
PrimPol: A Breakthrough among DNA Replication Enzymes and a Potential New Target for Cancer Therapy

 ,
,  , and
, and
Abstract
:1. Introduction: PrimPol, a New Enzyme Working on DNA Replication
2. Similarities and Differences among Polymerizing Enzymes of DNA Replication: RNA Primases, DNA Polymerases, and PrimPol
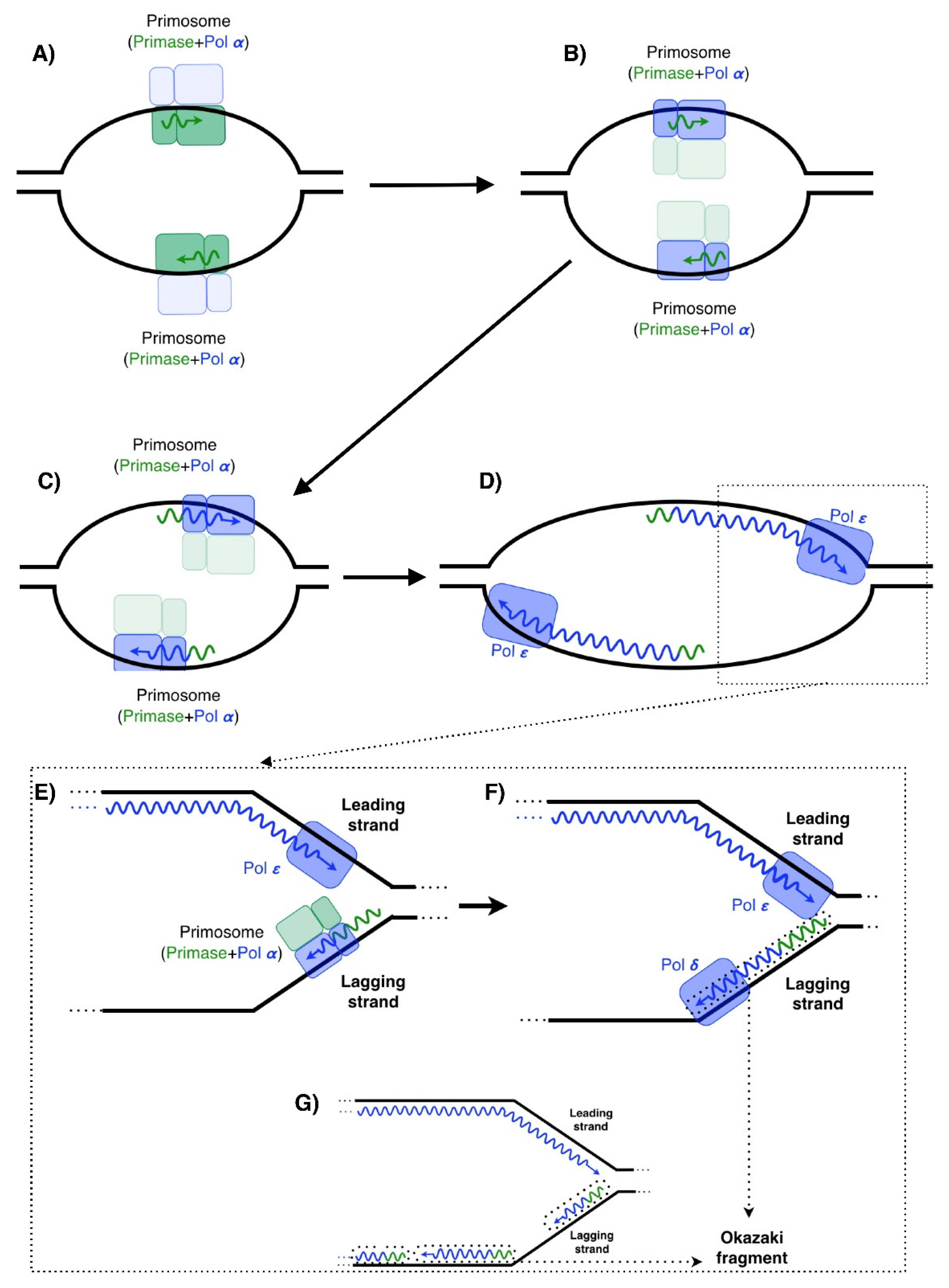
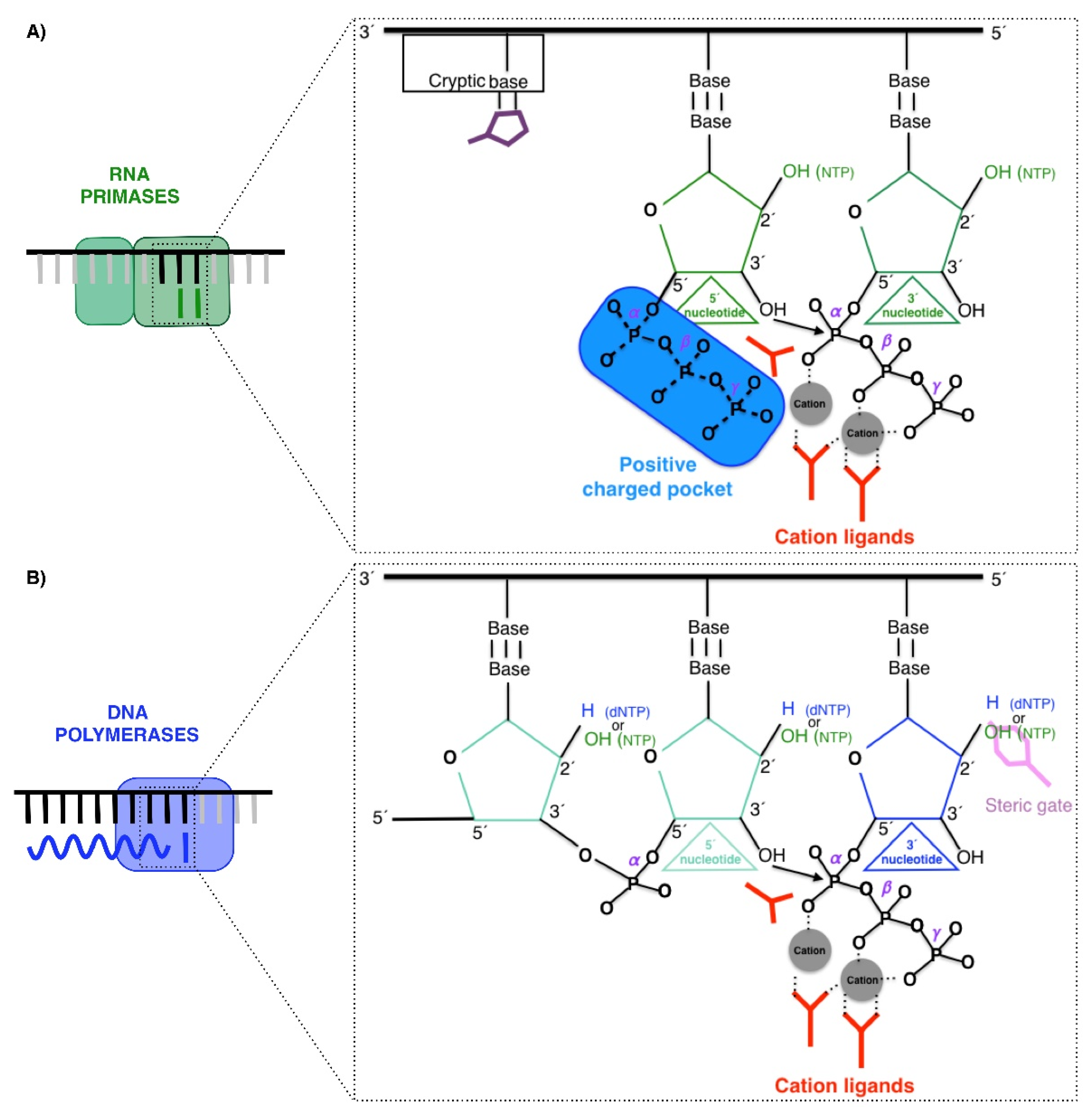
2.1. RNA Primases
2.2. Polymerases
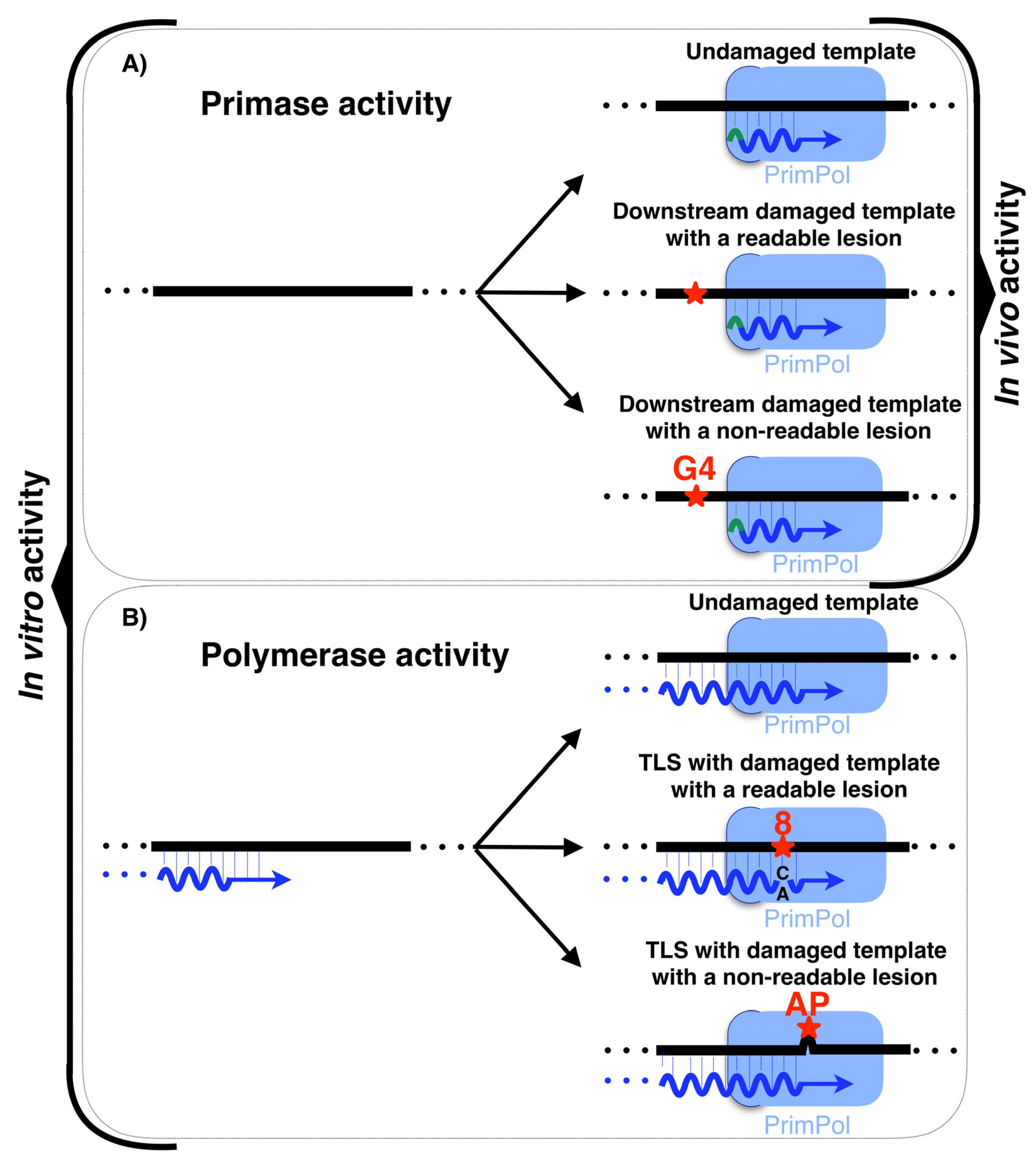
2.3. PrimPol
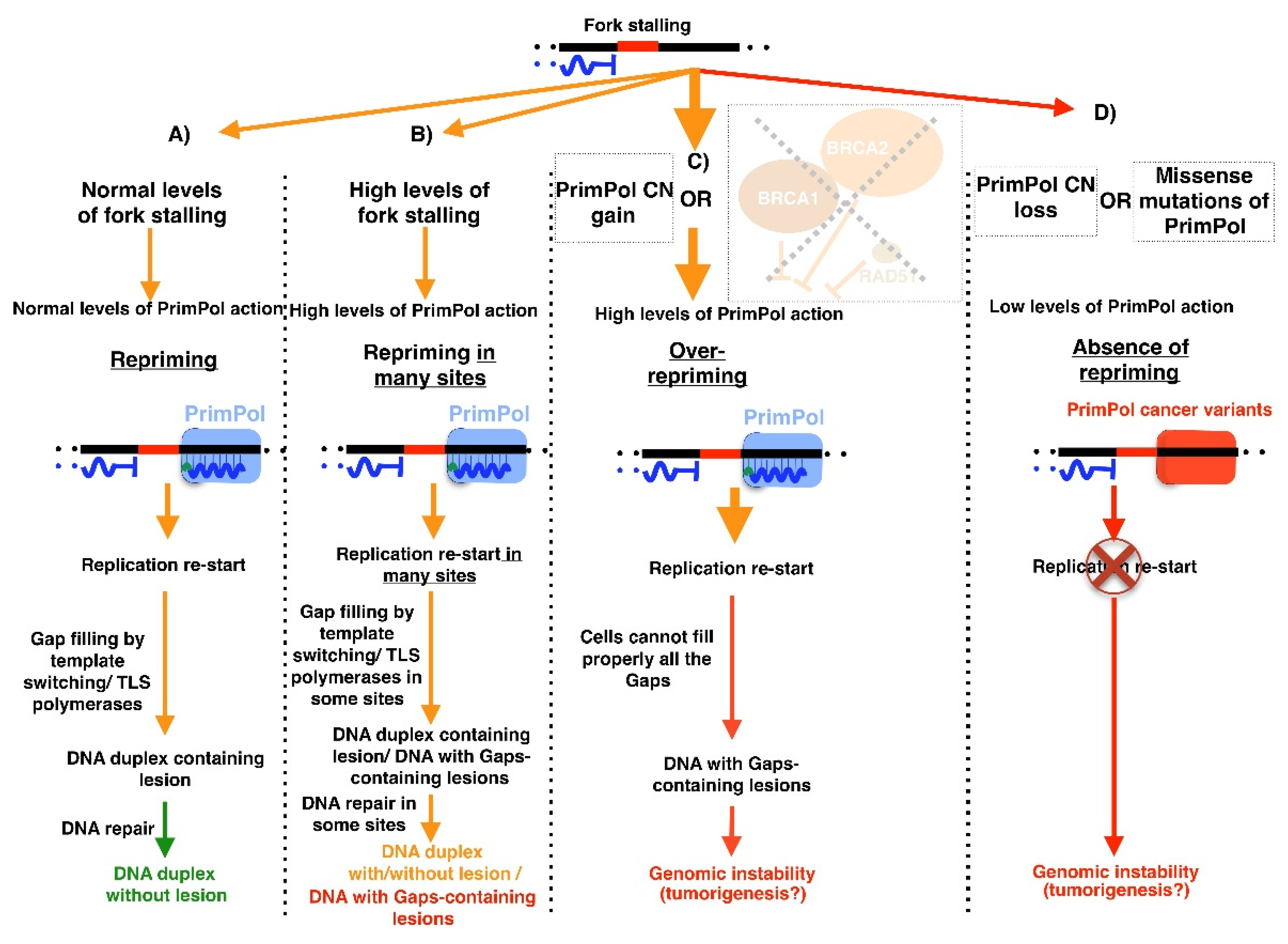
3. PrimPol: A New Player Alleviating Replication Stress
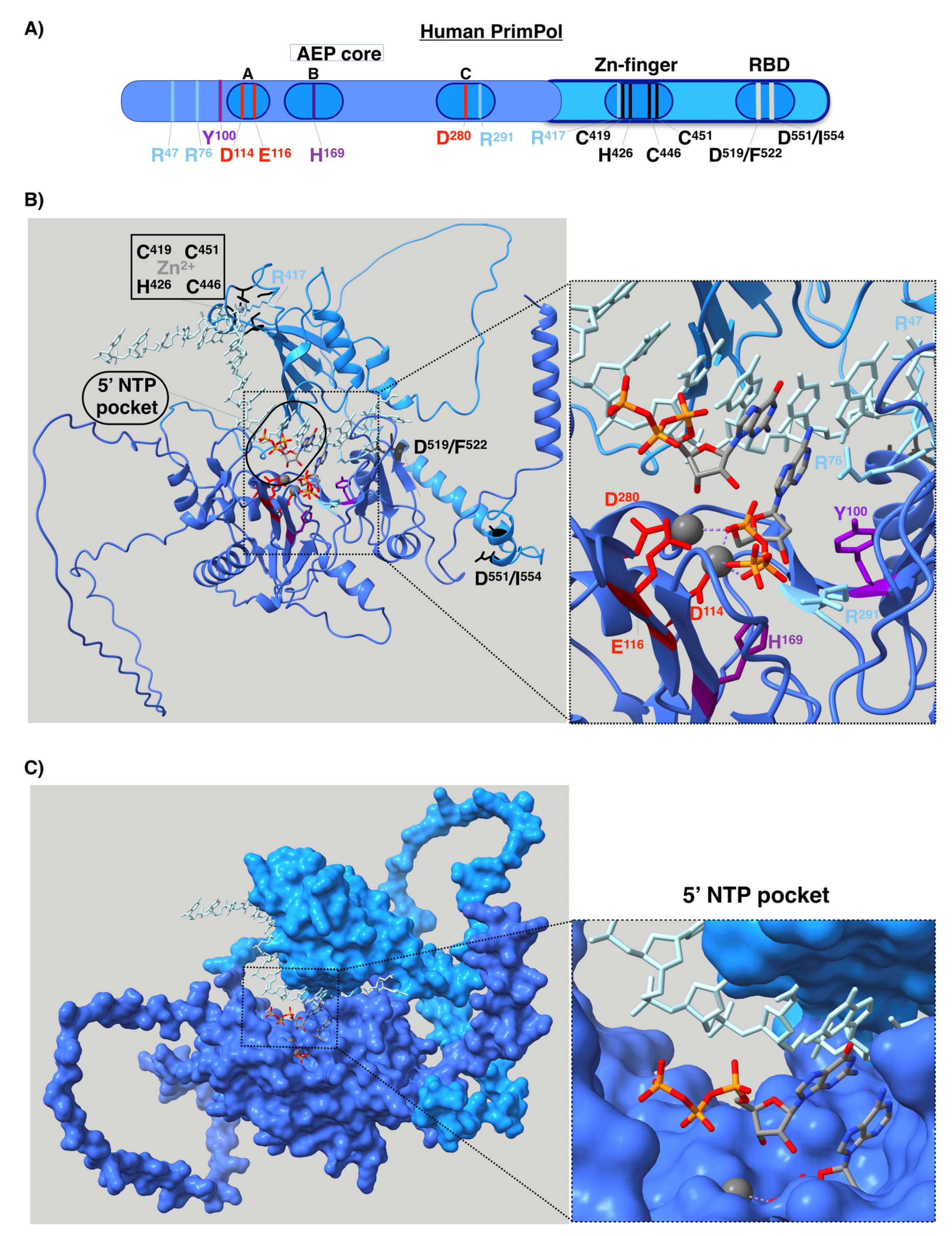
4. Structure and Regulation of Human PrimPol
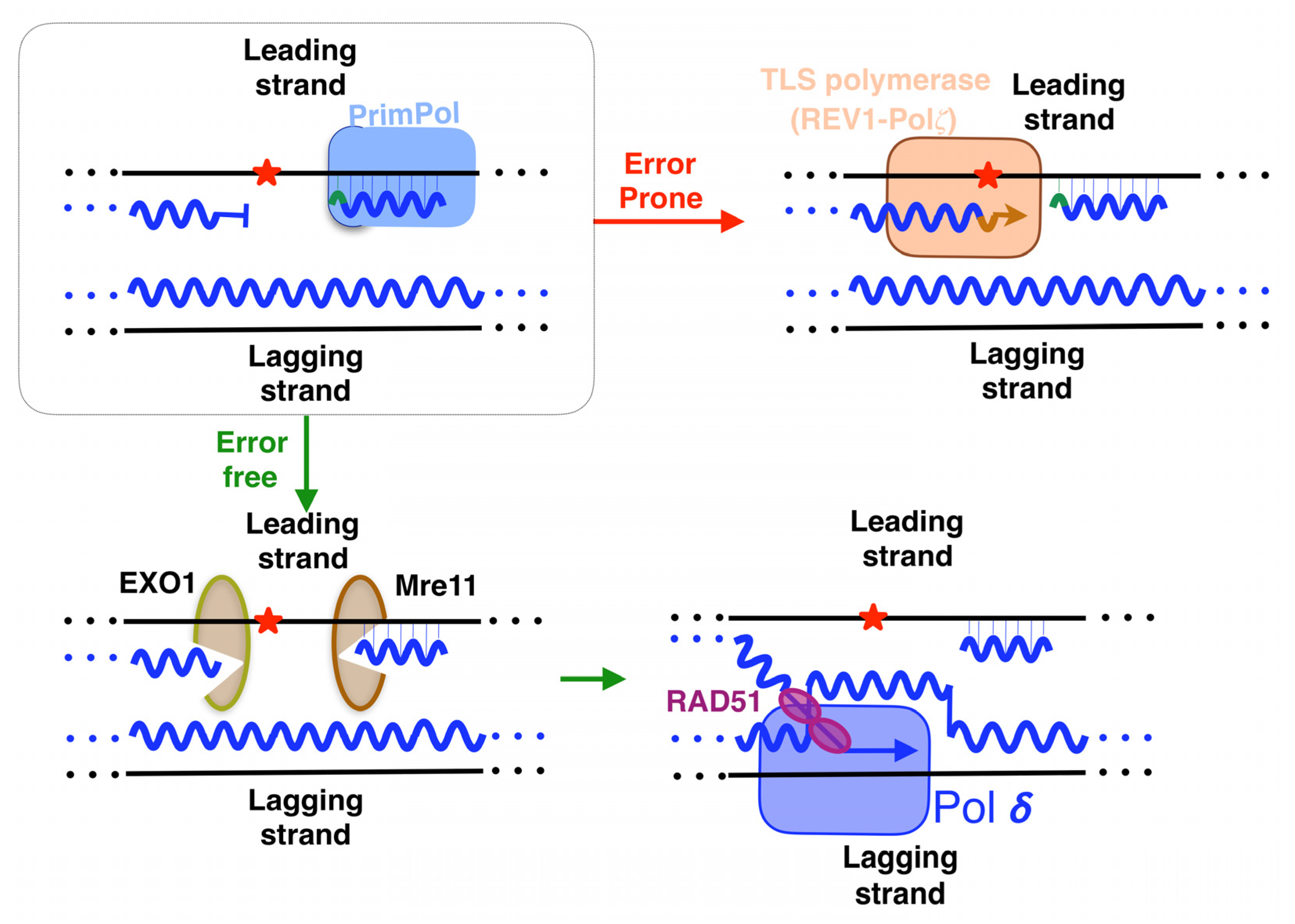
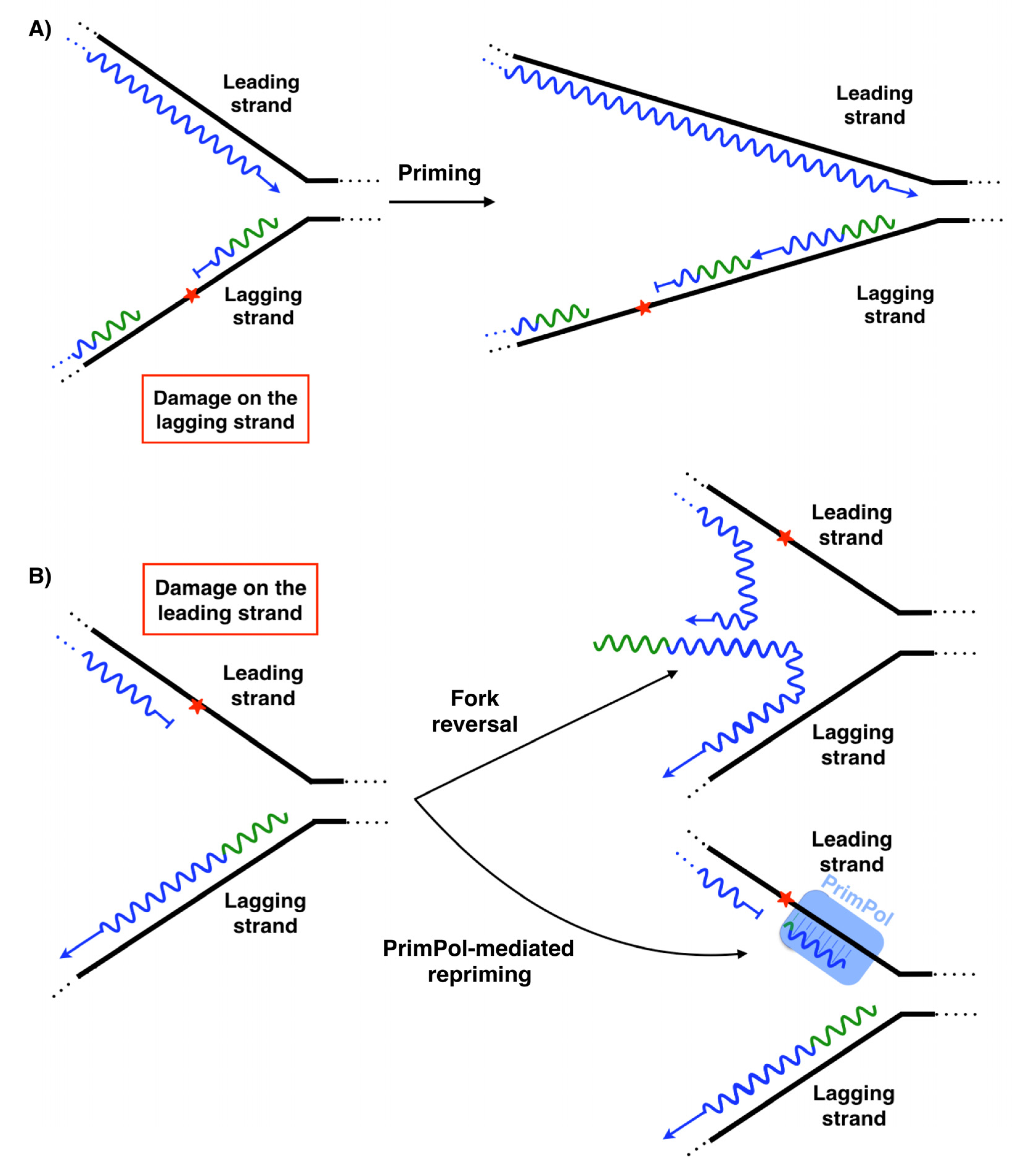
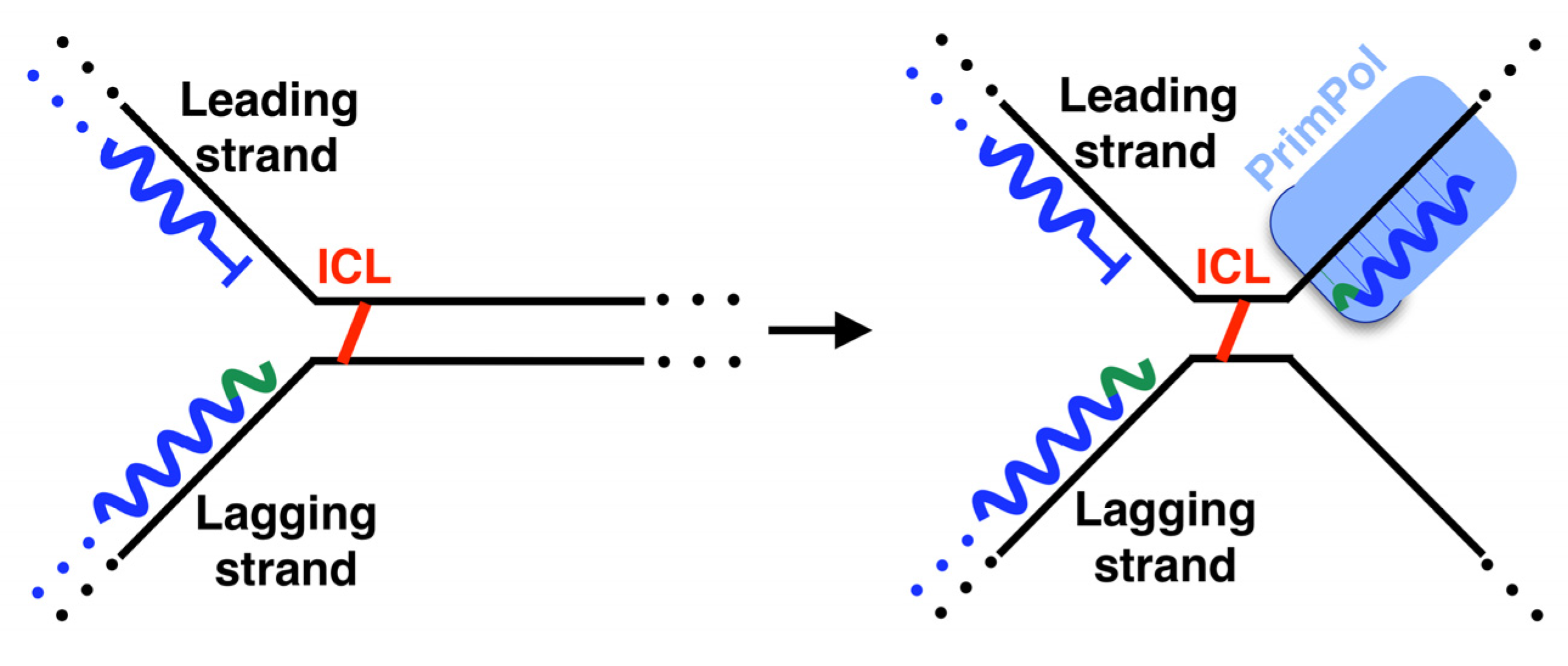
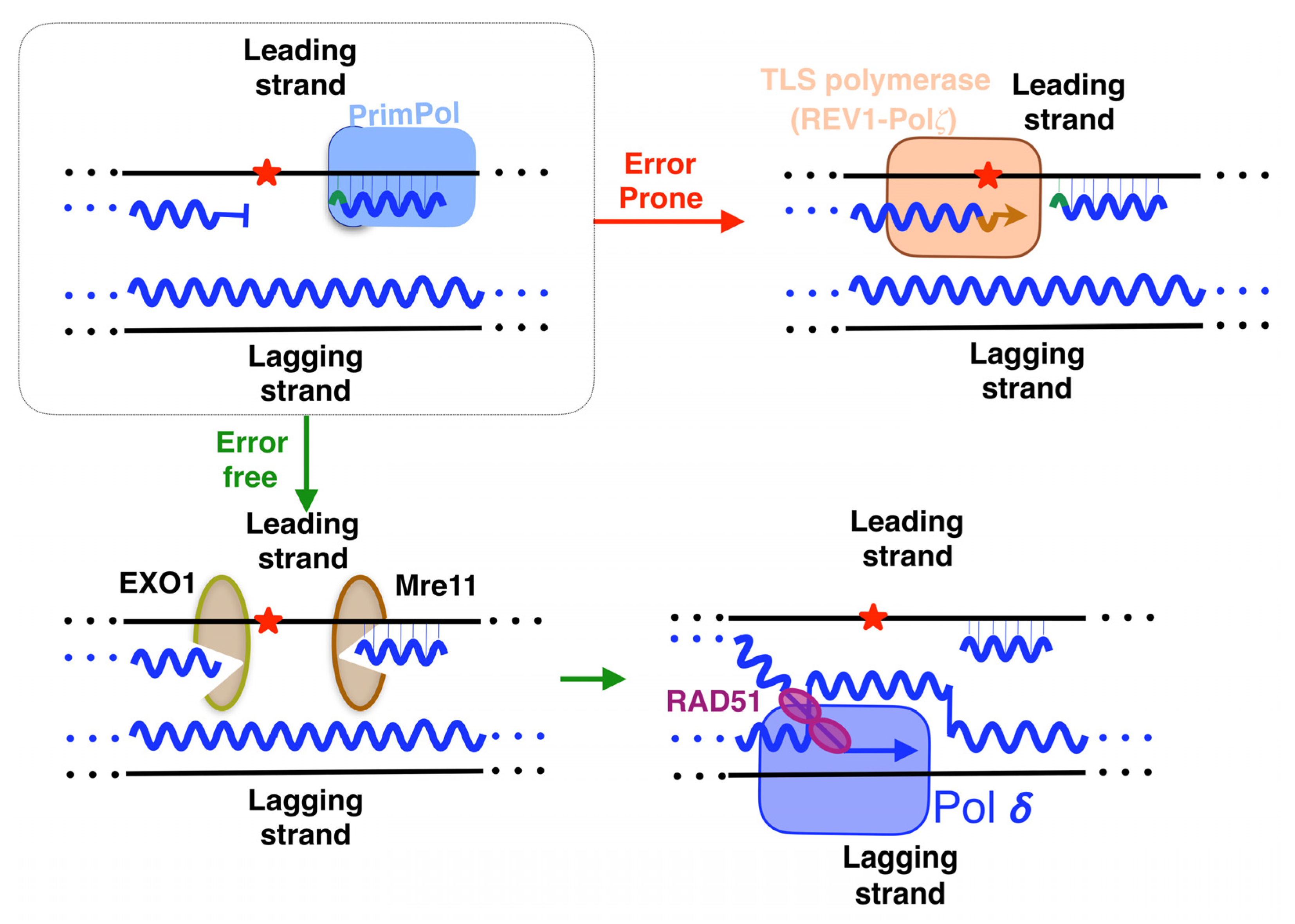
5. Lesion-Containing Gap Filling after the Repriming of PrimPol
6. Expression of Human PrimPol in Different Tissues
7. Role of PrimPol in Cancer and Its Potential Therapeutic Implication
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Structure Modeling of Human PrimPol
References
- Muzi-Falconi, M.; Giannattasio, M.; Foiani, M.; Plevani, P. The DNA Polymerase _-Primase Complex: Multiple Functions and Interactions. Sci. World J. 1900, 3, 575271. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L. The Pol α-Primase Complex. In The Eukaryotic Replisome: A Guide to Protein Structure and Function; Subcellular Biochemistry; MacNeill, S., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 157–169. ISBN 978-94-007-4572-8. [Google Scholar] [CrossRef]
- Baranovskiy, A.G.; Babayeva, N.D.; Zhang, Y.; Gu, J.; Suwa, Y.; Pavlov, Y.I.; Tahirov, T.H. Mechanism of Concerted RNA-DNA Primer Synthesis by the Human Primosome. J. Biol. Chem. 2016, 291, 10006–10020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranovskiy, A.G.; Zhang, Y.; Suwa, Y.; Gu, J.; Babayeva, N.D.; Pavlov, Y.I.; Tahirov, T.H. Insight into the Human DNA Primase Interaction with Template-Primer. J. Biol. Chem. 2016, 291, 4793–4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranovskiy, A.G.; Tahirov, T.H. Elaborated Action of the Human Primosome. Genes 2017, 8, 62. [Google Scholar] [CrossRef] [Green Version]
- Sinha, N.K.; Morris, C.F.; Alberts, B.M. Efficient In Vitro Replication of Double-Stranded DNA Templates by a Purified T4 Bacteriophage Replication System. J. Biol. Chem. 1980, 255, 4290–4303. [Google Scholar] [CrossRef]
- Chaffey NAlberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell. 4th Edn. Ann. Bot. 2003, 91, 401. [Google Scholar] [CrossRef]
- Miyabe, I.; Kunkel, T.A.; Carr, A.M. The Major Roles of DNA Polymerases Epsilon and Delta at the Eukaryotic Replication Fork Are Evolutionarily Conserved. PLoS Genet. 2011, 7, e1002407. [Google Scholar] [CrossRef] [Green Version]
- Lujan, S.A.; Williams, J.S.; Kunkel, T.A. DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol. 2016, 26, 640–654. [Google Scholar] [CrossRef] [Green Version]
- Nick McElhinny, S.A.; Gordenin, D.A.; Stith, C.M.; Burgers, P.M.J.; Kunkel, T.A. Division of Labor at the Eukaryotic Replication Fork. Mol. Cell 2008, 30, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, R.; Okazaki, T.; Sakabe, K.; Sugimoto, K.; Sugino, A. Mechanism of DNA Chain Growth. I. Possible Discontinuity and Unusual Secondary Structure of Newly Synthesized Chains. Proc. Natl. Acad. Sci. USA 1968, 59, 598–605. [Google Scholar] [CrossRef] [Green Version]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Mourón, S.; Rodriguez-Acebes, S.; Martínez-Jiménez, M.I.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Méndez, J. Repriming of DNA Synthesis at Stalled Replication Forks by Human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D.; et al. PrimPol Bypasses UV Photoproducts during Eukaryotic Chromosomal DNA Replication. Mol. Cell 2013, 52, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. HPrimpol1/CCDC111 Is a Human DNA Primase-Polymerase Required for the Maintenance of Genome Integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steitz, T.A.; Smerdon, S.J.; Jäger, J.; Joyce, C.M. A Unified Polymerase Mechanism for Nonhomologous DNA and RNA Polymerases. Science 1994, 266, 2022–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornberg, T.; Kornberg, A. 4. Bacterial DNA Polymerases. In The Enzymes; Protein Synthesis DNA Synthesis and Repair RNA Synthesis Energy-Linked ATPases Synthetases; Boyer, P.D., Ed.; Academic Press: Cambridge, MA, USA, 1974; Volume 10, pp. 119–144. [Google Scholar]
- Sawaya, M.R.; Pelletier, H.; Kumar, A.; Wilson, S.H.; Kraut, J. Crystal Structure of Rat DNA Polymerase β: Evidence for a Common Polymerase Mechanism. Science 1994, 264, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, H.; Sawaya, M.R.; Kumar, A.; Wilson, S.H.; Kraut, J. Structures of Ternary Complexes of Rat DNA Polymerase β, a DNA Template-Primer, and DdCTP. Science 1994, 264, 1891–1903. [Google Scholar] [CrossRef]
- Pelletier, H.; Sawaya, M.R.; Wolfle, W.; Wilson, S.H.; Kraut, J. Crystal Structures of Human DNA Polymerase β Complexed with DNA: Implications for Catalytic Mechanism, Processivity, and Fidelity. Biochemistry 1996, 35, 12742–12761. [Google Scholar] [CrossRef]
- Krayevsky, A.A. Chemical Reactions Catalyzed by DNA Polymerases. Russ. J. Bioorg. Chem. 2000, 26, 2–8. [Google Scholar] [CrossRef]
- Arezi, B.; Kuchta, R.D. Eukaryotic DNA Primase. Trends Biochem. Sci. 2000, 25, 572–576. [Google Scholar] [CrossRef]
- Kuchta, R.D.; Stengel, G. Mechanism and Evolution of DNA Primases. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 1180–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheaff, R.J.; Kuchta, R.D. Mechanism of Calf Thymus DNA Primase: Slow Initiation, Rapid Polymerization, and Intelligent Termination. Biochemistry 1993, 32, 3027–3037. [Google Scholar] [CrossRef] [PubMed]
- Traut, T.W. Physiological Concentrations of Purines and Pyrimidines. Mol. Cell Biochem 1994, 140, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Scherzinger, E.; Seiffert, D. Studies on Bacteriophage T7 DNA Synthesis In Vitro. Molec. Gen. Genet. 1975, 141, 213–232. [Google Scholar] [CrossRef]
- Scherzinger, E.; Lanka, E.; Morelli, G.; Seiffert, D.; Yuki, A. Bacteriophage-T7-Induced DNA-Priming Protein. Eur. J. Biochem. 1977, 72, 543–558. [Google Scholar] [CrossRef]
- Rowen, L.; Kornberg, A. Primase, the DnaG Protein of Escherichia Coli. An Enzyme Which Starts DNA Chains. J. Biol. Chem. 1978, 253, 758–764. [Google Scholar] [CrossRef]
- Garcia-Diaz, M.; Bebenek, K. Multiple Functions of DNA Polymerases. Crit. Rev. Plant Sci. 2007, 26, 105–122. [Google Scholar] [CrossRef] [Green Version]
- Brutlag, D.; Schekman, R.; Kornberg, A. A Possible Role for RNA Polymerase in the Initiation of M13 DNA Synthesis. Proc. Natl. Acad. Sci. USA 1971, 68, 2826–2829. [Google Scholar] [CrossRef] [Green Version]
- Kornberg, A.; Baker, T.A. DNA Replication; University Science Books; AIP Publishing LLC: Melville, NY, USA, 2005; ISBN 978-1-891389-44-3. [Google Scholar]
- Salas, M. Protein-Priming of Dna Replication. Annu. Rev. Biochem. 1991, 60, 39–71. [Google Scholar] [CrossRef]
- Zharkov, D.O. Base Excision DNA Repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef]
- Ramsden, D.A.; Asagoshi, K. DNA Polymerases in Nonhomologous End Joining: Are There Any Benefits to Standing out from the Crowd? Environ. Mol. Mutagenesis 2012, 53, 741–751. [Google Scholar] [CrossRef] [PubMed]
- McElhinny, S.A.N.; Kumar, D.; Clark, A.B.; Watt, D.L.; Watts, B.E.; Lundström, E.-B.; Johansson, E.; Chabes, A.; Kunkel, T.A. Genome Instability Due to Ribonucleotide Incorporation into DNA. Nat. Chem. Biol. 2010, 6, 774–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, A.R.; Zhang, S.; Burgers, P.M.; Lee, M.Y.; Kunkel, T.A. Ribonucleotide Incorporation, Proofreading and Bypass by Human DNA Polymerase δ. DNA Repair 2013, 12, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, C.M. Choosing the Right Sugar: How Polymerases Select a Nucleotide Substrate. Proc. Natl. Acad. Sci. USA 1997, 94, 1619–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.A.; Suo, Z. Unlocking the Sugar “Steric Gate” of DNA Polymerases. Biochemistry 2011, 50, 1135–1142. [Google Scholar] [CrossRef] [Green Version]
- Reijns, M.A.M.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic Removal of Ribonucleotides from DNA Is Essential for Mammalian Genome Integrity and Development. Cell 2012, 149, 1008–1022. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.S.; Clausen, A.R.; Nick McElhinny, S.A.; Watts, B.E.; Johansson, E.; Kunkel, T.A. Proofreading of Ribonucleotides Inserted into DNA by Yeast DNA Polymerase ɛ. DNA Repair 2012, 11, 649–656. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.S.; Lujan, S.A.; Kunkel, T.A. Processing Ribonucleotides Incorporated during Eukaryotic DNA Replication. Nat. Rev. Mol. Cell Biol. 2016, 17, 350–363. [Google Scholar] [CrossRef] [Green Version]
- Bernad, A.; Blanco, L.; Lázaro, J.M.; Martín, G.; Salas, M. A Conserved 3′→5′ Exonuclease Active Site in Prokaryotic and Eukaryotic DNA Polymerases. Cell 1989, 59, 219–228. [Google Scholar] [CrossRef]
- Bebenek, K.; Kunkel, T.A. Functions of DNA Polymerases. In Advances in Protein Chemistry; DNA Repair and Replication; Academic Press: Cambridge, MA, USA, 2004; Volume 69, pp. 137–165. [Google Scholar] [CrossRef]
- Skerra, A. Phosphorothioate Primers Improve the Amplification of DNA Sequences by DNA Polymerases with Proofreading Activity. Nucleic Acids Res. 1992, 20, 3551–3554. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, Y.I.; Frahm, C.; McElhinny, S.A.N.; Niimi, A.; Suzuki, M.; Kunkel, T.A. Evidence That Errors Made by DNA Polymerase α Are Corrected by DNA Polymerase δ. Curr. Biol. 2006, 16, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElhinny, S.A.N.; Kissling, G.E.; Kunkel, T.A. Differential Correction of Lagging-Strand Replication Errors Made by DNA Polymerases α and δ. Proc. Natl. Acad. Sci. USA 2010, 107, 21070–21075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lujan, S.A.; Williams, J.S.; Pursell, Z.F.; Abdulovic-Cui, A.A.; Clark, A.B.; McElhinny, S.A.N.; Kunkel, T.A. Mismatch Repair Balances Leading and Lagging Strand DNA Replication Fidelity. PLoS Genet. 2012, 8, e1003016. [Google Scholar] [CrossRef] [PubMed]
- Koc, K.N.; Stodola, J.L.; Burgers, P.M.; Galletto, R. Regulation of Yeast DNA Polymerase δ-Mediated Strand Displacement Synthesis by 5′-Flaps. Nucleic Acids Res. 2015, 43, 4179–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, H.-I.; Bambara, R.A. The Protein Components and Mechanism of Eukaryotic Okazaki Fragment Maturation. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 433–452. [Google Scholar] [CrossRef]
- Ayyagari, R.; Gomes, X.V.; Gordenin, D.A.; Burgers, P.M.J. Okazaki Fragment Maturation in Yeast: I. DISTRIBUTION OF FUNCTIONS BETWEEN FEN1 AND DNA2. J. Biol. Chem. 2003, 278, 1618–1625. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Shen, B. Okazaki Fragment Maturation: Nucleases Take Centre Stage. J. Mol. Cell Biol. 2011, 3, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Reijns, M.A.M.; Kemp, H.; Ding, J.; Marion de Procé, S.; Jackson, A.P.; Taylor, M.S. Lagging-Strand Replication Shapes the Mutational Landscape of the Genome. Nature 2015, 518, 502–506. [Google Scholar] [CrossRef]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056.e10. [Google Scholar] [CrossRef] [Green Version]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A Panoply of Errors: Polymerase Proofreading Domain Mutations in Cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA Polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-D.; Cuevas, I.; Zhang, M.; Lu, C.; Alam, M.M.; Fu, Y.-X.; You, M.J.; Akbay, E.A.; Zhang, H.; Castrillon, D.H. Polymerase-Mediated Ultramutagenesis in Mice Produces Diverse Cancers with High Mutational Load. J. Clin. Invest. 2018, 128, 4179–4191. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Nicolay, N.H.; Sharma, R.A. Biological and Therapeutic Relevance of Nonreplicative DNA Polymerases to Cancer. Antioxid. Redox Signal. 2013, 18, 851–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (Xeroderma Pigmentosum Variant) Gene Encodes Human DNA Polymerase η. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef]
- Johnson, R.E.; Kondratick, C.M.; Prakash, S.; Prakash, L. HRAD30 Mutations in the Variant Form of Xeroderma Pigmentosum. Science 1999, 285, 263–265. [Google Scholar] [CrossRef]
- McDonald, J.P.; Frank, E.G.; Plosky, B.S.; Rogozin, I.B.; Masutani, C.; Hanaoka, F.; Woodgate, R.; Gearhart, P.J. 129-Derived Strains of Mice Are Deficient in DNA Polymerase ι and Have Normal Immunoglobulin Hypermutation. J. Exp. Med. 2003, 198, 635–643. [Google Scholar] [CrossRef]
- Suzuki, N.; Ohashi, E.; Kolbanovskiy, A.; Geacintov, N.E.; Grollman, A.P.; Ohmori, H.; Shibutani, S. Translesion Synthesis by Human DNA Polymerase κ on a DNA Template Containing a Single Stereoisomer of DG-(+)- or DG-(−)-Anti-N2-BPDE (7,8-Dihydroxy-Anti-9,10-Epoxy-7,8,9,10-Tetrahydrobenzo[a]Pyrene). Biochemistry 2002, 41, 6100–6106. [Google Scholar] [CrossRef]
- Rechkoblit, O.; Zhang, Y.; Guo, D.; Wang, Z.; Amin, S.; Krzeminsky, J.; Louneva, N.; Geacintov, N.E. Trans-Lesion Synthesis Past Bulky Benzo[a]Pyrene Diol Epoxide N2-DG and N6-DA Lesions Catalyzed by DNA Bypass Polymerases. J. Biol. Chem. 2002, 277, 30488–30494. [Google Scholar] [CrossRef] [Green Version]
- Larimer, F.W.; Perry, J.R.; Hardigree, A.A. The REV1 Gene of Saccharomyces Cerevisiae: Isolation, Sequence, and Functional Analysis. J. Bacteriol. 1989, 171, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Deoxycytidyl Transferase Activity of Yeast REV1 Protein. Nature 1996, 382, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Mudrak, S.V.; Jinks-Robertson, S. The DCMP Transferase Activity of Yeast Rev1 Is Biologically Relevant during the Bypass of Endogenously Generated AP Sites. DNA Repair 2011, 10, 1262–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Thymine-Thymine Dimer Bypass by Yeast DNA Polymerase ζ. Science 1996, 272, 1646–1649. [Google Scholar] [CrossRef] [PubMed]
- García-Díaz, M.; Domínguez, O.; López-Fernández, L.A.; de Lera, L.T.; Saníger, M.L.; Ruiz, J.F.; Párraga, M.; García-Ortiz, M.J.; Kirchhoff, T.; del Mazo, J.; et al. DNA Polymerase Lambda (Pol λ), a Novel Eukaryotic DNA Polymerase with a Potential Role in Meiosis11Edited by M. Yaniv. J. Mol. Biol. 2000, 301, 851–867. [Google Scholar] [CrossRef]
- Domínguez, O.; Ruiz, J.F.; de Lera, T.L.; García-Díaz, M.; González, M.A.; Kirchhoff, T.; Martínez-A, C.; Bernad, A.; Blanco, L. DNA Polymerase Mu (Pol μ), Homologous to TdT, Could Act as a DNA Mutator in Eukaryotic Cells. EMBO J. 2000, 19, 1731–1742. [Google Scholar] [CrossRef]
- Ruiz, J.F.; Domínguez, O.; Laín de Lera, T.; Garcia-Díaz, M.; Bernad, A.; Blanco, L. DNA Polymerase Mu, a Candidate Hypermutase? Philos. Trans. R Soc. Lond. B Biol. Sci. 2001, 356, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Marini, F.; Wood, R.D. POLQ (Pol θ), a DNA Polymerase and DNA-dependent ATPase in Human Cells. Nucleic Acids Res. 2003, 31, 6117–6126. [Google Scholar] [CrossRef] [Green Version]
- Blanco, L.; Calvo, P.A.; Diaz-Talavera, A.; Carvalho, G.; Calero, N.; Martínez-Carrón, A.; Velázquez-Ruiz, C.; Villadangos, S.; Guerra, S.; Martínez-Jiménez, M.I. Chapter Nine-Mechanism of DNA Primer Synthesis by Human PrimPol. In The Enzymes; Zhao, L., Kaguni, L.S., Eds.; DNA Repair; Academic Press: Cambridge, MA, USA, 2019; Volume 45, pp. 289–310. [Google Scholar] [CrossRef]
- Boldinova, E.O.; Belousova, E.A.; Gagarinskaya, D.I.; Maltseva, E.A.; Khodyreva, S.N.; Lavrik, O.I.; Makarova, A.V. Strand Displacement Activity of PrimPol. Int. J. Mol. Sci. 2020, 21, 9027. [Google Scholar] [CrossRef]
- Díaz-Talavera, A.; Calvo, P.A.; González-Acosta, D.; Díaz, M.; Sastre-Moreno, G.; Blanco-Franco, L.; Guerra, S.; Martínez-Jiménez, M.I.; Méndez, J.; Blanco, L. A Cancer-Associated Point Mutation Disables the Steric Gate of Human PrimPol. Sci. Rep. 2019, 9, 1121. [Google Scholar] [CrossRef] [Green Version]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Jiménez, M.I.; García-Gómez, S.; Bebenek, K.; Sastre-Moreno, G.; Calvo, P.A.; Díaz-Talavera, A.; Kunkel, T.A.; Blanco, L. Alternative Solutions and New Scenarios for Translesion DNA Synthesis by Human PrimPol. DNA Repair 2015, 29, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.K.; Ketkar, A.; Lodeiro, M.F.; Cameron, C.E.; Eoff, R.L. Kinetic Analysis of Human PrimPol DNA Polymerase Activity Reveals a Generally Error-Prone Enzyme Capable of Accurately Bypassing 7,8-Dihydro-8-Oxo-2′-Deoxyguanosine. Biochemistry 2014, 53, 6584–6594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, A.V.; Boldinova, E.O.; Belousova, E.A.; Lavrik, O.I. In Vitro Lesion Bypass by Human PrimPol. DNA Repair 2018, 70, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Calvo, P.A.; Sastre-Moreno, G.; Perpiñá, C.; Guerra, S.; Martínez-Jiménez, M.I.; Blanco, L. The Invariant Glutamate of Human PrimPol DxE Motif Is Critical for Its Mn2+-Dependent Distinctive Activities. DNA Repair 2019, 77, 65–75. [Google Scholar] [CrossRef]
- Rechkoblit, O.; Johnson, R.E.; Gupta, Y.K.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural Basis of DNA Synthesis Opposite 8-Oxoguanine by Human PrimPol Primase-Polymerase. Nat. Commun. 2021, 12, 4020. [Google Scholar] [CrossRef]
- Shilkin, E.S.; Petrova, D.V.; Poltorachenko, V.A.; Boldinova, E.O.; Zharkov, D.O.; Makarova, A.V. Template Properties of 5-Methyl-2’-Deoxycytidine and 5-Hydroxymethyl-2’-Deoxycytidine in Reactions with Human Translesion and Reparative DNA Polymerases. Mol. Biol. 2021, 55, 267–272. [Google Scholar] [CrossRef]
- Boldinova, E.O.; Yudkina, A.V.; Shilkin, E.S.; Gagarinskaya, D.I.; Baranovskiy, A.G.; Tahirov, T.H.; Zharkov, D.O.; Makarova, A.V. Translesion Activity of PrimPol on DNA with Cisplatin and DNA–Protein Cross-Links. Sci. Rep. 2021, 11, 17588. [Google Scholar] [CrossRef]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication Stress and Cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Heller, R.C.; Marians, K.J. Replication Fork Reactivation Downstream of a Blocked Nascent Leading Strand. Nature 2006, 439, 557–562. [Google Scholar] [CrossRef]
- Heller, R.C.; Marians, K.J. Replisome Assembly and the Direct Restart of Stalled Replication Forks. Nat. Rev. Mol. Cell Biol. 2006, 7, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.P.; Poli, J.; Marians, K.J.; Pasero, P. Rescuing Stalled or Damaged Replication Forks. Cold Spring Harb. Perspect. Biol. 2013, 5, a012815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.R.G.; Yeeles, J.T.P. The Initial Response of a Eukaryotic Replisome to DNA Damage. Mol. Cell 2018, 70, 1067–1080.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication Fork Reversal in Eukaryotes: From Dead End to Dynamic Response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Fugger, K.; Mistrik, M.; Neelsen, K.J.; Yao, Q.; Zellweger, R.; Kousholt, A.N.; Haahr, P.; Chu, W.K.; Bartek, J.; Lopes, M.; et al. FBH1 Catalyzes Regression of Stalled Replication Forks. Cell Rep. 2015, 10, 1749–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork Reversal, Fork Protection, and Genome Stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef]
- Amunugama, R.; Willcox, S.; Wu, R.A.; Abdullah, U.B.; El-Sagheer, A.H.; Brown, T.; McHugh, P.J.; Griffith, J.D.; Walter, J.C. Replication Fork Reversal during DNA Interstrand Crosslink Repair Requires CMG Unloading. Cell Rep. 2018, 23, 3419–3428. [Google Scholar] [CrossRef]
- Torregrosa-Muñumer, R.; Forslund, J.M.E.; Goffart, S.; Pfeiffer, A.; Stojkovič, G.; Carvalho, G.; Al-Furoukh, N.; Blanco, L.; Wanrooij, S.; Pohjoismäki, J.L.O. PrimPol Is Required for Replication Reinitiation after MtDNA Damage. Proc. Natl. Acad. Sci. USA 2017, 114, 11398–11403. [Google Scholar] [CrossRef] [Green Version]
- Bailey, L.J.; Bianchi, J.; Doherty, A.J. PrimPol Is Required for the Maintenance of Efficient Nuclear and Mitochondrial DNA Replication in Human Cells. Nucleic Acids Res. 2019, 47, 4026–4038. [Google Scholar] [CrossRef]
- Bailey, L.J.; Doherty, A.J. Mitochondrial DNA Replication: A PrimPol Perspective. Biochem. Soc. Trans. 2017, 45, 513–529. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Guilliam, T.A.; Tsuda, M.; Yamamoto, J.; Bailey, L.J.; Iwai, S.; Takeda, S.; Doherty, A.J.; Hirota, K. Repriming by PrimPol Is Critical for DNA Replication Restart Downstream of Lesions and Chain-Terminating Nucleosides. Cell Cycle 2016, 15, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, D.; Jozwiakowski, S.K.; Romanello, M.; Guilbaud, G.; Guilliam, T.A.; Bailey, L.J.; Sale, J.E.; Doherty, A.J. PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol. Cell 2016, 61, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šviković, S.; Crisp, A.; Tan-Wong, S.M.; Guilliam, T.A.; Doherty, A.J.; Proudfoot, N.J.; Guilbaud, G.; Sale, J.E. R-Loop Formation during S Phase Is Restricted by PrimPol-Mediated Repriming. EMBO J. 2019, 38, e99793. [Google Scholar] [CrossRef] [PubMed]
- Duong, V.N.; Zhou, L.; Martínez-Jiménez, M.I.; He, L.; Cosme, M.; Blanco, L.; Paintsil, E.; Anderson, K.S. Identifying the Role of PrimPol in TDF-Induced Toxicity and Implications of Its Loss of Function Mutation in an HIV+ Patient. Sci. Rep. 2020, 10, 9343. [Google Scholar] [CrossRef]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef] [PubMed]
- Piberger, A.L.; Bowry, A.; Kelly, R.D.W.; Walker, A.K.; González-Acosta, D.; Bailey, L.J.; Doherty, A.J.; Méndez, J.; Morris, J.R.; Bryant, H.E.; et al. PrimPol-Dependent Single-Stranded Gap Formation Mediates Homologous Recombination at Bulky DNA Adducts. Nat. Commun. 2020, 11, 5863. [Google Scholar] [CrossRef] [PubMed]
- González-Acosta, D.; Blanco-Romero, E.; Ubieto-Capella, P.; Mutreja, K.; Míguez, S.; Llanos, S.; García, F.; Muñoz, J.; Blanco, L.; Lopes, M.; et al. PrimPol-Mediated Repriming Facilitates Replication Traverse of DNA Interstrand Crosslinks. EMBO J. 2021, 40, e106355. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Tirman, S.; Jackson, J.; Šviković, S.; Lemaçon, D.; Carvajal-Maldonado, D.; González-Acosta, D.; Vessoni, A.T.; Cybulla, E.; Wood, M.; et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 2020, 77, 461–474.e9. [Google Scholar] [CrossRef]
- Bai, G.; Kermi, C.; Stoy, H.; Schiltz, C.J.; Bacal, J.; Zaino, A.M.; Hadden, M.K.; Eichman, B.F.; Lopes, M.; Cimprich, K.A. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol. Cell 2020, 78, 1237–1251.e7. [Google Scholar] [CrossRef]
- Genois, M.-M.; Gagné, J.-P.; Yasuhara, T.; Jackson, J.; Saxena, S.; Langelier, M.-F.; Ahel, I.; Bedford, M.T.; Pascal, J.M.; Vindigni, A.; et al. CARM1 Regulates Replication Fork Speed and Stress Response by Stimulating PARP1. Mol. Cell 2021, 81, 784–800.e8. [Google Scholar] [CrossRef]
- Guilliam, T.A.; Jozwiakowski, S.K.; Ehlinger, A.; Barnes, R.P.; Rudd, S.G.; Bailey, L.J.; Skehel, J.M.; Eckert, K.A.; Chazin, W.J.; Doherty, A.J. Human PrimPol Is a Highly Error-Prone Polymerase Regulated by Single-Stranded DNA Binding Proteins. Nucleic Acids Res. 2015, 43, 1056–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rechkoblit, O.; Gupta, Y.K.; Malik, R.; Rajashankar, K.R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structure and Mechanism of Human PrimPol, a DNA Polymerase with Primase Activity. Sci. Adv. 2016, 2, e1601317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldinova, E.O.; Stojkovič, G.; Khairullin, R.; Wanrooij, S.; Makarova, A.V. Optimization of the Expression, Purification and Polymerase Activity Reaction Conditions of Recombinant Human PrimPol. PLoS ONE 2017, 12, e0184489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarsky, E.J.; Wallenmeyer, P.C.; Phi, K.K.; Suo, Z. Significant Impact of Divalent Metal Ions on the Fidelity, Sugar Selectivity, and Drug Incorporation Efficiency of Human PrimPol. DNA Repair 2017, 49, 51–59. [Google Scholar] [CrossRef]
- Xu, W.; Zhao, W.; Morehouse, N.; Tree, M.O.; Zhao, L. Divalent Cations Alter the Rate-Limiting Step of PrimPol-Catalyzed DNA Elongation. J. Mol. Biol. 2019, 431, 673–686. [Google Scholar] [CrossRef]
- Carvalho, G.; Díaz-Talavera, A.; Calvo, P.A.; Blanco, L.; Martínez-Jiménez, M.I. Human PrimPol Discrimination against Dideoxynucleotides during Primer Synthesis. Genes 2021, 12, 1487. [Google Scholar] [CrossRef]
- Boldinova, E.O.; Manukyan, А.А.; Makarova, А.V. The DNA Ligands Arg47 and Arg76 Are Crucial for Catalysis by Human PrimPol. DNA Repair 2021, 100, 103048. [Google Scholar] [CrossRef]
- Martínez-Jiménez, M.I.; Calvo, P.A.; García-Gómez, S.; Guerra-González, S.; Blanco, L. The Zn-Finger Domain of Human PrimPol Is Required to Stabilize the Initiating Nucleotide during DNA Priming. Nucleic Acids Res. 2018, 46, 4138–4151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Kasho, K.; Stojkovič, G.; Velázquez-Ruiz, C.; Martínez-Jiménez, M.I.; Doimo, M.; Laurent, T.; Berner, A.; Pérez-Rivera, A.E.; Jenninger, L.; Blanco, L.; et al. A Unique Arginine Cluster in PolDIP2 Enhances Nucleotide Binding and DNA Synthesis by PrimPol. Nucleic Acids Res. 2021, 49, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Guilliam, T.A.; Brissett, N.C.; Ehlinger, A.; Keen, B.A.; Kolesar, P.; Taylor, E.M.; Bailey, L.J.; Lindsay, H.D.; Chazin, W.J.; Doherty, A.J. Molecular Basis for PrimPol Recruitment to Replication Forks by RPA. Nat. Commun. 2017, 8, 15222. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Molinaro, C.; Martoriati, A.; Cailliau, K. Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies. Cancers 2021, 13, 3819. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, Z.; Huang, J.; Guo, G.; Gao, M.; Kim, W.; Zeng, X.; Kloeber, J.A.; Zhu, Q.; Zhao, F.; et al. The Deubiquitinase USP36 Regulates DNA Replication Stress and Confers Therapeutic Resistance through PrimPol Stabilization. Nucleic Acids Res. 2020, 48, 12711–12726. [Google Scholar] [CrossRef]
- Yoshimura, A.; Oikawa, M.; Jinbo, H.; Hasegawa, Y.; Enomoto, T.; Seki, M. WRNIP1 Controls the Amount of PrimPol. Biol. Pharm. Bull. 2019, 42, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Bailey, L.J.; Teague, R.; Kolesar, P.; Bainbridge, L.J.; Lindsay, H.D.; Doherty, A.J. PLK1 Regulates the PrimPol Damage Tolerance Pathway during the Cell Cycle. Sci. Adv. 2021, 7, eabh1004. [Google Scholar] [CrossRef]
- Martínez-Jiménez, M.I.; Lahera, A.; Blanco, L. Human PrimPol Activity Is Enhanced by RPA. Sci. Rep. 2017, 7, 783. [Google Scholar] [CrossRef] [Green Version]
- Guilliam, T.A.; Bailey, L.J.; Brissett, N.C.; Doherty, A.J. PolDIP2 Interacts with Human PrimPol and Enhances Its DNA Polymerase Activities. Nucleic Acids Res. 2016, 44, 3317–3329. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, M.; Ogawa, S.; Ooka, M.; Kobayashi, K.; Hirota, K.; Wakasugi, M.; Matsunaga, T.; Sakuma, T.; Yamamoto, T.; Chikuma, S.; et al. PDIP38/PolDIP2 Controls the DNA Damage Tolerance Pathways by Increasing the Relative Usage of Translesion DNA Synthesis over Template Switching. PLoS ONE 2019, 14, e0213383. [Google Scholar] [CrossRef]
- Gagarinskaya, D.I.; Makarova, A.V. A Multifunctional Protein PolDIP2 in DNA Translesion Synthesis. In Mechanisms of Genome Protection and Repair; Zharkov, D.O., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerlamd, 2020; pp. 35–45. ISBN 978-3-030-41283-8. [Google Scholar] [CrossRef]
- Wong, A.; Zhang, S.; Mordue, D.; Wu, J.M.; Zhang, Z.; Darzynkiewicz, Z.; Lee, E.Y.; Lee, M.Y. PDIP38 Is Translocated to the Spliceosomes/Nuclear Speckles in Response to UV-Induced DNA Damage and Is Required for UV-Induced Alternative Splicing of MDM2. Cell Cycle 2013, 12, 3373–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojkovič, G.; Makarova, A.V.; Wanrooij, P.H.; Forslund, J.; Burgers, P.M.; Wanrooij, S. Oxidative DNA Damage Stalls the Human Mitochondrial Replisome. Sci. Rep. 2016, 6, 28942. [Google Scholar] [CrossRef] [PubMed]
- Tirman, S.; Quinet, A.; Wood, M.; Meroni, A.; Cybulla, E.; Jackson, J.; Pegoraro, S.; Simoneau, A.; Zou, L.; Vindigni, A. Temporally Distinct Post-Replicative Repair Mechanisms Fill PRIMPOL-Dependent SsDNA Gaps in Human Cells. Mol. Cell 2021, 81, 4026–4040.e8. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R.; Niimi, A.; Ogi, T.; Brown, S.; Sabbioneda, S.; Wing, J.F.; Kannouche, P.L.; Green, C.M. Translesion Synthesis: Y-Family Polymerases and the Polymerase Switch. DNA Repair 2007, 6, 891–899. [Google Scholar] [CrossRef]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.-W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polζ Maintains the Viability of Homologous Recombination-Deficient Cancer Cells through Mutagenic Repair of PRIMPOL-Dependent SsDNA Gaps. Mol. Cell 2021, 81, 4008–4025.e7. [Google Scholar] [CrossRef]
- García-Medel, P.L.; Peralta-Castro, A.; Baruch-Torres, N.; Fuentes-Pascacio, A.; Pedroza-García, J.A.; Cruz-Ramirez, A.; Brieba, L.G. Arabidopsis Thaliana PrimPol Is a Primase and Lesion Bypass DNA Polymerase with the Biochemical Characteristics to Cope with DNA Damage in the Nucleus, Mitochondria, and Chloroplast. Sci. Rep. 2021, 11, 20582. [Google Scholar] [CrossRef]
- Bocquier, A.A.; Liu, L.; Cann, I.K.O.; Komori, K.; Kohda, D.; Ishino, Y. Archaeal Primase: Bridging the Gap between RNA and DNA Polymerases. Curr. Biol. 2001, 11, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Komori, K.; Ishino, S.; Bocquier, A.A.; Cann, I.K.O.; Kohda, D.; Ishino, Y. The Archaeal DNA Primase: BIOCHEMICAL CHARACTERIZATION OF THE P41-P46 COMPLEX FROMPYROCOCCUS FURIOSUS. J. Biol. Chem. 2001, 276, 45484–45490. [Google Scholar] [CrossRef] [Green Version]
- Picher, Á.J.; Budeus, B.; Wafzig, O.; Krüger, C.; García-Gómez, S.; Martínez-Jiménez, M.I.; Díaz-Talavera, A.; Weber, D.; Blanco, L.; Schneider, A. TruePrime Is a Novel Method for Whole-Genome Amplification from Single Cells Based on TthPrimPol. Nat. Commun. 2016, 7, 13296. [Google Scholar] [CrossRef]
- García-Quintans, N.; Baquedano, I.; Blesa, A.; Verdú, C.; Berenguer, J.; Mencía, M. A Thermostable DNA Primase-Polymerase from a Mobile Genetic Element Involved in Defence against Environmental DNA. Environ. Microbiol. 2020, 22, 4647–4657. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- THE GTEX CONSORTIUM; Ardlie, K.G.; Deluca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; et al. The Genotype-Tissue Expression (GTEx) Pilot Analysis: Multitissue Gene Regulation in Humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Richardson, R.B.; Allan, D.S.; Le, Y. Greater Organ Involution in Highly Proliferative Tissues Associated with the Early Onset and Acceleration of Ageing in Humans. Exp. Gerontol. 2014, 55, 80–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.K.; Castillo, C.; So, A.G.; Downey, K.M. An Auxiliary Protein for DNA Polymerase-Delta from Fetal Calf Thymus. J. Biol. Chem. 1986, 261, 12310–12316. [Google Scholar] [CrossRef]
- Prelich, G.; Tan, C.-K.; Kostura, M.; Mathews, M.B.; So, A.G.; Downey, K.M.; Stillman, B. Functional Identity of Proliferating Cell Nuclear Antigen and a DNA Polymerase-δ Auxiliary Protein. Nature 1987, 326, 517–520. [Google Scholar] [CrossRef]
- Dietrich, D.R. Toxicological and Pathological Applications of Proliferating Cell Nuclear Antigen (PCNA), A Novel Endogenous Marker for Cell Proliferation. Crit. Rev. Toxicol. 1993, 23, 77–109. [Google Scholar] [CrossRef]
- Iatropoulos, M.J.; Williams, G.M. Proliferation Markers. Exp. Toxicol. Pathol. 1996, 48, 175–181. [Google Scholar] [CrossRef]
- Zabrady, K.; Zabrady, M.; Kolesar, P.; Li, A.W.H.; Doherty, A.J. CRISPR-Associated Primase-Polymerases Are Implicated in Prokaryotic CRISPR-Cas Adaptation. Nat. Commun. 2021, 12, 3690. [Google Scholar] [CrossRef]
- Lange, S.S.; Takata, K.; Wood, R.D. DNA Polymerases and Cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, E.; Tomlinson, I. Replicative DNA Polymerase Mutations in Cancer. Curr. Opin. Genet. Dev. 2014, 24, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre-Moreno, G.; Pryor, J.M.; Díaz-Talavera, A.; Ruiz, J.F.; Ramsden, D.A.; Blanco, L. Polμ Tumor Variants Decrease the Efficiency and Accuracy of NHEJ. Nucleic Acids Res. 2017, 45, 10018–10031. [Google Scholar] [CrossRef] [PubMed]
- Makridakis, N.; Reichardt, J. Translesion DNA Polymerases and Cancer. Front. Genet. 2012, 3, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilzecker, B.; Buoninfante, O.A.; Pritchard, C.; Blomberg, O.S.; Huijbers, I.J.; van den Berk, P.C.M.; Jacobs, H. PrimPol Prevents APOBEC/AID Family Mediated DNA Mutagenesis. Nucleic Acids Res. 2016, 44, 4734–4744. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.; Fu, P.; Alcivar, A.L.; Fu, H.; Redon, C.; Foo, T.K.; Zuo, Y.; Ye, C.; Baxley, R.; Madireddy, A.; et al. BRCA2 Associates with MCM10 to Suppress PRIMPOL-Mediated Repriming and Single-Stranded Gap Formation after DNA Damage. Nat. Commun. 2021, 12, 5966. [Google Scholar] [CrossRef]
- Vallerga, M.B.; Mansilla, S.F.; Federico, M.B.; Bertolin, A.P.; Gottifredi, V. Rad51 Recombinase Prevents Mre11 Nuclease-Dependent Degradation and Excessive PrimPol-Mediated Elongation of Nascent DNA after UV Irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, E6624–E6633. [Google Scholar] [CrossRef] [Green Version]
- Kaluzhnaya, Y.G.; Bondarenko, K.A.; Boldinova, E.O.; Makarova, A.V. Dna Aptamers to Human Primpol. In Proceedings of the VII International Conference of Young Scientists: Biophysicists, Biotechnologists, Molecular Biologists and Virologists, Koltsovo Sciencie City, Russian, 27–29 October 2020; p. 461, eLIBRARY ID: 44476155. [Google Scholar]
- Fu, Z.; Xiang, J. Aptamers, the Nucleic Acid Antibodies, in Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 2793. [Google Scholar] [CrossRef]
- Mislak, A.C.; Anderson, K.S. Insights into the Molecular Mechanism of Polymerization and Nucleoside Reverse Transcriptase Inhibitor Incorporation by Human PrimPol. Antimicrob. Agents Chemother. 2015, 60, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I Poisoning Results in PARP-Mediated Replication Fork Reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, R.S.; Brissett, N.C.; Picher, A.J.; Andrade, P.; Juarez, R.; Thompson, D.; Fox, G.C.; Blanco, L.; Doherty, A.J. Structure and Function of a Mycobacterial NHEJ DNA Repair Polymerase. J. Mol. Biol. 2007, 366, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipps, G.; Weinzierl, A.O.; von Scheven, G.; Buchen, C.; Cramer, P. Structure of a Bifunctional DNA Primase-Polymerase. Nat. Struct. Mol. Biol. 2004, 11, 157–162. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Function | Activity Involved [Ref.] | Somatic Mutations in Cancer (COSMIC Database) |
|---|---|---|---|
| R47 | Contact the DNA template | Primase/polymerase [107,112] | - |
| *R76 | Contact the DNA template | Primase/polymerase [107,112] | R76H and R76C |
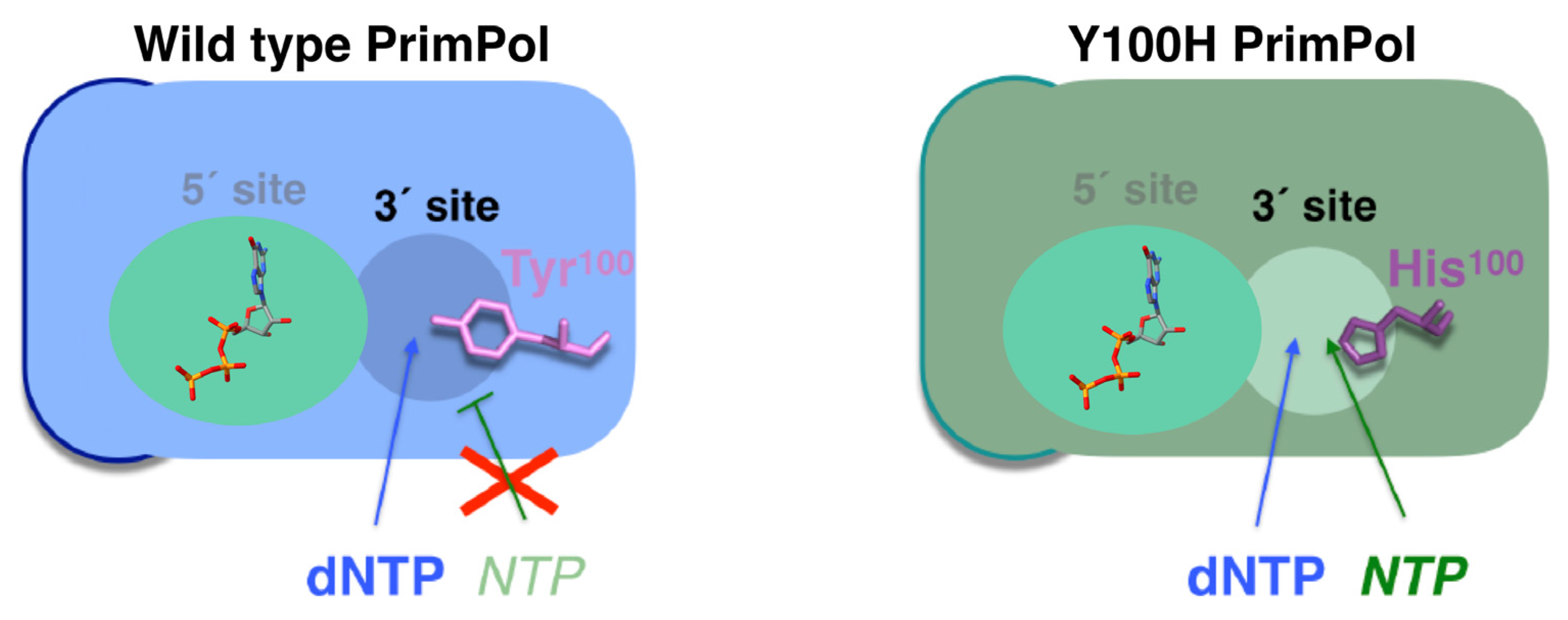
| Y100 | Steric gate (sugar selector) | Primase/polymerase [74] | Y100H |
| D114 | Cation ligand (Mn2+ or Mg2+) | Primase/polymerase [12,79,107] | - |
| E116 | Cation ligand (Mn2+ or Mg2+) | Primase/polymerase [12,79,107] | - |
| H169 | Stabilize the 3´ nucleotide | Primase/polymerase [12,107] | - |
| L200-S260 | PolDIP2 binding | Primase?/polymerase [116] | G201D, E203K, D204G, A208S, A208T, H214Y, P217S, P217L, H218Y, F219L, S220L, Q226L, K232T, M233I, T235R, W243S, T244A, G254W, SS59R |
| D280 | Cation ligand | Primase/polymerase [12,79,107] | - |
| R291 | Stabilize the 3´ nucleotide | Primase/polymerase [107,111] | R291W |
| *R417 | - | - | R417L, R417W and R417Q |
| C419, H426,C446 and C451 | Zn2+ ligand | Primase [12,113] | H426N and H426R |
| D519/F522 and D551/I554 | Binding of RPA | Primase/polymerase [117] | F522V |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Talavera, A.; Montero-Conde, C.; Leandro-García, L.J.; Robledo, M. PrimPol: A Breakthrough among DNA Replication Enzymes and a Potential New Target for Cancer Therapy. Biomolecules 2022, 12, 248. https://doi.org/10.3390/biom12020248
Díaz-Talavera A, Montero-Conde C, Leandro-García LJ, Robledo M. PrimPol: A Breakthrough among DNA Replication Enzymes and a Potential New Target for Cancer Therapy. Biomolecules. 2022; 12(2):248. https://doi.org/10.3390/biom12020248
Chicago/Turabian StyleDíaz-Talavera, Alberto, Cristina Montero-Conde, Luis Javier Leandro-García, and Mercedes Robledo. 2022. "PrimPol: A Breakthrough among DNA Replication Enzymes and a Potential New Target for Cancer Therapy" Biomolecules 12, no. 2: 248. https://doi.org/10.3390/biom12020248
APA StyleDíaz-Talavera, A., Montero-Conde, C., Leandro-García, L. J., & Robledo, M. (2022). PrimPol: A Breakthrough among DNA Replication Enzymes and a Potential New Target for Cancer Therapy. Biomolecules, 12(2), 248. https://doi.org/10.3390/biom12020248