
Leukotriene Signaling as a Target in α-Synucleinopathies

 ,
,
Abstract
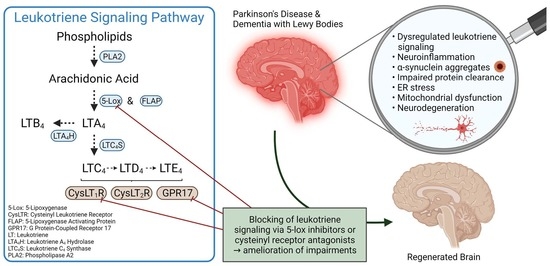
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
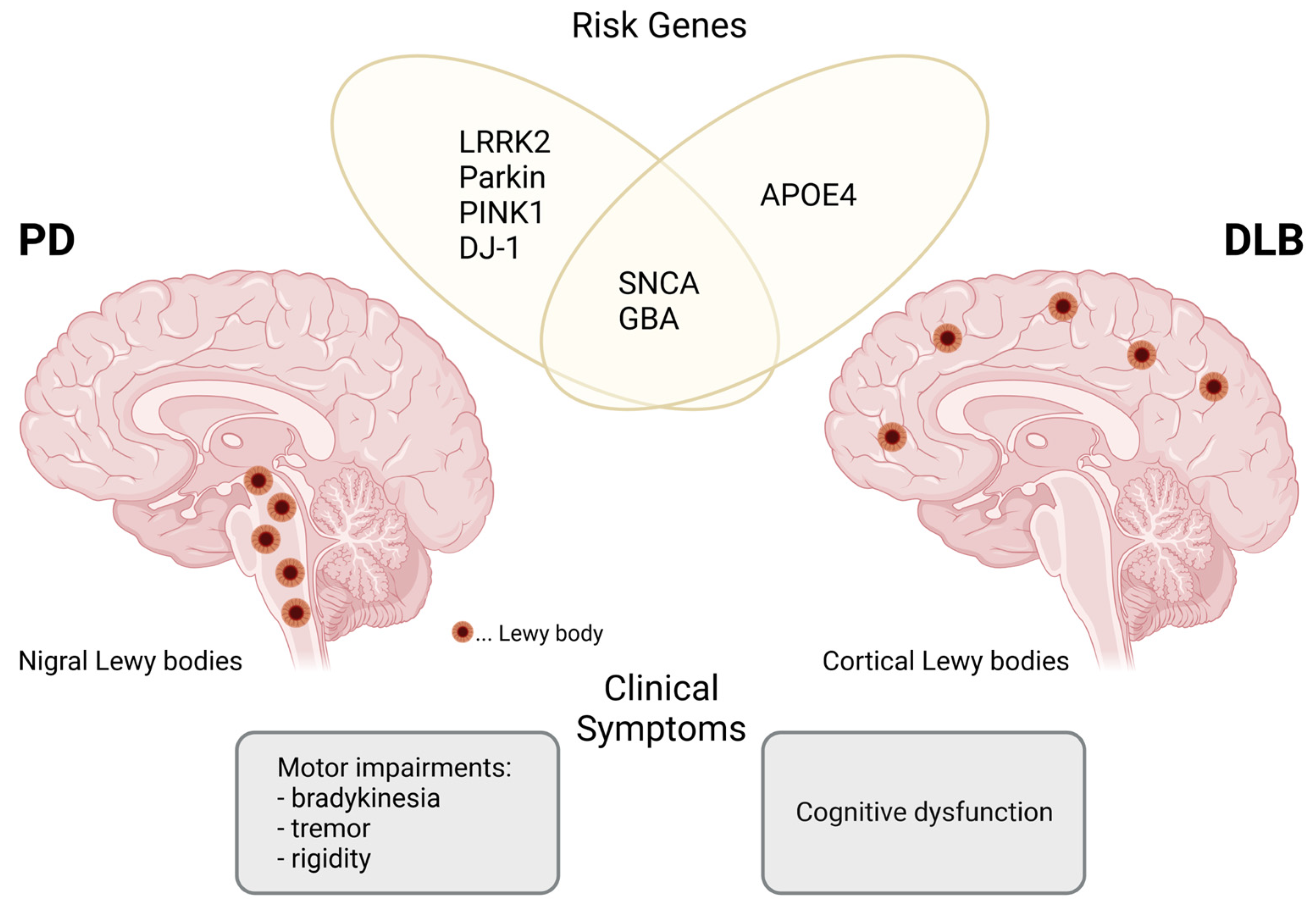
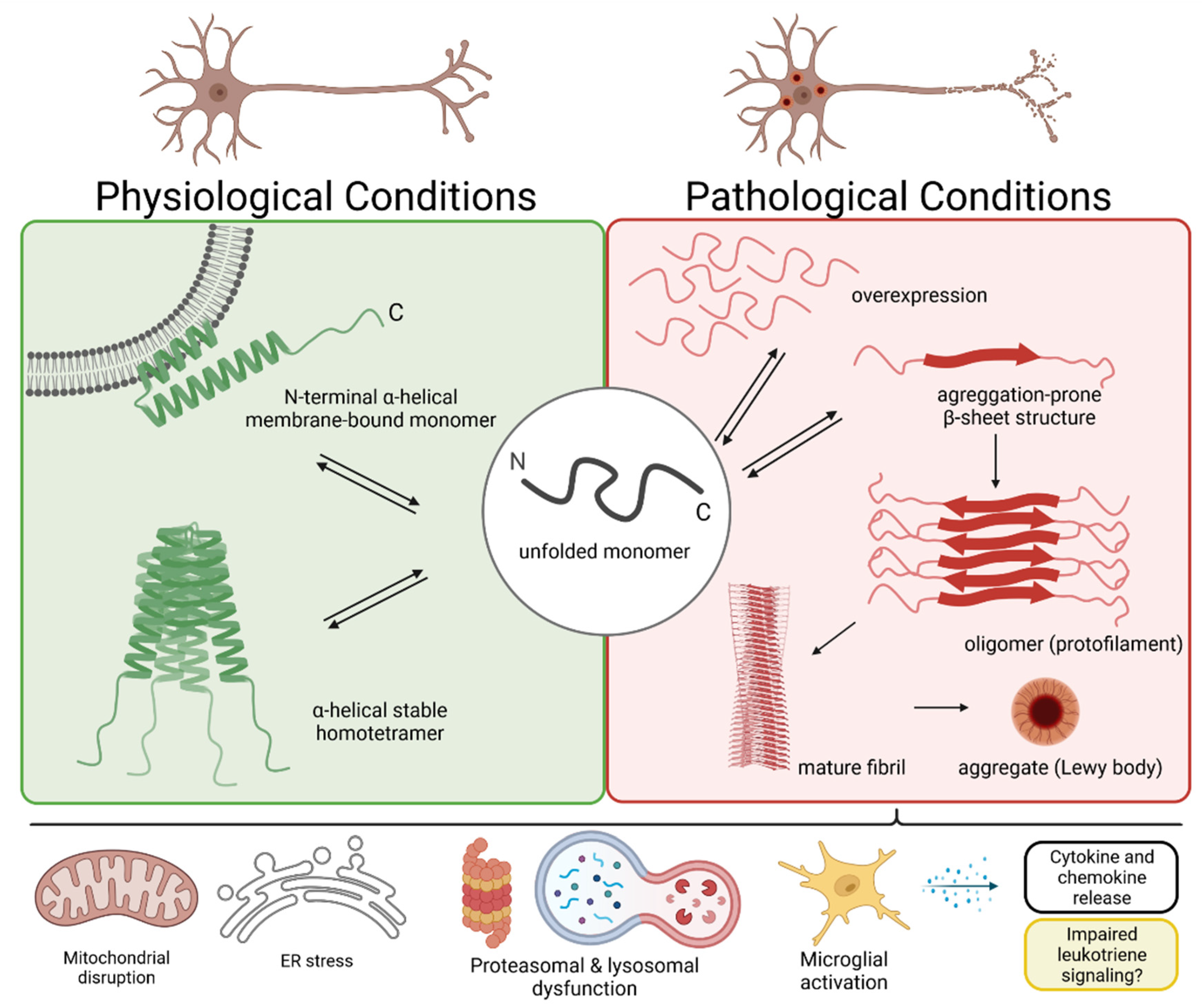
2. The α-Synuclein Protein and Lewy Body Formation
3. Mitochondrial Dysfunction in Neurons
4. Dysregulated Protein Clearance in PD and DLB
5. Microglia-Mediated Neuroinflammation in PD and DLB
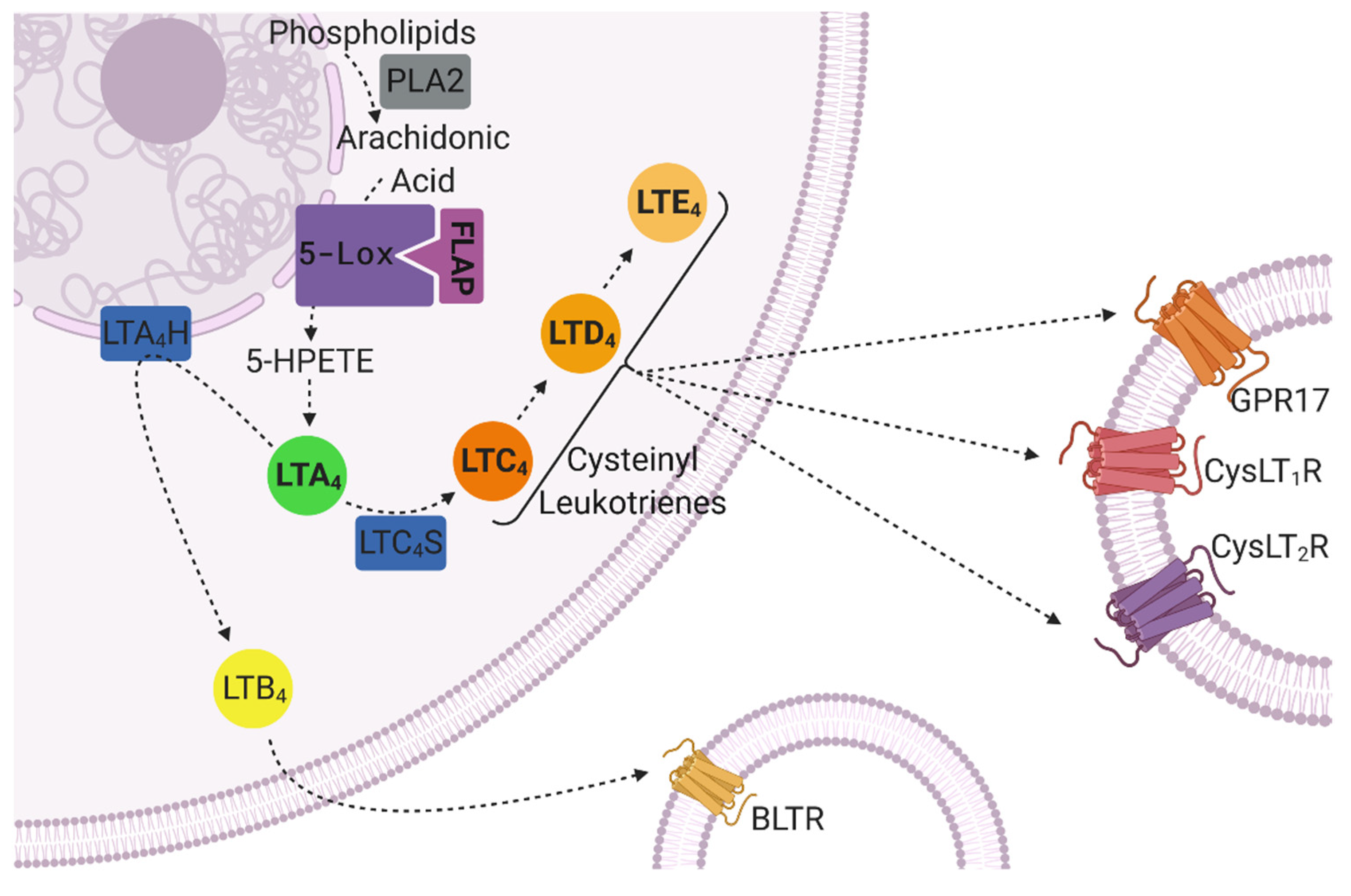
6. The Role of Leukotrienes in CNS Inflammation and Neurodegeneration
7. The Leukotriene Signaling Pathway in CNS Disorders
8. The Contribution of Leukotrienes in Autophagic Activity Impairment and Mitochondrial Dysfunction Outside the CNS
9. Targeting Leukotriene Signaling in Preclinical Studies
10. Montelukast as a Treatment Option for PD and DLB
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Gomperts, S.N. Lewy Body Dementias. Contin. Lifelong Learn. Neurol. 2016, 22, 435–463. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.P.M.; Surendranathan, A.; Bentley, A.; Barker, S.A.H.; Taylor, J.-P.; Thomas, A.J.; Allan, L.M.; McNally, R.J.; James, P.W.; McKeith, I.G.; et al. Clinical prevalence of Lewy body dementia. Alzheimer’s Res. Ther. 2018, 10, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salthouse, T.A. What and When of Cognitive Aging. Curr. Dir. Psychol. Sci. 2004, 13, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Orme, T.; Guerreiro, R.; Bras, J. The Genetics of Dementia with Lewy Bodies: Current Understanding and Future Directions. Curr. Neurol. Neurosci. Rep. 2018, 18, 67. [Google Scholar] [CrossRef] [Green Version]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [Green Version]
- Modi, P.; Mohamad, A.; Phom, L.; Koza, Z.; Das, A.; Chaurasia, R.; Samadder, S.; Achumi, B.; Muralidhara, R.S.P.; Yenisetti, S.C. Understanding Pathophysiology of Sporadic Parkinson’s Disease in Drosophila Model: Potential Opportunities and Notable Limitations. In Challenges in Parkinson’s Disease; IntechOpen: London, UK, 2016. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139, 59–74. [Google Scholar] [CrossRef]
- Yuan, H.; Zheng, J.-C.; Liu, P.; Zhang, S.-F.; Xu, J.-Y.; Bai, L.-M. Pathogenesis of Parkinson’s disease: Oxidative stress, environmental impact factors and inflammatory processes. Neurosci. Bull. 2007, 23, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Surguchev, A.A.; Surguchov, A. Synucleins and gene expression: Ramblers in a crowd or cops regulating traffic? Front. Mol. Neurosci. 2017. [Google Scholar] [CrossRef] [Green Version]
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 2018, 373, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Attrebi, O.N.; Ren, Y.; Qiao, W.; Sonustun, B.; Martens, Y.A.; Meneses, A.D.; Li, F.; Shue, F.; Zheng, J.; et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 2020, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Neurobiology and treatment of Parkinson’s disease. Trends Pharmacol. Sci. 2009, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging Treatment Approaches for Parkinson’s Disease. Front. Neurosci. 2018, 12, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershey, L.A.; Coleman-Jackson, R. Pharmacological Management of Dementia with Lewy Bodies. Drugs Aging 2019, 36, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Krainc, Y.C.W.D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Noori, A.; Mezlini, A.M.; Hyman, B.T.; Serrano-Pozo, A.; Das, S. Systematic review and meta-analysis of human transcriptomics reveals neuroinflammation, deficient energy metabolism, and proteostasis failure across neurodegeneration. Neurobiol. Dis. 2020, 149, 105225. [Google Scholar] [CrossRef]
- Michael, J.; Marschallinger, J.; Aigner, L. The leukotriene signaling pathway: A druggable target in Alzheimer’s disease. Drug Discov. Today 2018, 24, 505–516. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Zhang, S.; Li, C.; Zhang, L. Modulation of neuroinflammation by cysteinyl leukotriene 1 and 2 receptors: Implications for cerebral ischemia and neurodegenerative diseases. Neurobiol. Aging 2019, 87, 1–10. [Google Scholar] [CrossRef]
- Wallin, J.; Svenningsson, P. Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 5606. [Google Scholar] [CrossRef]
- Kim, W.S.; Kågedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane Curvature Induction and Tubulation Are Common Features of Synucleins and Apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Vilar, M.; Chou, H.-T.; Luhrs, T.; Maji, S.K.; Riek-Loher, D.; Verel, R.; Manning, G.; Stahlberg, H.; Riek, R. The fold of -synuclein fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 8637–8642. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhao, Y.-F.; Chen, Y.-X.; Li, Y.-M. Exploring the Roles of Post-Translational Modifications in the Pathogenesis of Parkinson’s Disease Using Synthetic and Semisynthetic Modified α-Synuclein. ACS Chem. Neurosci. 2019, 10, 910–921. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [Green Version]
- Uemura, N.; Uemura, M.; Luk, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Cell-to-Cell Transmission of Tau and α-Synuclein. Trends Mol. Med. 2020, 26, 936–952. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca2+ Signaling and Quality Control. Trends Mol. Med. 2019, 26, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, J.H.; Fuentes, F.; Vanasco, V.; Alvarez, S.; Alaimo, A.; Cassina, A.; Leskow, F.C.; Velazquez, F. Alpha-synuclein mitochondrial interaction leads to irreversible translocation and complex I impairment. Arch. Biochem. Biophys. 2018, 651, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, Prefibrillar α-Synuclein Oligomers Promote Complex I-dependent, Ca2+-induced Mitochondrial Dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschbach, J.; Von Einem, B.; Muller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of peroxisome proliferator-activated receptor γ coactivator 1α deregulation and α-synuclein oligomerization. Ann. Neurol. 2014, 77, 15–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and Protein Degradation Systems: A Reciprocal Relationship. Mol. Neurobiol. 2012, 47, 537–551. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and Monomeric α-Synuclein Bind to the S6′ Proteasomal Protein and Inhibit Proteasomal Function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef] [Green Version]
- St McNaught, K.P.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered Proteasomal Function in Sporadic Parkinson’s Disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef]
- Alghamdi, A.; Vallortigara, J.; Howlett, D.R.; Broadstock, M.; Hortobágyi, T.; Ballard, C.; Thomas, A.J.; O’Brien, J.T.; Aarsland, D.; Attems, J.; et al. Reduction of RPT6/S8 (a Proteasome Component) and Proteasome Activity in the Cortex is Associated with Cognitive Impairment in Lewy Body Dementia. J. Alzheimer’s Dis. 2017, 57, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, S.; Moore, D.; Minegishi, M.; Kasanuki, K.; Fujishiro, H.; Kabuta, T.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Localization of MAP1-LC3 in Vulnerable Neurons and Lewy Bodies in Brains of Patients With Dementia With Lewy Bodies. J. Neuropathol. Exp. Neurol. 2011, 70, 264–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohn, T.T.; Catlin, L.W. Immunolocalization of Influenza A Virus and Markers of Inflammation in the Human Parkinson’s Disease Brain. PLoS ONE 2011, 6, e20495. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Elenis, D.; Papasilekas, T.; Stranjalis, G.; Gerozissis, K.; Ioannou, P.; Vekrellis, K. Assessment of α-Synuclein Secretion in Mouse and Human Brain Parenchyma. PLoS ONE 2011, 6, e22225. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.-I.; et al. α-Synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Suk, J.-E.; Bae, E.-J.; Lee, S.-J. Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia Development and Function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [Green Version]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Schafer, D.P.; Stevens, B. Phagocytic glial cells: Sculpting synaptic circuits in the developing nervous system. Curr. Opin. Neurobiol. 2013, 23, 1034–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Teva, J.L.; Dusart, I.; Colin, C.; Gervais, A.; van Rooijen, N.; Mallat, M. Microglia Promote the Death of Developing Purkinje Cells. Neuron 2004, 41, 535–547. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. e-Neuroforum 2005, 11, 95. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98–2094. [Google Scholar] [CrossRef] [Green Version]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, G.A.; Baig, S.; Bednar, I.; Södersten, P.; Forsberg, G.; Siden, A. Increased cerebrospinal fluid concentration of nitrite in Parkinson’s disease. NeuroReport 1995, 6, 1642–1644. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Boissière, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E. Nitric oxide synthase and neuronal vulnerability in parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Kannarkat, G.T.; Boss, J.M.; Tansey, M.G. The Role of Innate and Adaptive Immunity in Parkinson’s Disease. J. Parkinson’s Dis. 2013, 3, 493–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iba, M.; Kim, C.; Sallin, M.; Kwon, S.; Verma, A.; Overk, C.; Rissman, R.A.; Sen, R.; Sen, J.M.; Masliah, E. Neuroinflammation is associated with infiltration of T cells in Lewy body disease and α-synuclein transgenic models. J. Neuroinflamm. 2020, 17, 214. [Google Scholar] [CrossRef] [PubMed]
- Gate, D.; Tapp, E.; Leventhal, O.; Shahid, M.; Nonninger, T.J.; Yang, A.C.; Strempfl, K.; Unger, M.S.; Fehlmann, T.; Oh, H.; et al. CD4 + T cells contribute to neurodegeneration in Lewy body dementia. Science 2021, 374, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Surendranathan, A.; Su, L.; Mak, E.; Passamonti, L.; Hong, Y.T.; Arnold, R.; Vázquez Rodríguez, P.; Bevan-Jones, W.R.; Brain, S.A.; Fryer, T.D.; et al. Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 2018, 141, 3415–3427. [Google Scholar] [CrossRef]
- Lavisse, S.; Goutal, S.; Wimberley, C.; Tonietto, M.; Bottlaender, M.; Gervais, P.; Kuhnast, B.; Peyronneau, M.-A.; Barret, O.; Lagarde, J.; et al. Increased microglial activation in patients with Parkinson disease using [18F]-DPA714 TSPO PET imaging. Park. Relat. Disord. 2020, 82, 29–36. [Google Scholar] [CrossRef]
- Varnäs, K.; Cselényi, Z.; Jucaite, A.; Halldin, C.; Svenningsson, P.; Farde, L.; Varrone, A. PET imaging of [11C]PBR28 in Parkinson’s disease patients does not indicate increased binding to TSPO despite reduced dopamine transporter binding. Eur. J. Pediatr. 2018, 46, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.A. Activated microglia in dementia with Lewy bodies. Neurology 2000, 55, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Xue, Q.-S. Microglia in dementia with Lewy bodies. Brain Behav. Immun. 2016, 55, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Amin, J.; Holmes, C.; Dorey, R.; Tommasino, E.; Casal, Y.R.; Williams, D.M.; Dupuy, C.; Nicoll, J.A.R.; Boche, D. Neuroinflammation in dementia with Lewy bodies: A human post-mortem study. Transl. Psychiatry 2020, 10, 267. [Google Scholar] [CrossRef]
- Feleke, R.; Reynolds, R.H.; Smith, A.M.; Tilley, B.; Taliun, S.A.G.; Hardy, J.; Matthews, P.M.; Gentleman, S.; Owen, D.R.; Johnson, M.R.; et al. Cross-platform transcriptional profiling identifies common and distinct molecular pathologies in Lewy body diseases. Acta Neuropathol. 2021, 142, 449–474. [Google Scholar] [CrossRef] [PubMed]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; Morris, C.M.; Antony, P.; et al. Single-cell sequencing of the human midbrain reveals glial activation and a neuronal state specific to Parkinson’s disease. Brain 2021. [Google Scholar]
- Muir, R. Review Article. Landscapes 2001, 2, 112–115. [Google Scholar] [CrossRef]
- Woods, J.W.; Evans, J.F.; Ethier, D.; Scott, S.; Vickers, P.J.; Hearn, L.; Heibein, J.A.; Charleson, S.; I Singer, I. 5-lipoxygenase and 5-lipoxygenase-activating protein are localized in the nuclear envelope of activated human leukocytes. J. Exp. Med. 1993, 178, 1935–1946. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Boyce, J.A. Cysteinyl Leukotrienes and Their Receptors: Cellular Distribution and Function in Immune and Inflammatory Responses. J. Immunol. 2004, 173, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.-S.; Haeggström, J.Z.; Hultenby, K.; Palmblad, J. Subcellular localization of leukotriene receptors in human endothelial cells. Exp. Cell Res. 2010, 316, 2790–2796. [Google Scholar] [CrossRef]
- Tager, A.M.; Luster, A.D. BLT1 and BLT2: The leukotriene B4 receptors. Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 123–134. [Google Scholar] [CrossRef]
- Bäck, M.; Dahlén, S.-E.; Drazen, J.M.; Evans, J.F.; Serhan, C.N.; Shimizu, T.; Yokomizo, T.; Rovati, G. International Union of Basic and Clinical Pharmacology. LXXXIV: Leukotriene Receptor Nomenclature, Distribution, and Pathophysiological Functions. Pharmacol. Rev. 2011, 63, 539–584. [Google Scholar] [CrossRef] [PubMed]
- Mellor, E.A.; Maekawa, A.; Austen, K.F.; Boyce, J.A. Cysteinyl leukotriene receptor 1 is also a pyrimidinergic receptor and is expressed by human mast cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7964–7969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciana, P.; Fumagalli, M.; Trincavelli, M.L.; Verderio, C.; Rosa, P.; Lecca, D.; Ferrario, S.; Parravicini, C.; Capra, V.; Gelosa, P.; et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006, 25, 4615–4627. [Google Scholar] [CrossRef]
- Shirasaki, H.; Kanaizumi, E.; Seki, N.; Himi, T. Leukotriene E4 induces MUC5AC release from human airway epithelial NCI-H292 cells. Allergol. Int. 2015, 64, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallstrand, T.S.; Henderson, W.R. An update on the role of leukotrienes in asthma. Curr. Opin. Allergy Clin. Immunol. 2010, 10, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laidlaw, T.M.; Boyce, J.A. Cysteinyl leukotriene receptors, old and new; implications for asthma. Clin. Exp. Allergy 2012, 42, 1313–1320. [Google Scholar] [CrossRef] [Green Version]
- Marschallinger, J.; Schäffner, I.; Klein, B.; Gelfert, R.; Rivera, F.J.; Illes, S.; Grassner, L.; Janssen, M.; Rotheneichner, P.; Schmuckermair, C.; et al. Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nat. Commun. 2015, 6, 8466. [Google Scholar] [CrossRef] [Green Version]
- Marschallinger, J.; Altendorfer, B.; Rockenstein, E.; Holztrattner, M.; Garnweidner-Raith, J.; Pillichshammer, N.; Leister, I.; Hutter-Paier, B.; Strempfl, K.; Unger, M.S.; et al. The Leukotriene Receptor Antagonist Montelukast Reduces Alpha-Synuclein Load and Restores Memory in an Animal Model of Dementia with Lewy Bodies. Neurotherapeutics 2020, 17, 1061–1074. [Google Scholar] [CrossRef] [Green Version]
- Michael, J.; Unger, M.S.; Poupardin, R.; Schernthaner, P.; Mrowetz, H.; Attems, J.; Aigner, L. Microglia depletion diminishes key elements of the leukotriene pathway in the brain of Alzheimer’s Disease mice. Acta Neuropathol. Commun. 2020, 8, 129. [Google Scholar] [CrossRef]
- Michael, J.; Zirknitzer, J.; Unger, M.; Poupardin, R.; Rieß, T.; Paiement, N.; Zerbe, H.; Hutter-Paier, B.; Reitsamer, H.; Aigner, L. The Leukotriene Receptor Antagonist Montelukast Attenuates Neuroinflammation and Affects Cognition in Transgenic 5xFAD Mice. Int. J. Mol. Sci. 2021, 22, 2782. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, H.; Zhang, L.; Ding, W.; Yao, H.-T.; Chen, K.-D.; Sheng, W.-W.; Chen, Z.; Wei, E.-Q. Expression of cysteinyl leukotriene receptor 1 in human traumatic brain injury and brain tumors. Neurosci. Lett. 2004, 363, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, B.; Zhang, X.; Huang, X.; Shi, W.; Liu, H.; Fang, S.; Lu, Y.; Zhang, W.; Tang, F.; et al. Cysteinyl leukotriene receptor 2 is spatiotemporally involved in neuron injury, astrocytosis and microgliosis after focal cerebral ischemia in rats. Neuroscience 2011, 189, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Hoshi, M.; Akazawa, C.; Nakamura, Y.; Tsuzuki, H.; Inoue, K.; Kohsaka, S. Selective expression of Gi/o-coupled ATP receptor P2Y12 in microglia in rat brain. Glia 2003, 44, 242–250. [Google Scholar] [CrossRef]
- Walker, D.G.; Tang, T.M.; Mendsaikhan, A.; Tooyama, I.; Serrano, G.E.; Sue, L.I.; Beach, T.G.; Lue, L.-F. Patterns of Expression of Purinergic Receptor P2RY12, a Putative Marker for Non-Activated Microglia, in Aged and Alzheimer’s Disease Brains. Int. J. Mol. Sci. 2020, 21, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.-Y.; Zhang, X.-Y.; Wang, X.-R.; Xu, D.-M.; Chen, L.; Zhang, L.-H.; Fang, S.-H.; Lu, Y.-B.; Zhang, W.-P.; Wei, E.-Q. Cysteinyl leukotriene receptor 1 mediates LTD4-induced activation of mouse microglial cells in vitro. Acta Pharmacol. Sin. 2013, 35, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; Wang, X.-R.; Xu, D.-M.; Yu, S.-Y.; Shi, Q.-J.; Zhang, L.-H.; Chen, L.; Fang, S.-H.; Lu, Y.-B.; Zhang, W.-P.; et al. HAMI 3379, a CysLT2 Receptor Antagonist, Attenuates Ischemia-Like Neuronal Injury by Inhibiting Microglial Activation. J. Pharmacol. Exp. Ther. 2013, 346, 328–341. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wang, H.; Li, C.; Song, S.; Fang, S.; Wei, E.; Shi, Q. GPR17 mediates ischemia-like neuronal injury via microglial activation. Int. J. Mol. Med. 2018, 42, 2750–2762. [Google Scholar] [CrossRef] [Green Version]
- Hijioka, M.; Futokoro, R.; Ohto-Nakanishi, T.; Nakanishi, H.; Katsuki, H.; Kitamura, Y. Microglia-released leukotriene B4 promotes neutrophil infiltration and microglial activation following intracerebral hemorrhage. Int. Immunopharmacol. 2020, 85, 106678. [Google Scholar] [CrossRef]
- Huang, X.-J.; Zhang, W.-P.; Li, C.-T.; Shi, W.-Z.; Fang, S.-H.; Lu, Y.-B.; Chen, Z.; Wei, E.-Q. Activation of CysLT receptors induces astrocyte proliferation and death after oxygen–glucose deprivation. Glia 2007, 56, 27–37. [Google Scholar] [CrossRef]
- Ciccarelli, R.; D’Alimonte, I.; Santavenere, C.; D’Auro, M.; Ballerini, P.; Nargi, E.; Buccella, S.; Nicosia, S.; Caciagli, F.; Di Iorio, P. Cysteinyl-leukotrienes are released from astrocytes and increase astrocyte proliferation and glial fibrillary acidic protein via cys-LT1 receptors and mitogen-activated protein kinase pathway. Eur. J. Neurosci. 2004, 20, 1514–1524. [Google Scholar] [CrossRef]
- Ballerini, P.; Di Iorio, P.; Ciccarelli, R.; Caciagli, F.; Polp, A.; Beraudi, A.; Buccella, S.; D’Alimonte, I.; D’Auro, M.; Nargi, E.; et al. P2Y1 and Cysteinyl Leukotriene Receptors Mediate Purine and Cysteinyl Leukotriene Co-Release in Primary Cultures of Rat Microglia. Int. J. Immunopathol. Pharmacol. 2005, 18, 255–268. [Google Scholar] [CrossRef]
- Farias, S.E.; Zarini, S.; Precht, T.; Murphy, R.C.; Heidenreich, K.A. Transcellular biosynthesis of cysteinyl leukotrienes in rat neuronal and glial cells. J. Neurochem. 2007, 103, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Ikonomovic, M.D.; Abrahamson, E.E.; Uz, T.; Manev, H.; DeKosky, S.T. Increased 5-Lipoxygenase Immunoreactivity in the Hippocampus of Patients With Alzheimer’s Disease. J. Histochem. Cytochem. 2008, 56, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Bsc, P.F.G.; Ceballos-Diaz, C.; Golde, T.E.; Praticò, D. 5-Lipoxygenase gene transfer worsens memory, amyloid, and tau brain pathologies in a mouse model of alzheimer disease. Ann. Neurol. 2012, 72, 442–454. [Google Scholar] [CrossRef] [Green Version]
- Firuzi, O.; Zhuo, J.; Chinnici, C.; Wisniewski, T.; Praricò, D. 5-Lipoxygenase gene disruption reduces amyloid-β pathology in a mouse model of Alzheimer’s disease. FASEB J. 2007, 22, 1169–1178. [Google Scholar] [CrossRef]
- Tang, S.-S.; Wang, X.-Y.; Hong, H.; Long, Y.; Li, Y.-Q.; Xiang, G.-Q.; Jiang, L.-Y.; Zhang, H.-T.; Liu, L.-P.; Miao, M.-X.; et al. Leukotriene D4 induces cognitive impairment through enhancement of CysLT1R-mediated amyloid-β generation in mice. Neuropharmacology 2013, 65, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.-H.; Liou, H.-H.; Hour, M.-J.; Liou, H.-C.; Fu, W.-M. Protection of dopaminergic neurons by 5-lipoxygenase inhibitor. Neuropharmacology 2013, 73, 380–387. [Google Scholar] [CrossRef]
- Chou, V.; Holman, T.; Manning-Bog, A. Differential contribution of lipoxygenase isozymes to nigrostriatal vulnerability. Neuroscience 2012, 228, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; Chen, L.; Yang, Y.; Xu, D.-M.; Zhang, S.-R.; Li, C.-T.; Zheng, W.; Yu, S.-Y.; Wei, E.-Q.; Zhang, L.-H. Regulation of rotenone-induced microglial activation by 5-lipoxygenase and cysteinyl leukotriene receptor 1. Brain Res. 2014, 1572, 59–71. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Chen, L.; Xu, D.-M.; Wang, X.-R.; Wang, Y.-F.; Li, C.-T.; Wei, E.-Q.; Zhang, L.-H. Zileuton, a 5-lipoxygenase inhibitor, attenuates mouse microglial cell-mediated rotenone toxicity in PC12 cells. Zhejiang Da Xue Xue Bao Yi Xue Ban 2014, 43, 273–280. [Google Scholar]
- Chen, L.; Yang, Y.; Li, C.-T.; Zhang, S.-R.; Zheng, W.; Wei, E.-Q.; Zhang, L.-H. CysLT 2 receptor mediates lipopolysaccharide-induced microglial inflammation and consequent neurotoxicity in vitro. Brain Res. 2015, 1624, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; He, Q.; Wang, H.; Hu, X.; Luo, Y.; Yang, Y.; Kuang, S.; Tian, X.; Ma, J.; Yang, J. 5-lipoxygenase activation is involved in the mechanisms of chronic hepatic injury in a rat model of chronic aluminum overload exposure. Toxicol. Appl. Pharmacol. 2016, 305, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yang, J.; He, Q.; Luo, Y.; Chen, Z.; Yang, L.; Yi, H.; Li, H.; Xia, H.; Ran, D.; et al. CysLTR1 Blockage Ameliorates Liver Injury Caused by Aluminum-Overload via PI3K/AKT/mTOR-Mediated Autophagy Activation in Vivo and in Vitro. Mol. Pharm. 2018, 15, 1996–2006. [Google Scholar] [CrossRef] [PubMed]
- Koller, A.; Bruckner, D.; Aigner, L.; Reitsamer, H.; Trost, A. Cysteinyl leukotriene receptor 1 modulates autophagic activity in retinal pigment epithelial cells. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wang, R.; Han, Q. Inhibition of leukotriene B4 receptor 1 attenuates lipopolysaccharide-induced cardiac dysfunction: Role of AMPK-regulated mitochondrial function. Sci. Rep. 2017, 7, srep44352. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cheng, Y.; Liu, Y.; Shi, J.; Cheng, Z. Montelukast promotes mitochondrial biogenesis via CREB/PGC-1α in human bronchial epithelial cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 4234–4239. [Google Scholar] [CrossRef]
- Ren, P.; Gong, F.; Chang, L.; Hong, X.; Xing, L.; Zhang, H. Zafirlukast promotes mitochondrial respiration by stimulating mitochondrial biogenesis in human bronchial epithelial cells. Histochem. J. 2021, 52, 643–650. [Google Scholar] [CrossRef]
- Chu, J.; Li, J.-G.; Pratico, M. Zileuton Improves Memory Deficits, Amyloid and Tau Pathology in a Mouse Model of Alzheimer’s Disease with Plaques and Tangles. PLoS ONE 2013, 8, e70991. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.-S.; Ji, M.-J.; Chen, L.; Hu, M.; Long, Y.; Li, Y.-Q.; Miao, M.-X.; Li, J.-C.; Li, N.; Ji, H.; et al. Protective effect of pranlukast on Aβ 1–42-induced cognitive deficits associated with downregulation of cysteinyl leukotriene receptor 1. Int. J. Neuropsychopharmacol. 2013, 17, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Kalra, J.; Kumar, P.; Majeed, A.B.A.; Prakash, A. Modulation of LOX and COX pathways via inhibition of amyloidogenesis contributes to mitoprotection against β-amyloid oligomer-induced toxicity in an animal model of Alzheimer’s disease in rats. Pharmacol. Biochem. Behav. 2016, 146–147, 1–12. [Google Scholar] [CrossRef]
- Kumar, A.; Prakash, A.; Pahwa, D.; Mishra, J. Montelukast potentiates the protective effect of rofecoxib against kainic acid-induced cognitive dysfunction in rats. Pharmacol. Biochem. Behav. 2012, 103, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kalonia, H.; Kumar, P.; Kumar, A.; Nehru, B. Protective effect of montelukast against quinolinic acid/malonic acid induced neurotoxicity: Possible behavioral, biochemical, mitochondrial and tumor necrosis factor-α level alterations in rats. Neuroscience 2010, 171, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Prasad, N.; Raghavendra, R.; Lokesh, B.; Naidu, K. Spice phenolics inhibit human PMNL 5-lipoxygenase. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, O.M.; Sleem, A.A.; Youness, E.R.; Yassen, N.N.Y.N.; Shaffie, N.; El-Toumy, S.A. Capsicum Protects Against Rotenone-Induced Toxicity in Mice Brain Via Reduced Oxidative Stress and 5-Lipoxygenase Activation. J. Pharm. Pharmacol. Res. 2018, 2, 60–77. [Google Scholar] [CrossRef]
- Ramires, R.; Caiaffa, M.F.; Tursi, A.; Haeggström, J.Z.; Macchia, L. Novel inhibitory effect on 5-lipoxygenase activity by the anti-asthma drug montelukast. Biochem. Biophys. Res. Commun. 2004, 324, 815–821. [Google Scholar] [CrossRef]
- Anderson, R.; Theron, A.J.; Gravett, C.M.; Steel, H.C.; Tintinger, G.R.; Feldman, C. Montelukast inhibits neutrophil pro-inflammatory activity by a cyclic AMP-dependent mechanism. J. Cereb. Blood Flow Metab. 2009, 156, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Tintinger, G.R.; Feldman, C.; Theron, A.J.; Anderson, R. Montelukast: More than a Cysteinyl Leukotriene Receptor Antagonist? Sci. World J. 2010, 10, 2403–2413. [Google Scholar] [CrossRef]
- Grinde, B.; Engdahl, B. Prescription database analyses indicates that the asthma medicine montelukast might protect against dementia: A hypothesis to be verified. Immun. Ageing 2017, 14, 20. [Google Scholar] [CrossRef]
- Grinde, B.; Schirmer, H.; Eggen, A.E.; Aigner, L.; Engdahl, B. A possible effect of montelukast on neurological aging examined by the use of register data. Int. J. Clin. Pharm. 2020, 43, 541–548. [Google Scholar] [CrossRef]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ho, G.; Xu, L.; Powell, W.; Martin, J. The β2-agonist Salbutamol Inhibits Bronchoconstriction and Leukotriene D4Synthesis After Dry Gas Hyperpnea in the Guinea-pig. Pulm. Pharmacol. Ther. 1999, 12, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Kim, S.; Lee, J.M.; Oh, Y.-S.; Park, S.M.; Kim, S.R. Montelukast treatment protects nigral dopaminergic neurons against microglial activation in the 6-hydroxydopamine mouse model of Parkinson’s disease. NeuroReport 2017, 28, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, O.; Medhat, D.; Sleem, A.; Shaffie, N. Neuroprotection by Montelukast against Rotenone-Induced Rat Brain Damage. React. Oxyg. Species 2018. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, V.B.; Marathe, P.A. Effect of montelukast in experimental model of Parkinson’s disease. Neurosci. Lett. 2018, 682, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic Loss and Inclusion Body Formation in α-Synuclein Mice: Implications for Neurodegenerative Disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive Immunization Reduces Behavioral and Neuropathological Deficits in an Alpha-Synuclein Transgenic Model of Lewy Body Disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef] [Green Version]
- Potashkin, J.A.; Blume, S.R.; Runkle, N.K. Limitations of Animal Models of Parkinson’s Disease. Park. Dis. 2011, 2011, 658083. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.-S.; Heng, Y.; Mou, Z.; Huang, J.-Y.; Yuan, Y.-H.; Chen, N.-H. Reassessment of subacute MPTP-treated mice as animal model of Parkinson’s disease. Acta Pharmacol. Sin. 2017, 38, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J. Animal models of α-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef]
- Rabl, R.; Breitschaedel, C.; Flunkert, S.; Duller, S.; Amschl, D.; Neddens, J.; Niederkofler, V.; Rockenstein, E.; Masliah, E.; Roemer, H.; et al. Early start of progressive motor deficits in Line 61 α-synuclein transgenic mice. BMC Neurosci. 2017, 18, 22. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strempfl, K.; Unger, M.S.; Flunkert, S.; Trost, A.; Reitsamer, H.A.; Hutter-Paier, B.; Aigner, L. Leukotriene Signaling as a Target in α-Synucleinopathies. Biomolecules 2022, 12, 346. https://doi.org/10.3390/biom12030346
Strempfl K, Unger MS, Flunkert S, Trost A, Reitsamer HA, Hutter-Paier B, Aigner L. Leukotriene Signaling as a Target in α-Synucleinopathies. Biomolecules. 2022; 12(3):346. https://doi.org/10.3390/biom12030346
Chicago/Turabian StyleStrempfl, Katharina, Michael S. Unger, Stefanie Flunkert, Andrea Trost, Herbert A. Reitsamer, Birgit Hutter-Paier, and Ludwig Aigner. 2022. "Leukotriene Signaling as a Target in α-Synucleinopathies" Biomolecules 12, no. 3: 346. https://doi.org/10.3390/biom12030346