
The Impact of NRF2 Inhibition on Drug-Induced Colon Cancer Cell Death and p53 Activity: A Pilot Study


 , and
, and 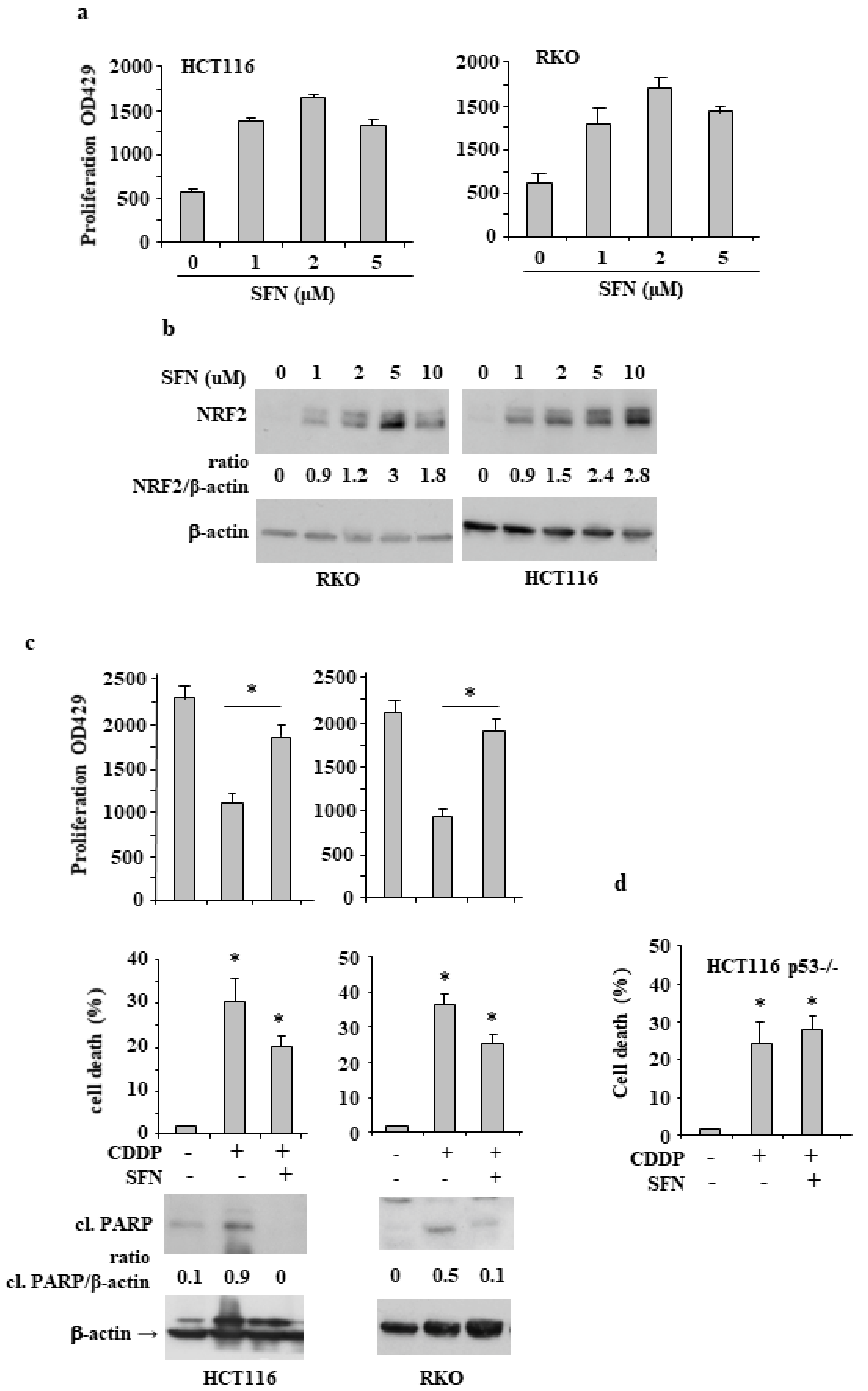{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Generation of CRISPR/Cas9-Based NRF2- Knockout HCT116 Cells (NRF2 KO Cells)
2.3. Cell Viability Assays
2.4. Proliferation Assay (XTT)
2.5. Western Blotting
2.6. RNA Extraction and Semiquantitative Reverse Transcription (RT)-Polymerase Chain Reaction (PCR) Analysis
2.7. Statistical Analysis
3. Results
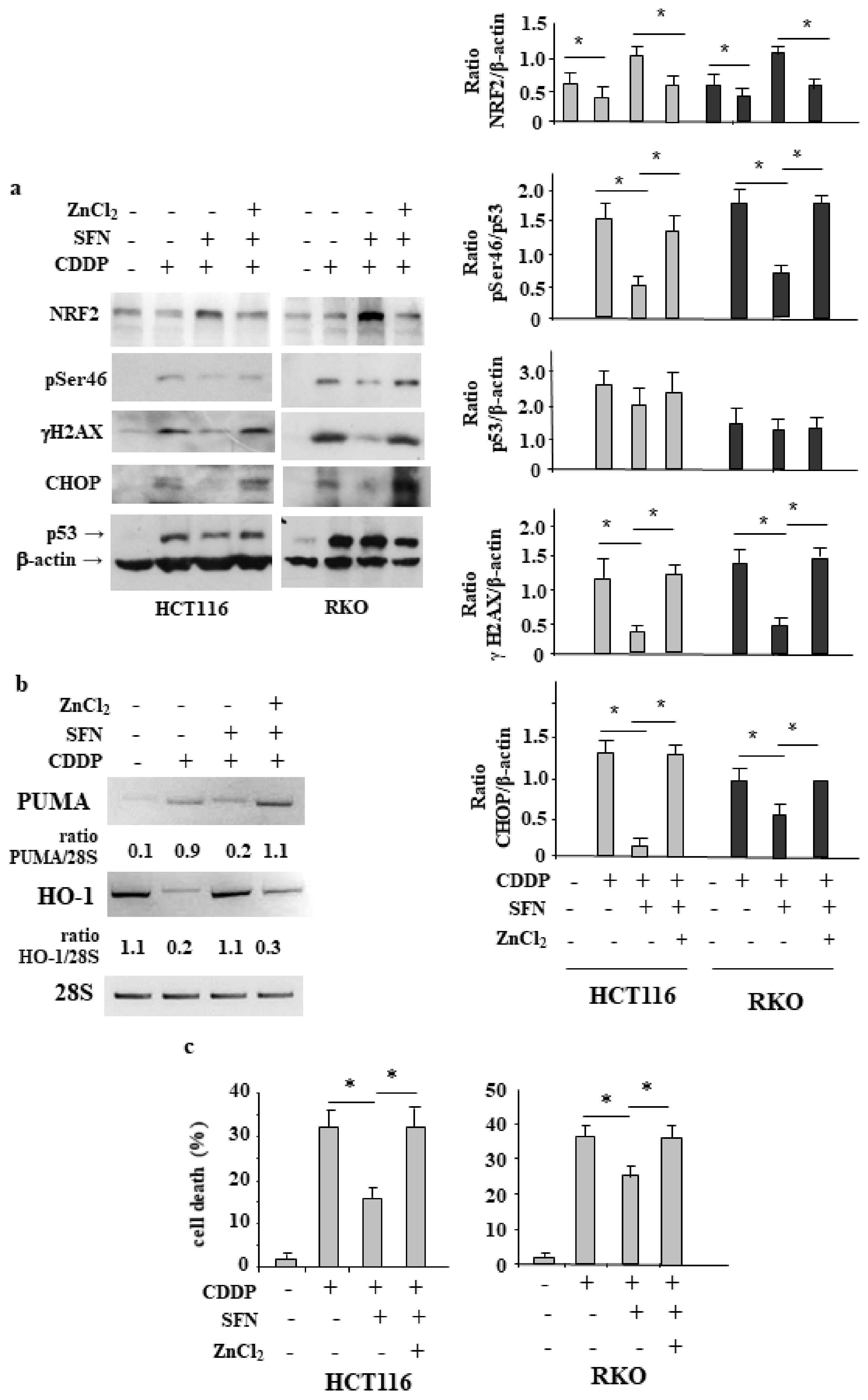
3.1. Sulforaphane Reduces the Cisplatin (CDDP)-Induced Colon CancerCcell Death
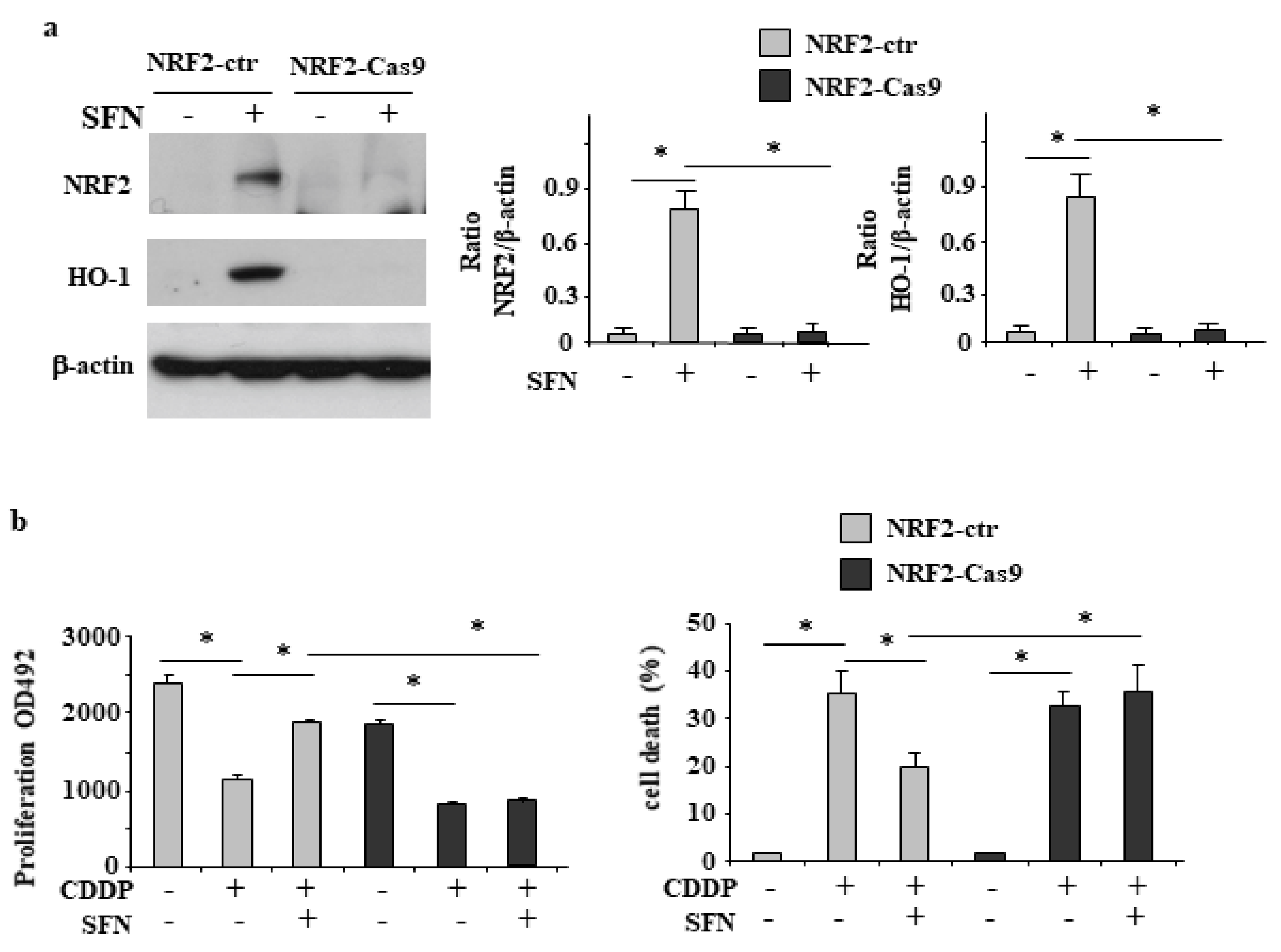
3.2. NRF2 Is Involved in Reduction of CDDP Citotoxycity
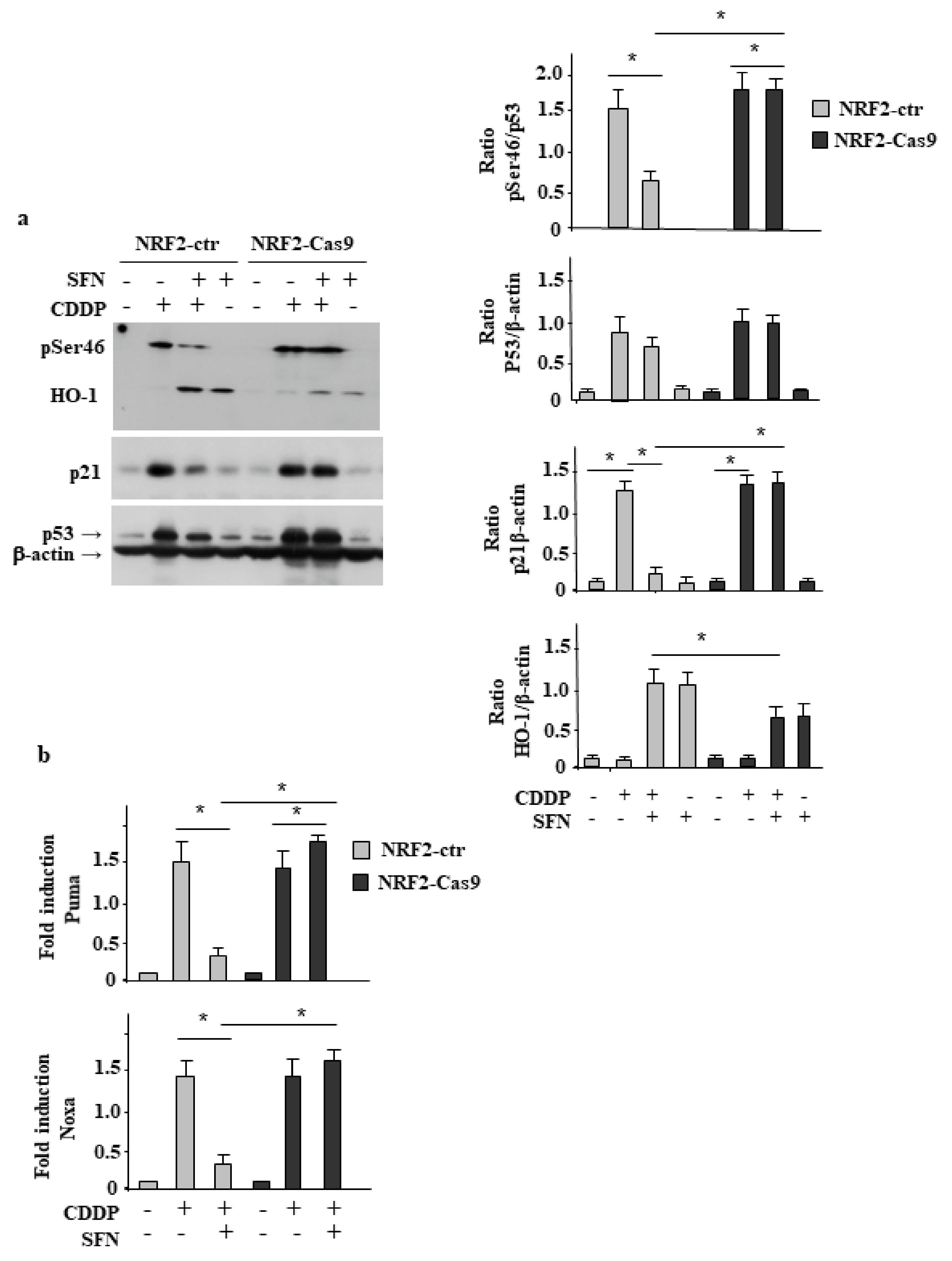
3.3. NRF2 Inhibition Restores CDDP-Induced p53 Activity Impaired by SFN
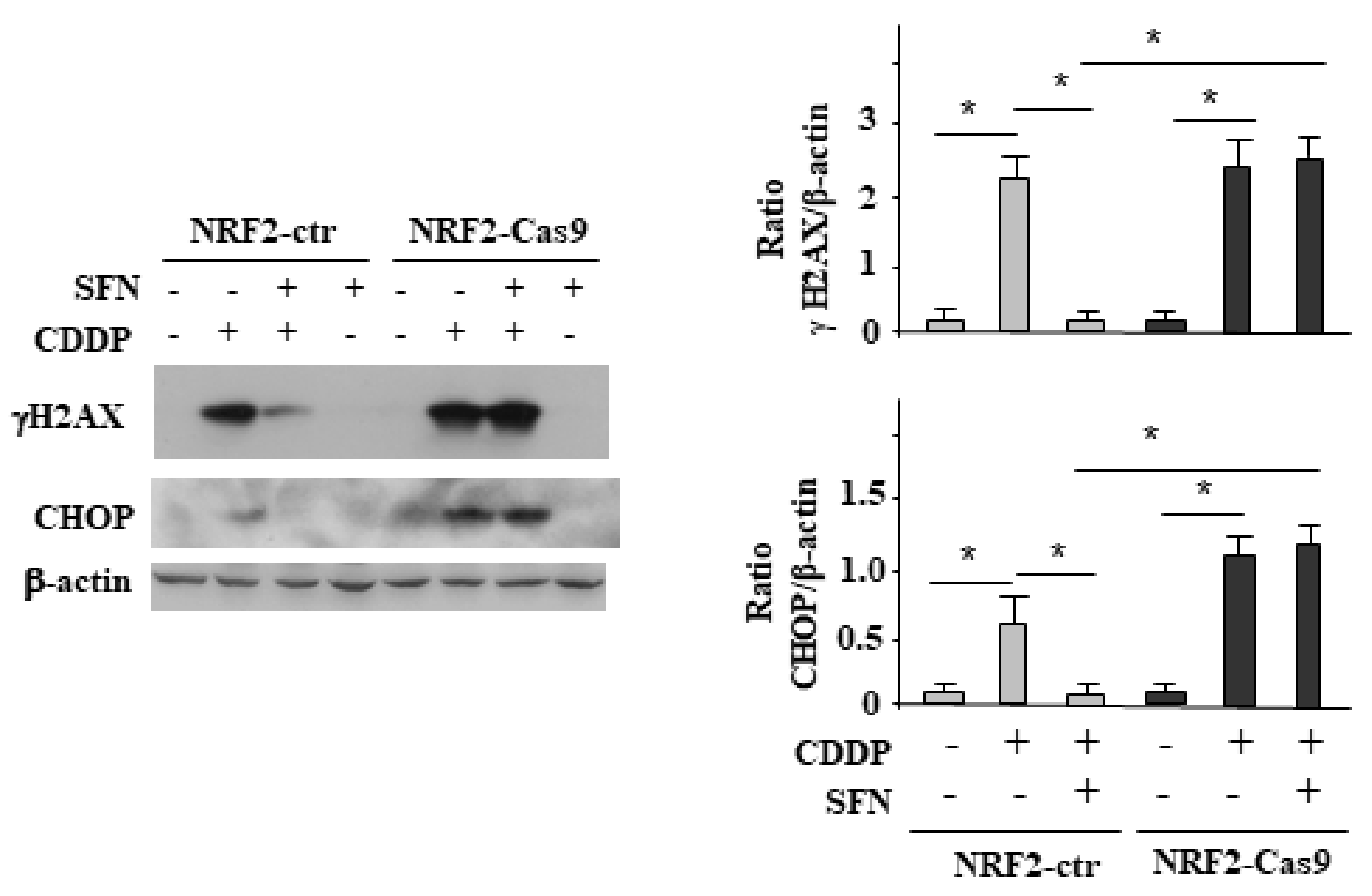
3.4. NRF2 Activation Impairs the CDDP-Induced DNA Damage
3.5. ZnCl2 Supplementation Rescues p53 Activity Inhibited by SFN
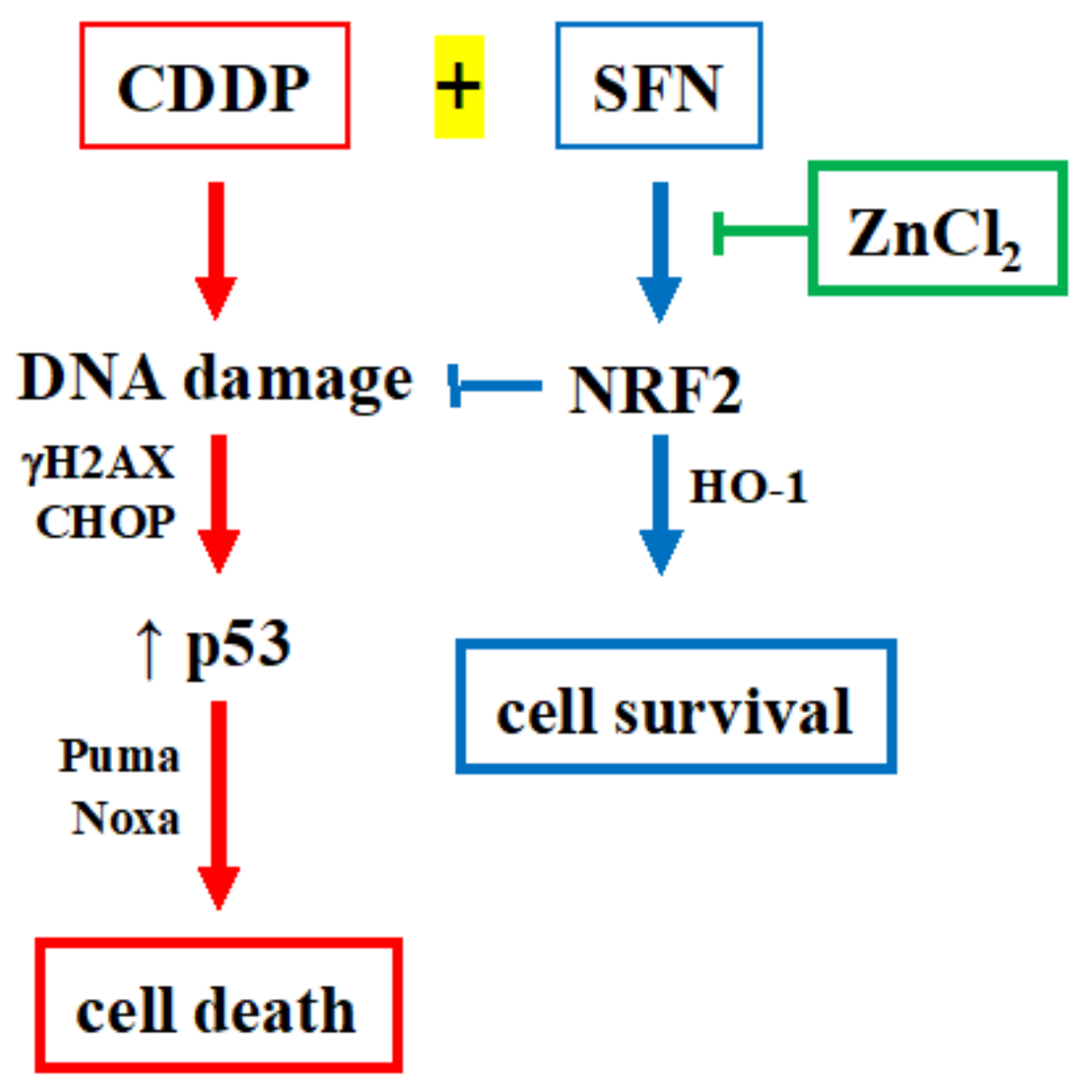
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and kallmarks of cancer. Cancer Cell. 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Torrente, L.; Maan, G.; Rezig, A.O.; Quinn, J.; Jackson, A.; Grilli, A.; Casares, L.; Zhang, Y.; Kulesskiy, E.; Saarela, J.; et al. High NRF2 levels correlates with poor prognosis in colorectal cancer patients and sensitivity to the kinase inhibitor AT9283 in vitro. Biomolecules 2020, 10, 1365. [Google Scholar] [CrossRef]
- No, J.H.; Kim, Y.B.; Song, Y.S. Targeting Nrf2 signaling to combat chemoresistance. J. Cancer. Prev. 2014, 19, 111–117. [Google Scholar] [CrossRef] [PubMed]
- McMahonm, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. P62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; RamaRao, M. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the light the growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnokova, O.A.; Solier, S.; Pommier, Y. gH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Gilardoni Montani, M.S.; Santarelli, R.; DOrazi, G.; Faggioni, A.; Cirone, M. Apigenin, by activating p53 and inhibiting STAT3, modulates the balance between pro-apoptotic and pro-survival pathways to induce PEL cell death. J. Exp. Clin. Cancer Res. 2017, 36, 167. [Google Scholar] [CrossRef]
- Garufi, A.; Pistritto, G.; Baldari, S.; Toietta, G.; Cirone, M.; D’Orazi, G. p53-dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell-fate decision in normal/high glucose conditions. J. Exp. Clin. Cancer Res. 2017, 36, 126. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; Porru, M.; Simon, A.J.; Rechavi, G.; Leonetti, C.; Givol, D.; DOrazi, G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011, 10, 1679–1689. [Google Scholar] [CrossRef]
- Garufi, A.; Trisciuoglio, D.; Cirone, M.; D’Orazi, G. ZnCl2 sustains the adriamycin-induced cell death inhibited by high glucose. Cell Death Dis. 2016, 7, e2280. [Google Scholar] [CrossRef][Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Gonnella, R.; Guttieri, L.; Gilardini Montani, M.S.; Santarelli, R.; Bassetti, E.; D’Orazi, G.; Cirone, M. Zinc supplementation enhances the pro-death function of UPR in lymphoma cells exposed to radiation. Biology 2022, 11, 132. [Google Scholar] [CrossRef]
- Gonzalez-Quiroz, M.; Blondel, A.; Sagredo, A.; Hetz, C.; Chevet, E.; Pedeux, R. When edoplasmic reticulum protostasis meets the DNA damage response. Trends. Cell. Biol. 2020, 30, 881–891. [Google Scholar] [CrossRef]
- Benedetti, R.; Gilardini Montani, M.S.; Romeo, M.A.; Arena, A.; Santarelli, R.; D’Orazi, G.; Cirone, M. Role of UPR Sensor Activation in Cell Death–Survival Decision of Colon Cancer Cells Stressed by DPE Treatment. Biomedicines 2021, 9, 1262. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; DOrazi, G. High glucose dephosphorylates serine 46 and inhibits p53 apoptotic activity. J. Exp. Clin. Cancer Res. 2014, 33, 79. [Google Scholar] [CrossRef]
- Garufi, A.; Traversi, G.; Gilardini Montani, M.S.; DOrazi, V.; Pistritto, G.; Cirone, M.; DOrazi, G. Reduced chemotherapeutic sensitivity in high glucose condition: Implication of antioxidant response. Oncotarget 2019, 10, 4691–4702. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garufi, A.; Baldari, S.; Pettinari, R.; Gilardini Montani, M.S.; D’Orazi, V.; Pistritto, G.; Crispini, E.; Giorno, E.; Toietta, G.; Marchetti, F.; et al. A ruthenium(II) curcumin compound modulates NRF2 expression balancing the cell death/survival outcome in both wild-type and mutant p53-carrying cancer cells. J. Exp. Clin. Cancer Res. 2020, 39, 122. [Google Scholar] [CrossRef]
- Garufi, A.; Giorno, E.; Gilardini Montani, M.S.; Pistrito, G.; Crispini, A.; Cirone, M.; D’Orazi, G. P62/SQSTM1/Keap1/NRF2 axis reduces cancer cells death-sensitivity in response to Zn(II)- curcumin complex. Biomolecules 2021, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Garufi, A.; Ubertini, V.; Mancini, F.; D’Orazi, V.; Baldari, S.; Moretti, F.; Bossi, G.; D’Orazi, G. The beneficial effect of Zinc(II) on low-dose chemotherapeutic sensitivity involves p53 activation in wild-type p53 cancer cells. J. Exp. Clin. Cancer Res. 2015, 34, 87. [Google Scholar] [CrossRef] [PubMed]
- Chian, S.; Li, Y.Y.; Wang, X.J.; Tang, X.W. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the Nrf2 pathway. Asian Pac. J. Cancer Prev. 2014, 15, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- DOrazi, G.; Givol, D. p53 reactivation: The link to zinc. Cell Cycle 2012, 11, 2581–2582. [Google Scholar] [CrossRef]
- Margalit, O.; Simon, A.J.; Yabukov, E.; Puca, R.; Yosepovich, A.; Avivi, C.; Jacob-Hirsch, J.; Gelernter, I.; Harmelin, A.; Barshack, I.; et al. Zinc supplement augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int. J. Cancer 2012, 131, E562–E568. [Google Scholar] [CrossRef]
- Cirone, M.; Garufi, A.; Di Renzo, L.; Granato, M.; Faggioni, A.; D’Orazi, G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunology 2013, 2, e26198. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Huang, M.T.; Prawan, A.; Liu, Y.; Hao, X.; Yu, S.; Lung Chen, W.K.; Chan, J.Y.; Reddy, B.S.; Yang, C.S.; et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. 2008, 1, 187–191. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, A.M.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Long. 2016, 2016, 1958174. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef]
- Mayo, L.D.; Rok Seo, Y.; Jackson, M.W.; Smith, M.L.; Rivera Guzman, J.R.; Koegaonkar, C.K.; Donner, D.B. Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J. Biol. Chem. 2005, 280, 25953–25959. [Google Scholar] [CrossRef]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-interacting protein kinase 2 phosphorylates p53 at Ser46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef]
- Di Stefano, V.; Soddu, S.; Sacchi, A.; D’Orazi, G. HIPK2 contributes to PCAF-mediated p53 acetylation and selective transactivation of p21 Waf1 after non-apoptotic DNA damage. Oncogene 2005, 24, 5431–5544. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, T.; Wang, H.; Lau, A.; Fang, D.; Zhang, D.D. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid. Redox Signal. 2012, 17, 1670–1675. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Givol, D.; D’Orazi, G. Regulation of p53 activity by HIPK2: Molecular mechanisms and therapeutical implications in human cancer cells. Oncogene 2010, 29, 4378–4387. [Google Scholar] [CrossRef]
- Torrente, L.; Sanchez, C.; Moreno, R.; Chowdhry, S.; Cabello, P.; Isono, K.; Koseki, H.; Honda, T.; Hayes, J.D.; Dinkova-Kostova, A.T.; et al. Crosstalk between NRF2 and HIPK2 shapes cytoprotective responses. Oncogene 2017, 36, 6204–6212. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Garufi, A.; Cirone, M. NRF2 interferes with HIPK2/p53 activity to impair solid tumors chemosensitivity. IUBMB Life 2020, 72, 1634–1639. [Google Scholar] [CrossRef] [PubMed]
- Verdina, A.; Di Segni, M.; Amoreo, C.A.; Sperduti, I.; Buglioni, S.; Mottolese, M.; Di Rocco, G.; Soddu, S. HIPK2 is a potential predictive marker of a favorable response for adjuvant chemotherapy in stage II colorectal cancer. Oncol. Rep. 2021, 45, 899–910. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Long. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- McMahonm, M.; Swift, S.R.; Hayes, J.D. Zinc-binding triggers a conformational switch in the cullin-3 substrate adaptor protein KEAP1 that controls transcription factor NRF2. Toxicol. Appl. Pharmacol. 2018, 360, 45–57. [Google Scholar] [CrossRef]
- Dhawan, D.K.; Chadha, V.D. Zinc: A promising agent in dietary chemoprevention of cancer. Indian, J. Med. Res. 2010, 132, 676–682. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garufi, A.; Pistritto, G.; D’Orazi, V.; Cirone, M.; D’Orazi, G. The Impact of NRF2 Inhibition on Drug-Induced Colon Cancer Cell Death and p53 Activity: A Pilot Study. Biomolecules 2022, 12, 461. https://doi.org/10.3390/biom12030461
Garufi A, Pistritto G, D’Orazi V, Cirone M, D’Orazi G. The Impact of NRF2 Inhibition on Drug-Induced Colon Cancer Cell Death and p53 Activity: A Pilot Study. Biomolecules. 2022; 12(3):461. https://doi.org/10.3390/biom12030461
Chicago/Turabian StyleGarufi, Alessia, Giuseppa Pistritto, Valerio D’Orazi, Mara Cirone, and Gabriella D’Orazi. 2022. "The Impact of NRF2 Inhibition on Drug-Induced Colon Cancer Cell Death and p53 Activity: A Pilot Study" Biomolecules 12, no. 3: 461. https://doi.org/10.3390/biom12030461
APA StyleGarufi, A., Pistritto, G., D’Orazi, V., Cirone, M., & D’Orazi, G. (2022). The Impact of NRF2 Inhibition on Drug-Induced Colon Cancer Cell Death and p53 Activity: A Pilot Study. Biomolecules, 12(3), 461. https://doi.org/10.3390/biom12030461