
Evidence for Enhanced Efficacy of Passive Immunotherapy against Beta-Amyloid in CD33-Negative 5xFAD Mice

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Animal Treatment Studies
2.2.1. Ethical Statement
2.2.2. Experimental Animals and Housing
2.2.3. Study Design
2.2.4. Sample Collection
2.2.5. Preparation of T-Per and 5 M Guanidine Hydrochloride (5 M GdmCl) Brain Fractions
2.2.6. Elevated plus Maze (EPM) Test
2.3. Immunohistochemical Readout
2.3.1. Tissue Preparation and Immunohistochemistry
2.3.2. Quantification of Immunohistochemical Labeling
2.3.3. Immunohistochemical Quantification of Plaque-Surrounding Microglia
2.3.4. Determination of Median Aβ Plaque Size and Aβ Plaque Counts
2.4. ELISA Readout
3. Results
3.1. Identification of Minimal Antibody Treatment Dose—Pilot Study
3.2. Treatment of 5xFAD and 5xFAD/CD33KO Mice with K11—Treatment Study
3.2.1. CD33KO Decreases Amyloid Plaque Load in 5xFAD Mice and Leads to an Add-On Effect in Passive Immunotherapy
3.2.2. CD33KO Ameliorates Behavioral Deficits in 5xFAD Mice
3.2.3. CD33KO Increases Number of Plaque-Surrounding Microglia under K11 Treatment
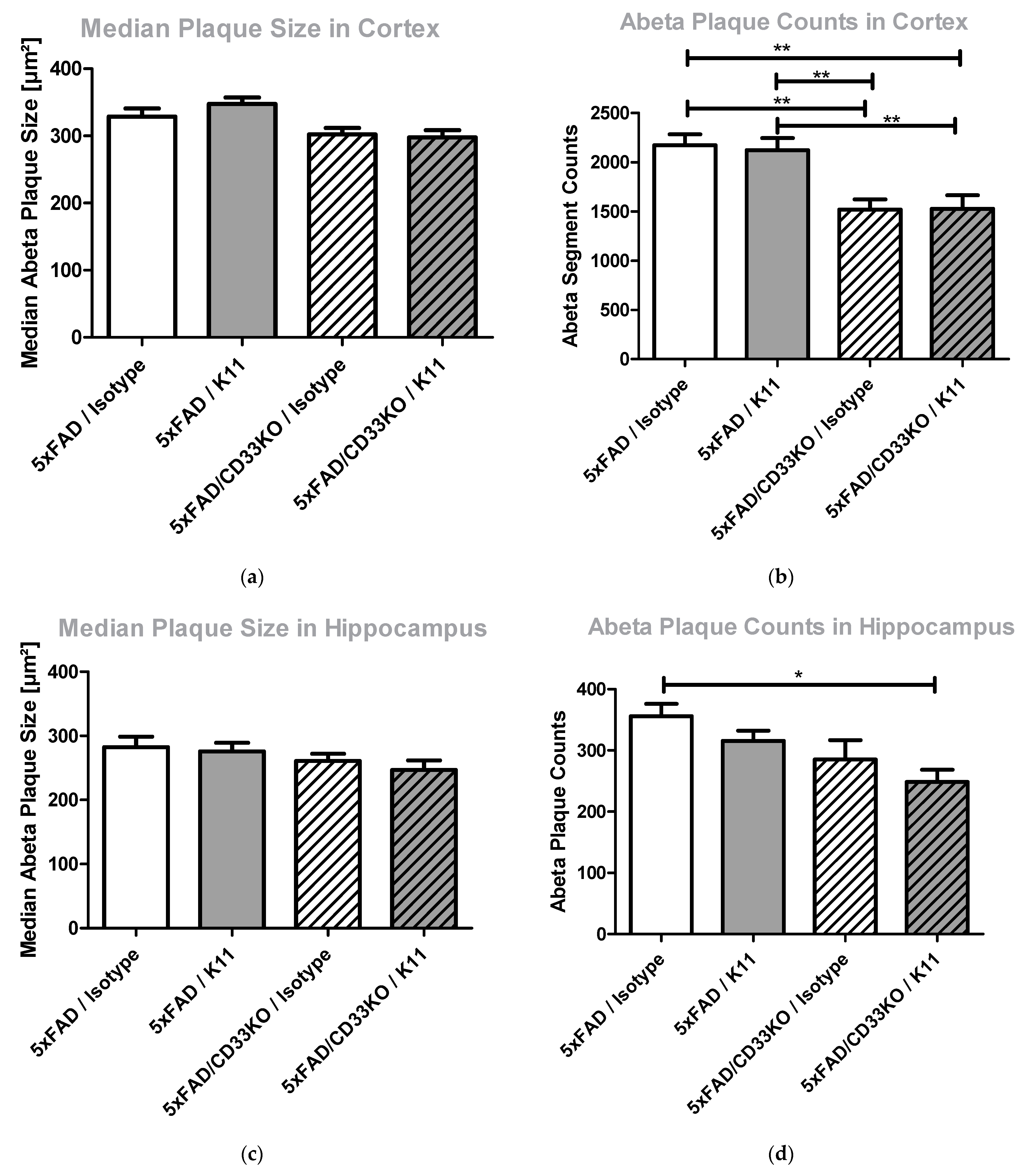
3.2.4. CD33KO as Well as Antibody Treatment Lead to a Reduction in Amyloid Plaque Counts Rather Than Amyloid Plaque Size
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Mondragón-Rodríguez, S.; Perry, G.; Luna-Muñoz, J.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein at sites Ser(396-404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Rahfeld, J.-U.; Lues, I.; Lemere, C.A. Passive Aβ Immunotherapy: Current Achievements and Future Perspectives. Molecules 2018, 23, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usman, M.B.; Bhardwaj, S.; Roychoudhury, S.; Kumar, D.; Alexiou, A.; Kumar, P.; Ambasta, R.K.; Prasher, P.; Shukla, S.; Upadhye, V.; et al. Immunotherapy for Alzheimer’s Disease: Current Scenario and Future Perspectives. J. Prev. Alzheimer’s Dis. 2021, 8, 534–551. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Iqbal, K. Thinking beyond the Aducanumab Controversy. Ann. Neurol. 2021, 90, 1003–1004. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.L.; Liu, B.; Kleinschmidt, M.; Schilling, S.; Demuth, H.-U.; Lemere, C.A. Passive immunization against pyroglutamate-3 amyloid-β reduces plaque burden in Alzheimer-like transgenic mice: A pilot study. Neurodegener. Dis. 2012, 10, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Demattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; Delong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnoth, K.; Piechotta, A.; Kleinschmidt, M.; Konrath, S.; Schenk, M.; Taudte, N.; Ramsbeck, D.; Rieckmann, V.; Geissler, S.; Eichentopf, R.; et al. Targeting isoaspartate-modified Aβ rescues behavioral deficits in transgenic mice with Alzheimer’s disease-like pathology. Alzheimer’s Res. Ther. 2020, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Meyer, A.; Heiser, U.; Kurat, S.; Böhme, L.; Kleinschmidt, M.; Bühring, K.-U.; Hutter-Paier, B.; Farcher, M.; Demuth, H.-U.; et al. Glutaminyl Cyclase Inhibitor PQ912 Improves Cognition in Mouse Models of Alzheimer’s Disease-Studies on Relation to Effective Target Occupancy. J. Pharmacol. Exp. Ther. 2017, 362, 119–130. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Lowe, S.L.; Willis, B.A.; Hawdon, A.; Natanegara, F.; Chua, L.; Foster, J.; Shcherbinin, S.; Ardayfio, P.; Sims, J.R. Donanemab (LY3002813) dose-escalation study in Alzheimer’s disease. Alzheimer’s Dement. 2021, 7, e12112. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Logue, M.W.; Schu, M.; Vardarajan, B.N.; Buros, J.; Green, R.C.; Go, R.C.P.; Griffith, P.; Obisesan, T.O.; Shatz, R.; Borenstein, A.; et al. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch. Neurol. 2011, 68, 1569–1579. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.-L.; Liu, L.-H.; Wang, Y.; Tang, H.-D.; Ren, R.-J.; Xu, W.; Ma, J.-F.; Wang, L.-L.; Zhuang, J.-P.; Wang, G.; et al. The prevalence of CD33 and MS4A6A variant in Chinese Han population with Alzheimer’s disease. Hum. Genet. 2012, 131, 1245–1249. [Google Scholar] [CrossRef]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mähler Convenor, M.; Berard, M.; Feinstein, R.; Gallagher, A.; Illgen-Wilcke, B.; Pritchett-Corning, K.; Raspa, M. FELASA recommendations for the health monitoring of mouse, rat, hamster, guinea pig and rabbit colonies in breeding and experimental units. Lab. Anim. 2014, 48, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Brinkman-Van der Linden, E.C.M.; Angata, T.; Reynolds, S.A.; Powell, L.D.; Hedrick, S.M.; Varki, A. CD33/Siglec-3 binding specificity, expression pattern, and consequences of gene deletion in mice. Mol. Cell. Biol. 2003, 23, 4199–4206. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Gordon, M.N.; Morgan, D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat. Protoc. 2006, 1, 1591–1595. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.L.; Liu, B.; Rahfeld, J.-U.; Kleinschmidt, M.; O’Nuallain, B.; Le, K.X.; Lues, I.; Caldarone, B.J.; Schilling, S.; Demuth, H.-U.; et al. An anti-pyroglutamate-3 Aβ vaccine reduces plaques and improves cognition in APPswe/PS1ΔE9 mice. Neurobiol. Aging 2015, 36, 3187–3199. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, D.M.; Munireddy, S.K.; Rosenthal, A.; Ugen, K.E.; Gordon, M.N.; Morgan, D. Microglial activation facilitates Abeta plaque removal following intracranial anti-Abeta antibody administration. Neurobiol. Dis. 2004, 15, 11–20. [Google Scholar] [CrossRef]
- Bard, F.; Barbour, R.; Cannon, C.; Carretto, R.; Fox, M.; Games, D.; Guido, T.; Hoenow, K.; Hu, K.; Johnson-Wood, K.; et al. Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc. Natl. Acad. Sci. USA 2003, 100, 2023–2028. [Google Scholar] [CrossRef] [Green Version]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.-L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.-H.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Panza, F.; Lozupone, M.; Seripa, D.; Imbimbo, B.P. Amyloid-β immunotherapy for alzheimer disease: Is it now a long shot? Ann. Neurol. 2019, 85, 303–315. [Google Scholar] [CrossRef]
- Crehan, H.; Liu, B.; Kleinschmidt, M.; Rahfeld, J.-U.; Le, K.X.; Caldarone, B.J.; Frost, J.L.; Hettmann, T.; Hutter-Paier, B.; O’Nuallain, B.; et al. Effector function of anti-pyroglutamate-3 Aβ antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alzheimer’s Res. Ther. 2020, 12, 12. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Greco, W.R.; Bravo, G.; Parsons, J.C. The search for synergy: A critical review from a response surface perspective. Pharmacol. Rev. 1995, 47, 331–385. [Google Scholar]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Caudle, R.M.; Williams, G.M. The misuse of analysis of variance to detect synergy in combination drug studies. Pain 1993, 55, 313–317. [Google Scholar] [CrossRef]
- Berenbaum, M.C. Synergy, additivism and antagonism in immunosuppression. A critical review. Clin. Exp. Immunol. 1977, 28, 1–18. [Google Scholar]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Malik, M.; Chiles, J.; Xi, H.S.; Medway, C.; Simpson, J.; Potluri, S.; Howard, D.; Liang, Y.; Paumi, C.M.; Mukherjee, S.; et al. Genetics of CD33 in Alzheimer’s disease and acute myeloid leukemia. Hum. Mol. Genet. 2015, 24, 3557–3570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Purdon, T.J.; Schultz, L.M.; Brentjens, R.J. CD33-Directed Chimeric Antigen Receptor (CAR) T Cells for the Treatment of Acute Myeloid Leukemia (AML). Blood 2016, 128, 2825. [Google Scholar] [CrossRef]
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.N.; Gooi, J.H.; Doughty, L.; Nero, T.L.; Markulić, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Aβ Phagocytosis. iScience 2019, 19, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Keith, C.T.; Borisy, A.A.; Stockwell, B.R. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 2005, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnoth, K.; Geissler, S.; Feldhaus, J.; Taudte, N.; Ilse, V.; Zürner, S.; Greiser, S.; Braumann, U.-D.; Rahfeld, J.-U.; Cynis, H.; et al. Evidence for Enhanced Efficacy of Passive Immunotherapy against Beta-Amyloid in CD33-Negative 5xFAD Mice. Biomolecules 2022, 12, 399. https://doi.org/10.3390/biom12030399
Gnoth K, Geissler S, Feldhaus J, Taudte N, Ilse V, Zürner S, Greiser S, Braumann U-D, Rahfeld J-U, Cynis H, et al. Evidence for Enhanced Efficacy of Passive Immunotherapy against Beta-Amyloid in CD33-Negative 5xFAD Mice. Biomolecules. 2022; 12(3):399. https://doi.org/10.3390/biom12030399
Chicago/Turabian StyleGnoth, Kathrin, Stefanie Geissler, Julia Feldhaus, Nadine Taudte, Victoria Ilse, Sebastian Zürner, Sebastian Greiser, Ulf-Dietrich Braumann, Jens-Ulrich Rahfeld, Holger Cynis, and et al. 2022. "Evidence for Enhanced Efficacy of Passive Immunotherapy against Beta-Amyloid in CD33-Negative 5xFAD Mice" Biomolecules 12, no. 3: 399. https://doi.org/10.3390/biom12030399