, B.P.; De Iuliis, G.N.; Redgrove, K.A.; McLaughlin, E.A.; Nixon, B. The lipid peroxidation product 4-hydroxynonenal contributes to oxidative stress-mediated deterioration of the ageing oocyte. Sci. Rep. 2017, 7, 6247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Sharma, R.; Sharma, A.; Awasthi, S.; Awasthi, Y.C. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim. Pol. 2003, 50, 319–336. [Google Scholar] [CrossRef]
- Ji, Y.; Dai, Z.L.; Wu, G.Y.; Wu, Z.L. 4-Hydroxy-2-nonenal induces apoptosis by activating ERK1/2 signaling and depleting intracellular glutathione in intestinal epithelial cells. Sci. Rep. 2016, 6, 32929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, G.; Pizzimenti, S.; Dianzani, M.U. 4-hydroxynonenal and regulation of cell cycle: Effects on the pRb/E2F pathway. Free Radic. Biol. Med. 2004, 37, 597–606. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhang, S.; Chan, J.Y.; Zhang, D.D. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell Biol. 2007, 27, 6334–6349. [Google Scholar] [CrossRef] [Green Version]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sano, M.; Shinmura, K.; Tamaki, K.; Katsumata, Y.; Matsuhashi, T.; Morizane, S.; Ito, H.; Hishiki, T.; Endo, J.; et al. 4-hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. J. Mol. Cell Cardiol. 2010, 49, 576–586. [Google Scholar] [CrossRef]
- Shoeb, M.; H Ansari, N.; K Srivastava, S.; V Ramana, K. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Forman, H.J. 4-hydroxynonenal-mediated signaling and aging. Free Radic. Biol. Med. 2017, 111, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Kutuk, O.; Basaga, H. Apoptosis signalling by 4-hydroxynonenal: A role for JNK-c-Jun/AP-1 pathway. Redox Rep. 2007, 12, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Usatyuk, P.V.; Natarajan, V. Hydroxyalkenals and oxidized phospholipids modulation of endothelial cytoskeleton, focal adhesion and adherens junction proteins in regulating endothelial barrier function. Microvasc. Res. 2012, 83, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usatyuk, P.V.; Parinandi, N.L.; Natarajan, V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J. Biol. Chem. 2006, 281, 35554–35566. [Google Scholar] [CrossRef] [Green Version]
- Vatsyayan, R.; Lelsani, P.C.R.; Chaudhary, P.; Kumar, S.; Awasthi, S.; Awasthi, Y.C. The expression and function of vascular endothelial growth factor in retinal pigment epithelial (RPE) cells is regulated by 4-hydroxynonenal (HNE) and glutathione S-transferaseA4-4. Biochem. Biophys. Res. Commun. 2012, 417, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Gargiulo, S.; Gamba, P.; Testa, G.; Rossin, D.; Biasi, F.; Poli, G.; Leonarduzzi, G. Relation between TLR4/NF-κB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal, and atherosclerotic plaque instability. Aging Cell 2015, 4, 569–581. [Google Scholar] [CrossRef]
- Dolinsky, V.W.; Chan, A.Y.; Robillard Frayne, I.; Light, P.E.; Des Rosiers, C.; Dyck, J.R. Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation 2009, 119, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Calamaras, T.D.; Lee, C.; Lan, F.; Ido, Y.; Siwik, D.A.; Colucci, W.S. Post-translational modification of serine/threonine kinase LKB1 via Adduction of the Reactive Lipid Species 4-Hydroxy-trans-2-nonenal (HNE) at lysine residue 97 directly inhibits kinase activity. J. Biol. Chem. 2012, 287, 42400–42406. [Google Scholar] [CrossRef] [Green Version]
- Calamaras, T.D.; Lee, C.; Lan, F.; Ido, Y.; Siwik, D.A.; Colucci, W.S. The lipid peroxidation product 4-hydroxy-trans-2-nonenal causes protein synthesis in cardiac myocytes via activated mTORC1-p70S6K-RPS6 signaling. Free Radic. Biol. Med. 2015, 82, 137–146. [Google Scholar] [CrossRef]
- Wang, X.; Proud, C.G. The mTOR Pathway in the Control of Protein Synthesis. Physiology 2006, 21, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Invest. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Rodrigues, G.A. Differential roles of AMPKα1 and AMPKα2 in regulating 4-HNE-induced RPE cell death and permeability. Exp. Eye Res. 2010, 91, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Sahin, K.; Tuzcu, M.; Gencoglu, H.; Dogukan, A.; Timurkan, M.; Sahin, N.; Aslan, A.; Kucuk, O. Epigallocatechin-3-gallate activates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Life Sci. 2010, 87, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Swiader, A.; Camare, C.; Guerby, P.; Salvayre, R.; Negre-Salvayre, A. 4-Hydroxynonenal Contributes to Fibroblast Senescence in Skin Photoaging Evoked by UV-A Radiation. Antioxidants 2021, 10, 365. [Google Scholar] [CrossRef]
- Cherubini, A.; Ruggiero, C.; Polidori, M.C.; Mecocci, P. Potential markers of oxidative stress in stroke. Free Radic. Biol. Med. 2005, 39, 841–852. [Google Scholar] [CrossRef]
- Bhattarai, S.; Sharma, S.; Ara, H.; Subedi, U.; Sun, G.; Li, C.; Bhuiyan, M.S.; Kevil, C.; Armstrong, W.P.; Minvielle, M.T.; et al. Disrupted Blood-Brain Barrier and Mitochondrial Impairment by Autotaxin-Lysophosphatidic Acid Axis in Postischemic Stroke. J. Am. Heart Assoc. 2021, 10, e021511. [Google Scholar] [CrossRef]
- Re, G.; Azzimondi, G.; Lanzarini, C.; Bassein, L.; Vaona, I.; Guarnieri, C. Plasma lipoperoxidative markers in ischaemic stroke suggest brain embolism. Eur. J. Emerg. Med. 1997, 4, 5–9. [Google Scholar]
- Lee, W.C.; Wong, H.Y.; Chai, Y.Y.; Shi, C.W.; Amino, N.; Kikuchi, S.; Huang, S.H. Lipid peroxidation dysregulation in ischemic stroke: Plasma 4-HNE as a potential biomarker? Biochem. Biophys. Res. Commun. 2012, 425, 842–847. [Google Scholar] [CrossRef]
- Sun, A.; Ren, J. ALDH2, a novel protector against stroke? Cell Res. 2013, 23, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Karlhuber, G.M.; Bauer, H.C.; Eckl, P.M. Cytotoxic and genotoxic effects of 4-hydroxynonenal in cerebral endothelial cells. Mutat. Res. 1997, 381, 209–216. [Google Scholar] [CrossRef]
- Eckl, P.M. Genotoxicity of HNE. Mol. Asp. Med. 2003, 24, 161–165. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.L.; Qi, S.H. ALDH2 Protects Against Ischemic Stroke in Rats by Facilitating 4-HNE Clearance and AQP4 Down-Regulation. Neurochem. Res. 2018, 43, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Mihalas, B.P.; Bromfield, E.G.; Sutherland, J.M.; De Iuliis, G.N.; McLaughlin, E.A.; Aitken, R.J.; Nixon, B. Oxidative damage in naturally aged mouse oocytes is exacerbated by dysregulation of proteasomal activity. J. Biol. Chem. 2018, 293, 18944–18964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.A.; Weinberg, A.; Hetherington, L.; Villaverde, A.I.; Velkov, T.; Baell, J.; Gordon, C.P. Defining the Mechanisms by Which the Reactive Oxygen Species By-Product, 4-Hydroxynonenal, Affects Human Sperm Cell Function. Biol. Reprod. 2015, 92, 1–10. [Google Scholar] [CrossRef]
- Aitken, R.J.; Whiting, S.; De Iuliis, G.N.; McClymont, S.; Mitchell, L.A.; Baker, M.A. Electrophilic aldehydes generated by sperm metabolism activate mitochondrial reactive oxygen species generation and apoptosis by targeting succinate dehydrogenase. J. Biol. Chem. 2012, 287, 33048–33060. [Google Scholar] [CrossRef] [Green Version]
- Bergheim, I.; Wolfgarten, E.; Bollschweiler, E.; Holscher, A.H.; Bode, C.; Parlesak, A. Cytochrome P450 levels are altered in patients with esophageal squamous-cell carcinoma. World J. Gastroenterol. 2007, 13, 997–1002. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef]
- Warnakulasuriya, S.; Parkkila, S.; Nagao, T.; Preedy, V.R.; Pasanen, M.; Koivisto, H.; Niemela, O. Demonstration of ethanol-induced protein adducts in oral leukoplakia (pre-cancer) and cancer. J. Oral Pathol. Med. 2008, 37, 157–165. [Google Scholar] [CrossRef]
- Xu, T.; Liu, S.; Ma, T.; Jia, Z.; Zhang, Z.; Wang, A. Aldehyde dehydrogenase 2 protects against oxidative stress associated with pulmonary arterial hypertension. Redox. Biol. 2017, 11, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Chen, H.M.; Zhao, X.Y.; Liu, M.; Jin, W.; Yan, W.; Wu, Y.F.; Tan, Z.B.; Fan, H.J.; Wu, Y.T.; et al. Alda-1, an aldehyde dehydrogenase-2 agonist, improves long-term survival in rats with chronic heart failure following myocardial infarction. Mol. Med. Rep. 2018, 18, 3159–3166. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Zhang, Q.; Luo, Q.; Ying, Y.; Liu, Y.; Li, Y.; Wei, W.; Yan, F.; Zhang, H. Alda-1 Attenuates Lung Ischemia-Reperfusion Injury by Reducing 4-Hydroxy-2-Nonenal in Alveolar Epithelial Cells. Crit. Care Med. 2016, 44, e544–e552. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, R.; Li, W.; Zhao, Y.; Yang, H.; Chen, H.; Jiang, H.; Dong, Z.; Hu, J.; Liu, J.; et al. Alda-1 treatment promotes the therapeutic effect of mitochondrial transplantation for myocardial ischemia-reperfusion injury. Bioact. Mater. 2021, 6, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, M.; Ushimaru, N.; Osada, N.; Oda, T.; Ishige, K.; Ito, Y. N-acetylcysteine selectively protects cerebellar granule cells from 4-hydroxynonenal-induced cell death. Neurosci. Res. 2006, 55, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 162. [Google Scholar] [CrossRef]
- Batinic-Haberle, I.; Tome, M.E. Thiol regulation by Mn porphyrins, commonly known as SOD mimics. Redox. Biol. 2019, 25, 101139. [Google Scholar] [CrossRef]
- Anselmo, W.; Branchetti, E.; Grau, J.B.; Li, G.; Ayoub, S.; Lai, E.K.; Rioux, N.; Tovmasyan, A.; Fortier, J.H.; Sacks, M.S. Porphyrin-Based SOD Mimic MnTnBu OE-2-PyP5+ Inhibits Mechanisms of Aortic Valve Remodeling in Human and Murine Models of Aortic Valve Sclerosis. J. Am. Heart Association. 2018, 7, e007861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Carroll, D.W.; You, Y.; Chaiswing, L.; Wen, R.; Batinic-Haberle, I.; Bondada, S.; Liang, Y.; St Clair, D.K. A novel redox regulator, MnTnBuOE-2-PyP(5+), enhances normal hematopoietic stem/progenitor cell function. Redox. Biol. 2017, 12, 129–138. [Google Scholar] [CrossRef]
- Batinic-Haberle, I.; Tovmasyan, A.; Huang, Z.; Duan, W.; Du, L.; Siamakpour-Reihani, S.; Cao, Z.; Sheng, H.; Spasojevic, I.; Alvarez Secord, A. H2O2-Driven Anticancer Activity of Mn Porphyrins and the Underlying Molecular Pathways. Oxid. Med. Cell Longev. 2021, 2021, 6653790. [Google Scholar] [CrossRef] [PubMed]
- Bromfield, E.G.; Mihalas, B.P.; Dun, M.D.; Aitken, R.J.; McLaughlin, E.A.; Walters, J.L.; Nixon, B. Inhibition of arachidonate 15-lipoxygenase prevents 4-hydroxynonenal-induced protein damage in male germ cells. Biol. Reprod. 2017, 96, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowicka-Bauer, K.; Nixon, B. Molecular Changes Induced by Oxidative Stress that Impair Human Sperm Motility. Antioxidants 2020, 9, 134. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Cell or Tissue | Mitochondria Affected | Adducted Proteins, Mitochondria | References |
|---|---|---|---|
| Myocardial tissue | Calcium accumulation inside mitochondria | VDAC and MCU | Santin, 2020 [66] |
| Myocardial tissue | Decrease in mitochondrial respiratory chain complex I and II activity | SDH | Lashin, 2006 [68] |
| Liver tissue | HMG-CoA synthase inactivation | HMG-CoA synthase | Patel, 2007 [69] |
| RV myocardial tissue | Decreased oxidative phosphorylation | NADH dehydrogenase [ubiquinone] iron–sulfur protein 2, elongation factor Tu, dihydrolipoyl dehydrogenase, ES1 protein homolog, fumarate hydratase, creatine kinase S-type, cytochrome b–c1 complex subunit 1, aconitate hydratase, NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 10, cytochrome c1, heme protein, stress-70 protein, superoxide dismutase | Hwang, 2020 [67] |
| Murine MII-stage oocyte | Loss of mitochondrial membrane potential | SDHA | Lord, 2015 [70] |
| Cardiac tissue | Inactivates the oxidoreductase activity of AIFm2 | AIFm2 | Miriyala, 2017 [7] |
| Cardiac tissue | Decreased oxygen consumption rate | NADH ATP synthase subunit, dihydrolipoyl dehydrogenase, succinate dehydrogenase [ubiquinone] flavoprotein subunit, trifunctional enzyme subunit α, creatine kinase S-type, cytoplasmic isoform of fumarate hydratase, succinyl-CoA:3-ketoacid–coenzyme A transferase 1 | Zhao, 2014 [71] |
| Myocardial tissue | Decreased oxygen consumption rate, decreased complex I and complex V activity | NDUFS2, SDHA, ATP5B, and DLD | Sharma, 2020 [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Sharma, P.; Bailey, T.; Bhattarai, S.; Subedi, U.; Miller, C.; Ara, H.; Kidambi, S.; Sun, H.; Panchatcharam, M.; et al. Electrophilic Aldehyde 4-Hydroxy-2-Nonenal Mediated Signaling and Mitochondrial Dysfunction. Biomolecules 2022, 12, 1555. https://doi.org/10.3390/biom12111555
Sharma S, Sharma P, Bailey T, Bhattarai S, Subedi U, Miller C, Ara H, Kidambi S, Sun H, Panchatcharam M, et al. Electrophilic Aldehyde 4-Hydroxy-2-Nonenal Mediated Signaling and Mitochondrial Dysfunction. Biomolecules. 2022; 12(11):1555. https://doi.org/10.3390/biom12111555
Chicago/Turabian StyleSharma, Sudha, Papori Sharma, Tara Bailey, Susmita Bhattarai, Utsab Subedi, Chloe Miller, Hosne Ara, Srivatsan Kidambi, Hong Sun, Manikandan Panchatcharam, and et al. 2022. "Electrophilic Aldehyde 4-Hydroxy-2-Nonenal Mediated Signaling and Mitochondrial Dysfunction" Biomolecules 12, no. 11: 1555. https://doi.org/10.3390/biom12111555
APA StyleSharma, S., Sharma, P., Bailey, T., Bhattarai, S., Subedi, U., Miller, C., Ara, H., Kidambi, S., Sun, H., Panchatcharam, M., & Miriyala, S. (2022). Electrophilic Aldehyde 4-Hydroxy-2-Nonenal Mediated Signaling and Mitochondrial Dysfunction. Biomolecules, 12(11), 1555. https://doi.org/10.3390/biom12111555