
Propofol, an Anesthetic Agent, Inhibits HCN Channels through the Allosteric Modulation of the cAMP-Dependent Gating Mechanism

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Complementary DNA, Cell Culture, and Transfection
2.2. Whole-Cell Patch-Clamp Recording and Data Analysis
2.3. Model Simulation of Electrophysiological Data
2.4. Statistical Analyses
3. Results
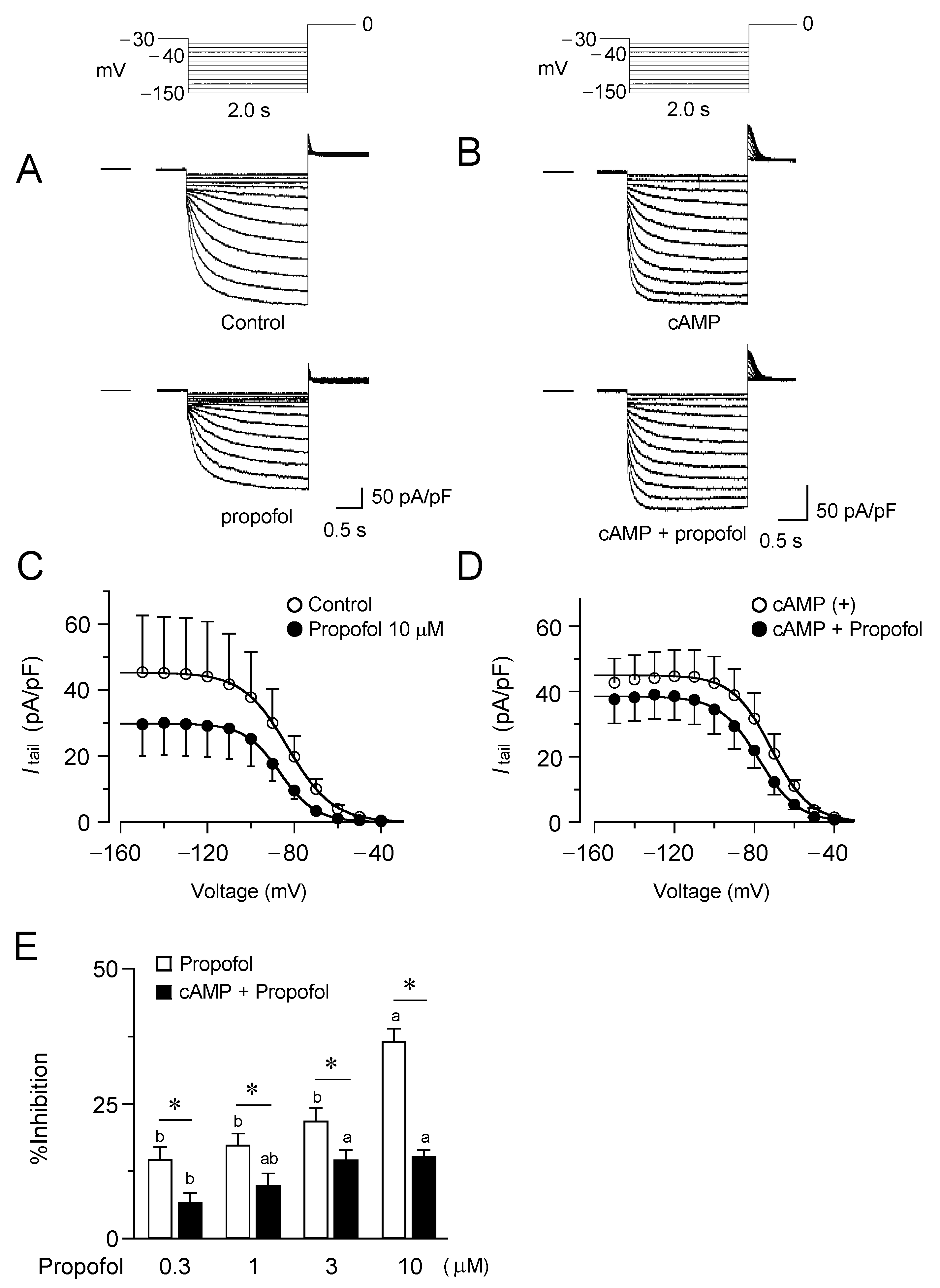
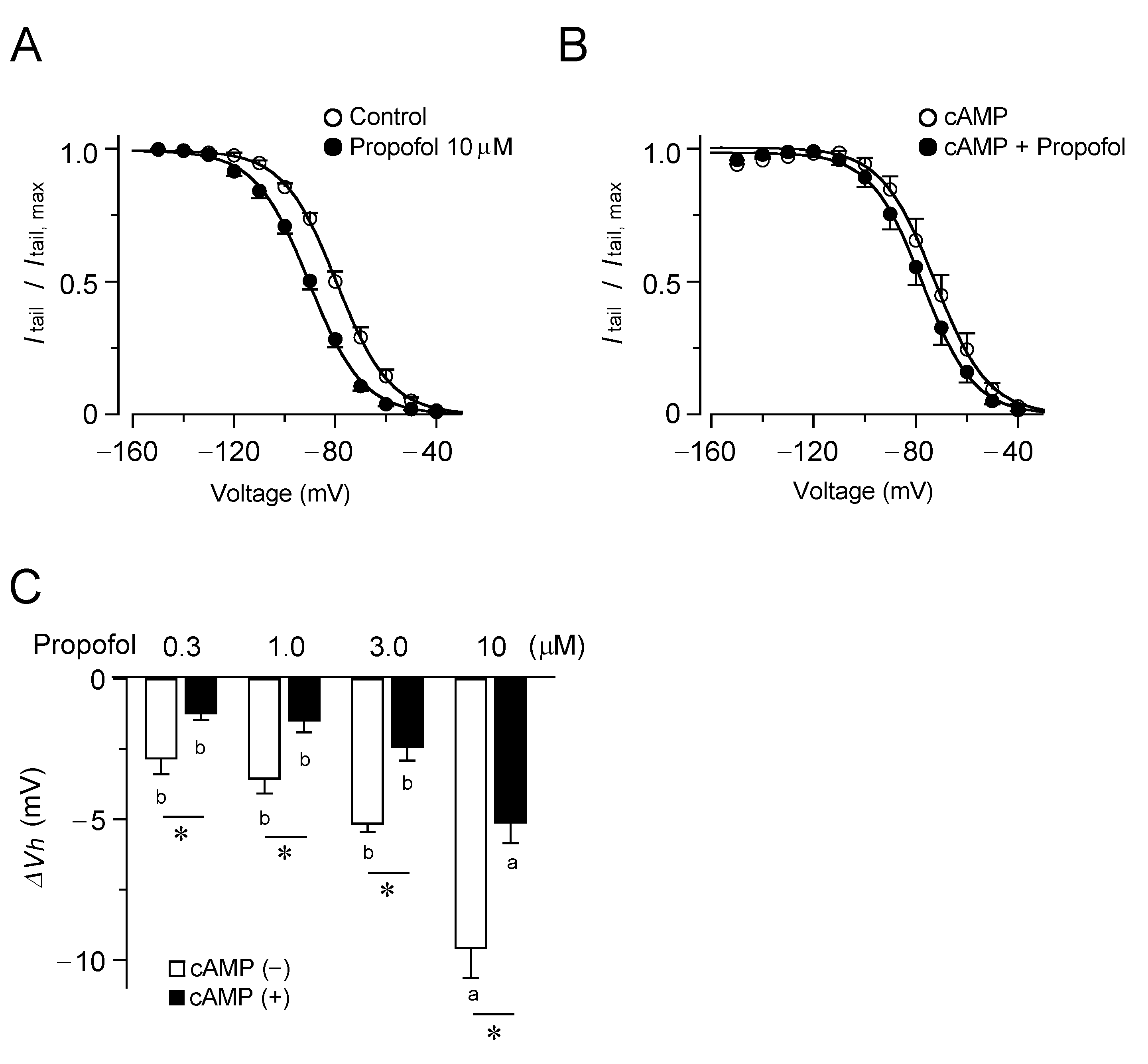
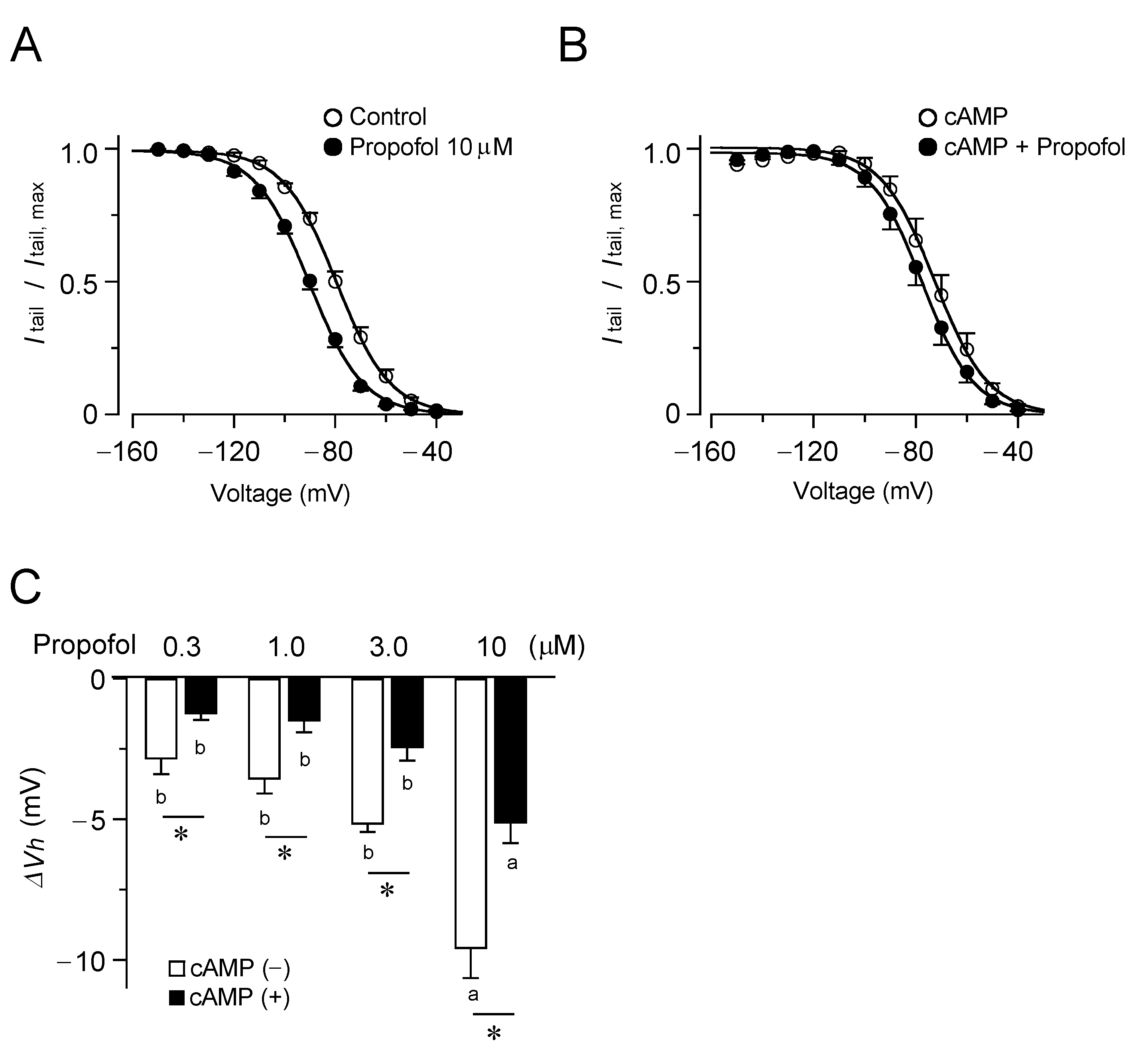
3.1. Inhibitory Effects of Propofol on HCN4 Channels Expressed in CHO Cells
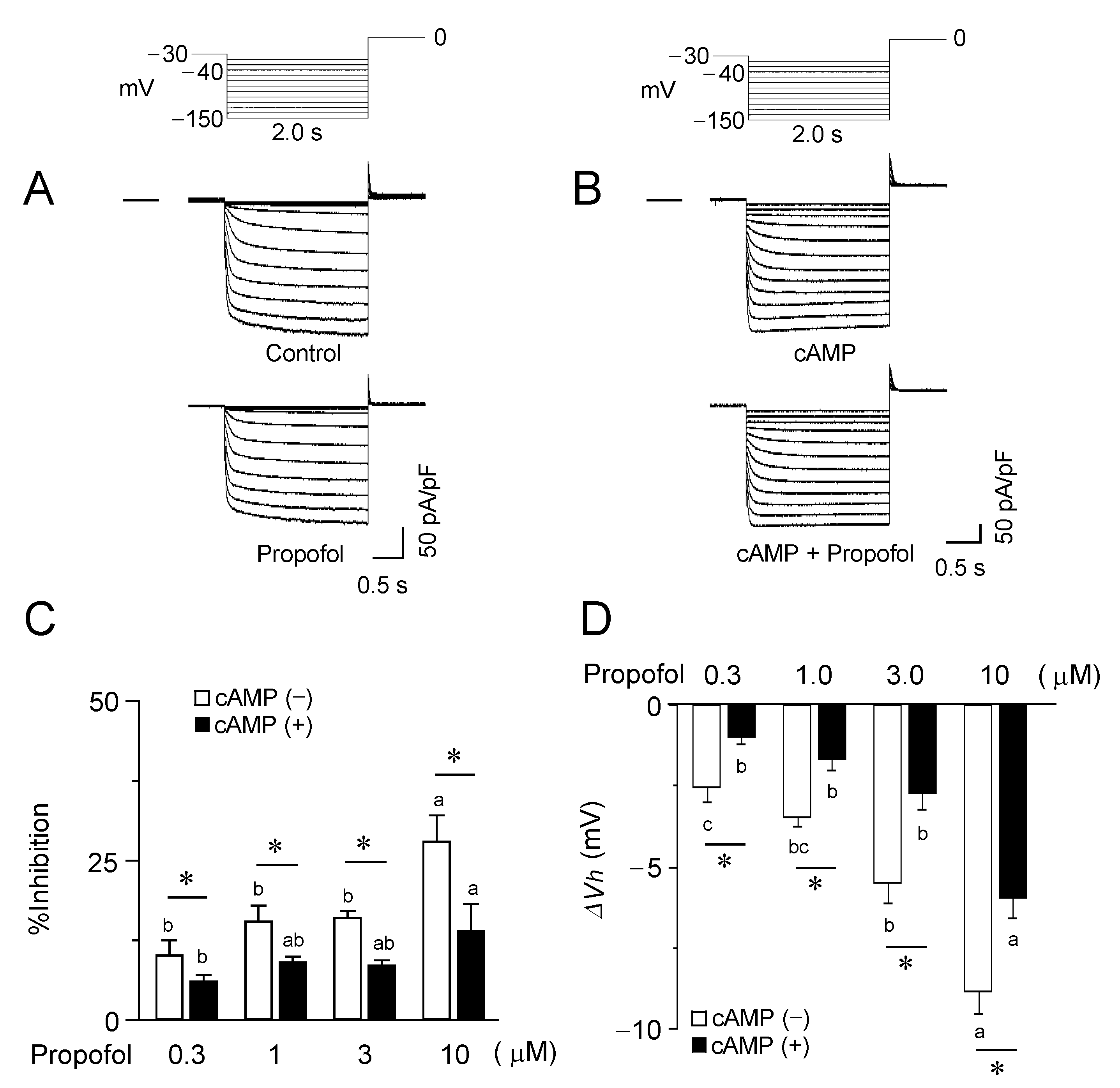
3.2. Inhibitory Effects of Propofol on HCN2 Channels Expressed in CHO Cells
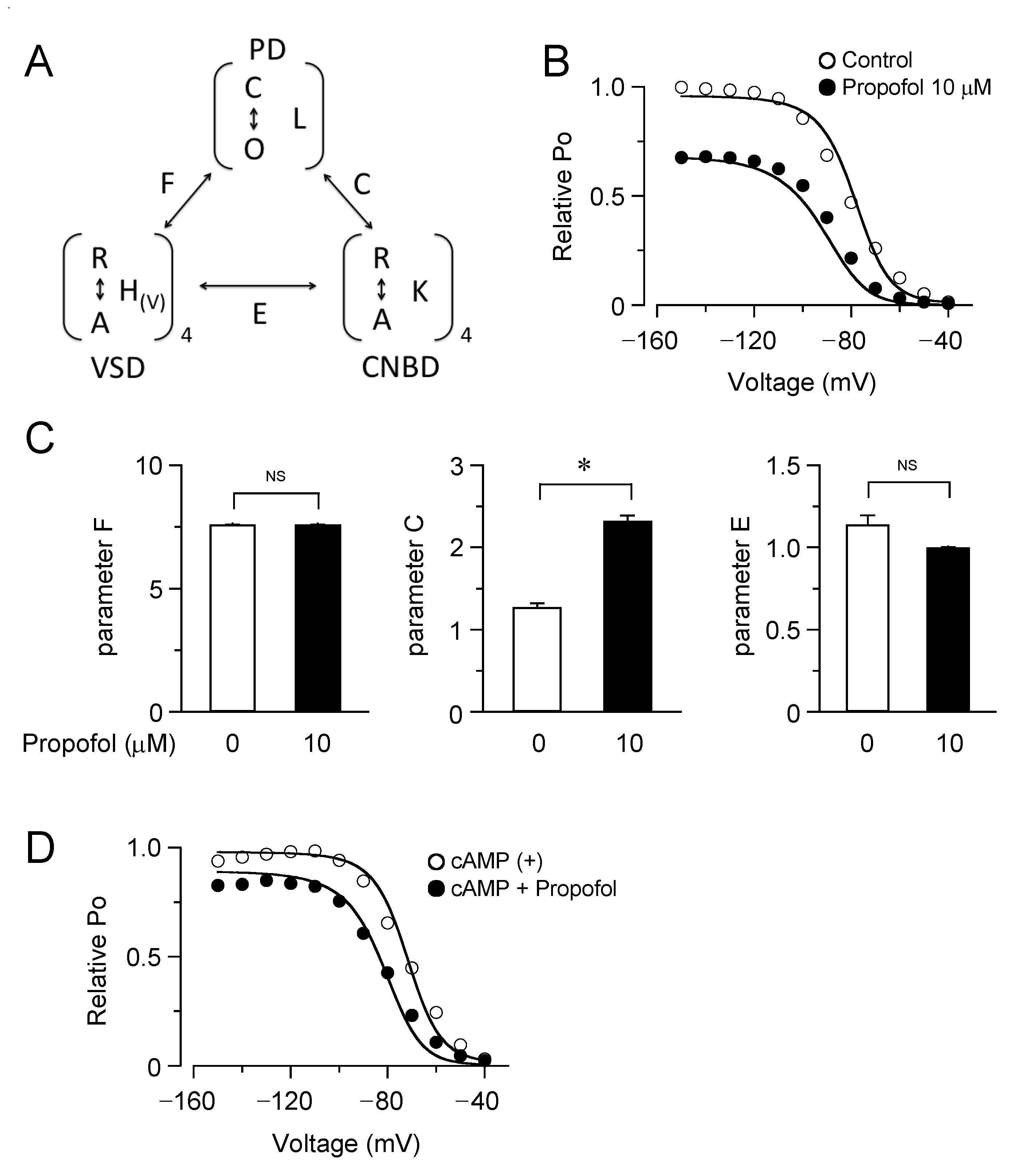
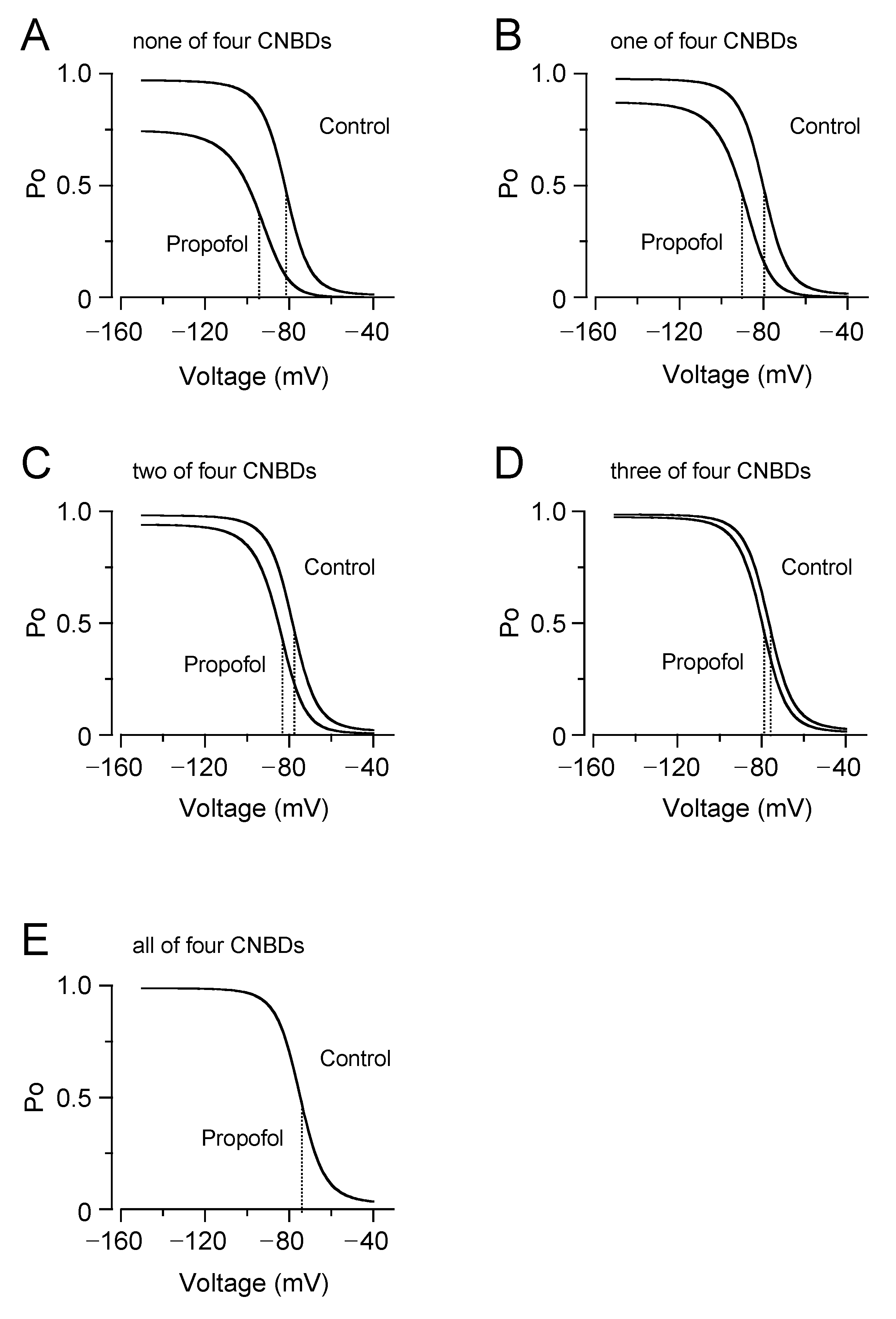
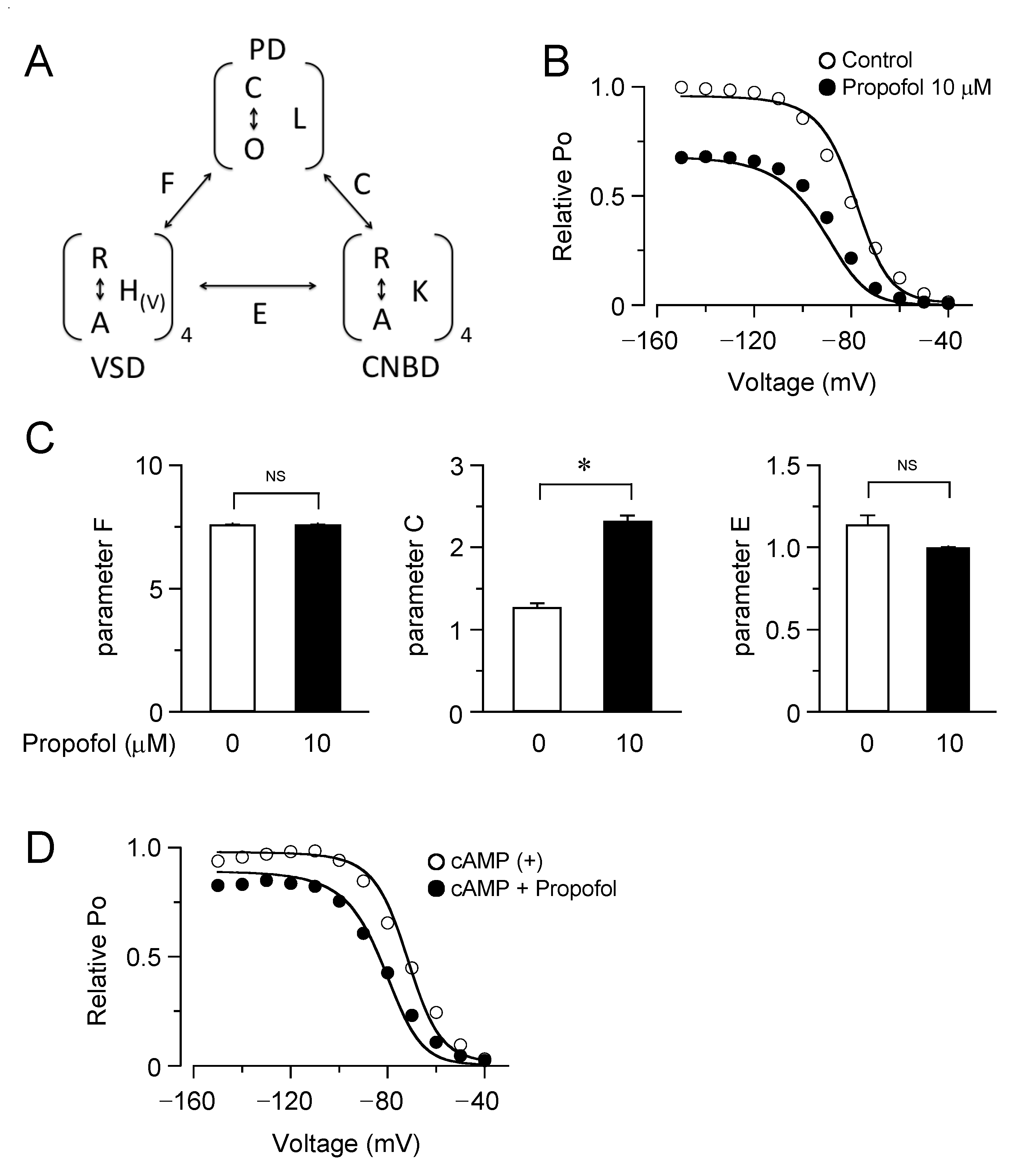
3.3. Computational Simulation of Propofol Effects on HCN4 Channels Using an Allosteric Gate Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, I.; White, P.F.; Nathanson, M.; Gouldson, R. Propofol: An update on its clinical use. Anesthesiology 1994, 81, 1005–1043. [Google Scholar] [PubMed]
- Franks, N.P. General anaesthesia: From molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 2008, 9, 370–386. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, H.C., Jr.; Akabas, M.H.; Goldstein, P.A.; Trudell, J.R.; Orser, B.A.; Harrison, N.L. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol. Sci. 2005, 26, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, U.; Antkowiak, B. Molecular and neuronal substrates for general anaesthetics. Nat. Rev. Neurosci. 2004, 5, 709–720. [Google Scholar] [CrossRef]
- Chen, X.; Shu, S.; Bayliss, D.A. Suppression of ih contributes to propofol-induced inhibition of mouse cortical pyramidal neurons. J. Neurophysiol. 2005, 94, 3872–3883. [Google Scholar] [CrossRef]
- Higuchi, H.; Funahashi, M.; Miyawaki, T.; Mitoh, Y.; Kohjitani, A.; Shimada, M.; Matsuo, R. Suppression of the hyperpolarization-activated inward current contributes to the inhibitory actions of propofol on rat CA1 and CA3 pyramidal neurons. Neurosci. Res. 2003, 45, 459–472. [Google Scholar] [CrossRef]
- Ying, S.W.; Abbas, S.Y.; Harrison, N.L.; Goldstein, P.A. Propofol block of I(h) contributes to the suppression of neuronal excitability and rhythmic burst firing in thalamocortical neurons. Eur. J. Neurosci. 2006, 23, 465–480. [Google Scholar] [CrossRef]
- Krajčová, A.; Waldauf, P.; Anděl, M.; Duška, F. Propofol infusion syndrome: A structured review of experimental studies and 153 published case reports. Crit. Care 2015, 19, 398. [Google Scholar] [CrossRef]
- Kevin, L.G.; Novalija, E.; Stowe, D.F. Reactive oxygen species as mediators of cardiac injury and protection: The relevance to anesthesia practice. Anesth. Analg. 2005, 101, 1275–1287. [Google Scholar] [CrossRef]
- Robinson, R.B.; Siegelbaum, S.A. Hyperpolarization-activated cation currents: From molecules to physiological function. Annu. Rev. Physiol. 2003, 65, 453–480. [Google Scholar] [CrossRef]
- Milanesi, R.; Baruscotti, M.; Gnecchi-Ruscone, T.; DiFrancesco, D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 2006, 354, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Tosaki, A. ArrhythmoGenoPharmacoTherapy. Front. Pharmacol. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.S.; Chetkovich, D.M. HCN channels in behavior and neurological disease: Too hyper or not active enough? Mol. Cell. Neurosci. 2011, 46, 357–367. [Google Scholar] [CrossRef]
- Zhou, C.; Liang, P.; Liu, J.; Ke, B.; Wang, X.; Li, F.; Li, T.; Bayliss, D.A.; Chen, X. HCN1 Channels Contribute to the Effects of Amnesia and Hypnosis but not Immobility of Volatile Anesthetics. Anesth. Analg. 2015, 121, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Kojima, A.; Ito, Y.; Kitagawa, H.; Matsuura, H. Ionic mechanisms underlying the negative chronotropic action of propofol on sinoatrial node automaticity in guinea pig heart. Br. J. Pharmacol. 2015, 172, 799–814. [Google Scholar] [CrossRef]
- Kojima, A.; Ito, Y.; Kitagawa, H.; Matsuura, H.; Nosaka, S. Direct negative chronotropic action of desflurane on sinoatrial node pacemaker activity in the guinea pig heart. Anesthesiology 2014, 120, 1400–1413. [Google Scholar] [CrossRef]
- Lee, C.H.; MacKinnon, R. Structures of the Human HCN1 Hyperpolarization-Activated Channel. Cell 2017, 168, 111–120.e111. [Google Scholar] [CrossRef]
- VanSchouwen, B.; Akimoto, M.; Sayadi, M.; Fogolari, F.; Melacini, G. Role of Dynamics in the Autoinhibition and Activation of the Hyperpolarization-activated Cyclic Nucleotide-modulated (HCN) Ion Channels. J. Biol. Chem. 2015, 290, 17642–17654. [Google Scholar] [CrossRef]
- Wainger, B.J.; DeGennaro, M.; Santoro, B.; Siegelbaum, S.A.; Tibbs, G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 2001, 411, 805–810. [Google Scholar] [CrossRef]
- Xu, X.; Vysotskaya, Z.V.; Liu, Q.; Zhou, L. Structural basis for the cAMP-dependent gating in the human HCN4 channel. J. Biol. Chem. 2010, 285, 37082–37091. [Google Scholar] [CrossRef]
- Dai, G.; Aman, T.K.; DiMaio, F.; Zagotta, W.N. Electromechanical coupling mechanism for activation and inactivation of an HCN channel. Nat. Commun. 2021, 12, 2802. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.E.; Zagotta, W.N. Insights into the molecular mechanism for hyperpolarization-dependent activation of HCN channels. Proc. Natl. Acad. Sci. USA 2018, 115, E8086–E8095. [Google Scholar] [CrossRef] [PubMed]
- James, Z.M.; Borst, A.J.; Haitin, Y.; Frenz, B.; DiMaio, F.; Zagotta, W.N.; Veesler, D. CryoEM structure of a prokaryotic cyclic nucleotide-gated ion channel. Proc. Natl. Acad. Sci. USA 2017, 114, 4430–4435. [Google Scholar] [CrossRef] [PubMed]
- Kojima, A.; Fukushima, Y.; Ito, Y.; Ding, W.G.; Ueda, R.; Seto, T.; Kitagawa, H.; Matsuura, H. Interactions of Propofol With Human Voltage-gated Kv1.5 Channel Determined by Docking Simulation and Mutagenesis Analyses. J. Cardiovasc. Pharmacol. 2018, 71, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, M.; Kojima, A.; Mi, X.; Ding, W.G.; Omatsu-Kanbe, M.; Kitagawa, H.; Matsuura, H. Characterization and functional role of rapid- and slow-activating delayed rectifier K(+) currents in atrioventricular node cells of guinea pigs. Pflug. Arch. Eur. J. Physiol. 2021, 473, 1885–1898. [Google Scholar] [CrossRef]
- Horrigan, F.T.; Aldrich, R.W. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 2002, 120, 267–305. [Google Scholar] [CrossRef]
- Tibbs, G.R.; Rowley, T.J.; Sanford, R.L.; Herold, K.F.; Proekt, A.; Hemmings, H.C., Jr.; Andersen, O.S.; Goldstein, P.A.; Flood, P.D. HCN1 channels as targets for anesthetic and nonanesthetic propofol analogs in the amelioration of mechanical and thermal hyperalgesia in a mouse model of neuropathic pain. J. Pharmacol. Exp. Ther. 2013, 345, 363–373. [Google Scholar] [CrossRef]
- Müller-Wirtz, L.M.; Maurer, F.; Brausch, T.; Kiefer, D.; Floss, M.; Doneit, J.; Volk, T.; Sessler, D.I.; Fink, T.; Lehr, T.; et al. Exhaled Propofol Concentrations Correlate With Plasma and Brain Tissue Concentrations in Rats. Anesth. Analg. 2021, 132, 110–118. [Google Scholar] [CrossRef]
- Stieber, J.; Thomer, A.; Much, B.; Schneider, A.; Biel, M.; Hofmann, F. Molecular basis for the different activation kinetics of the pacemaker channels HCN2 and HCN4. J. Biol. Chem. 2003, 278, 33672–33680. [Google Scholar] [CrossRef]
- Cacheaux, L.P.; Topf, N.; Tibbs, G.R.; Schaefer, U.R.; Levi, R.; Harrison, N.L.; Abbott, G.W.; Goldstein, P.A. Impairment of hyperpolarization-activated, cyclic nucleotide-gated channel function by the intravenous general anesthetic propofol. J. Pharmacol. Exp. Ther. 2005, 315, 517–525. [Google Scholar] [CrossRef]
- Ludwig, A.; Zong, X.; Stieber, J.; Hullin, R.; Hofmann, F.; Biel, M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999, 18, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Kojima, A.; Ding, W.G.; Kitagawa, H.; Matsuura, H. Dexmedetomidine Exerts a Negative Chronotropic Action on Sinoatrial Node Cells Through the Activation of Imidazoline Receptors. J. Cardiovasc. Pharmacol. 2021, 78, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Novella Romanelli, M.; Sartiani, L.; Masi, A.; Mannaioni, G.; Manetti, D.; Mugelli, A.; Cerbai, E. HCN Channels Modulators: The Need for Selectivity. Curr. Top. Med. Chem. 2016, 16, 1764–1791. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Zúñiga, R.; Concha, G.; Zúñiga, L. HCN Channels: New Therapeutic Targets for Pain Treatment. Molecules 2018, 23, 2094. [Google Scholar] [CrossRef] [PubMed]
- Lyashchenko, A.K.; Redd, K.J.; Yang, J.; Tibbs, G.R. Propofol inhibits HCN1 pacemaker channels by selective association with the closed states of the membrane embedded channel core. J. Physiol. 2007, 583, 37–56. [Google Scholar] [CrossRef]
- Craven, K.B.; Zagotta, W.N. CNG and HCN channels: Two peas, one pod. Annu. Rev. Physiol. 2006, 68, 375–401. [Google Scholar] [CrossRef]
- Lolicato, M.; Bucchi, A.; Arrigoni, C.; Zucca, S.; Nardini, M.; Schroeder, I.; Simmons, K.; Aquila, M.; DiFrancesco, D.; Bolognesi, M.; et al. Cyclic dinucleotides bind the C-linker of HCN4 to control channel cAMP responsiveness. Nat. Chem. Biol. 2014, 10, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Weißgraeber, S.; Saponaro, A.; Thiel, G.; Hamacher, K. A reduced mechanical model for cAMP-modulated gating in HCN channels. Sci. Rep. 2017, 7, 40168. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, A.; Cantini, F.; Porro, A.; Bucchi, A.; DiFrancesco, D.; Maione, V.; Donadoni, C.; Introini, B.; Mesirca, P.; Mangoni, M.E.; et al. A synthetic peptide that prevents cAMP regulation in mammalian hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. eLife 2018, 7, e35753. [Google Scholar] [CrossRef]
- Chen, X.; Sirois, J.E.; Lei, Q.; Talley, E.M.; Lynch, C., 3rd; Bayliss, D.A. HCN subunit-specific and cAMP-modulated effects of anesthetics on neuronal pacemaker currents. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 5803–5814. [Google Scholar] [CrossRef]
- Wang, Z.J.; Blanco, I.; Hayoz, S.; Brelidze, T.I. The HCN domain is required for HCN channel cell-surface expression and couples voltage- and cAMP-dependent gating mechanisms. J. Biol. Chem. 2020, 295, 8164–8173. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, F.M.; Schoenfeld, M.H.; Barrett, C.; Edgerton, J.R.; Ellenbogen, K.A.; Gold, M.R.; Goldschlager, N.F.; Hamilton, R.M.; Joglar, J.A.; Kim, R.J.; et al. 2018 ACC/AHA/HRS Guideline on the Evaluation and Management of Patients With Bradycardia and Cardiac Conduction Delay: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2019, 140, e382–e482. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bucchi, A.; Viscomi, C.; Mandelli, G.; Consalez, G.; Gnecchi-Rusconi, T.; Montano, N.; Casali, K.R.; Micheloni, S.; Barbuti, A.; et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. USA 2011, 108, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Stieber, J.; Stöckl, G.; Hofmann, F.; Ludwig, A. HCN4 provides a ’depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007, 26, 4423–4432. [Google Scholar] [CrossRef]
- Hoesl, E.; Stieber, J.; Herrmann, S.; Feil, S.; Tybl, E.; Hofmann, F.; Feil, R.; Ludwig, A. Tamoxifen-inducible gene deletion in the cardiac conduction system. J. Mol. Cell. Cardiol. 2008, 45, 62–69. [Google Scholar] [CrossRef]
- Kozasa, Y.; Nakashima, N.; Ito, M.; Ishikawa, T.; Kimoto, H.; Ushijima, K.; Makita, N.; Takano, M. HCN4 pacemaker channels attenuate the parasympathetic response and stabilize the spontaneous firing of the sinoatrial node. J. Physiol. 2018, 596, 809–825. [Google Scholar] [CrossRef]
- Mesirca, P.; Bidaud, I.; Briec, F.; Evain, S.; Torrente, A.G.; Le Quang, K.; Leoni, A.L.; Baudot, M.; Marger, L.; Chung You Chong, A.; et al. G protein-gated IKACh channels as therapeutic targets for treatment of sick sinus syndrome and heart block. Proc. Natl. Acad. Sci. USA 2016, 113, E932–E941. [Google Scholar] [CrossRef]
- Horiguchi, T.; Nishikawa, T. Propofol-nitrous oxide anesthesia enhances the heart rate response to intravenous isoproterenol infusion. Anesth. Analg. 2003, 96, 132–135. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Mende, U.; Diederich, M.; Hertle, B.; Memmesheimer, C.; Pohl, A.; Schmitz, W.; Scholz, H.; Steinfath, M.; Böhm, M.; et al. Chronic treatment with carbachol sensitizes the myocardium to cAMP-induced arrhythmia. Circulation 1996, 93, 763–771. [Google Scholar] [CrossRef]
- Ukai, M.; Ogawa, K. Cyclic nucleotides concentrations in the canine heart with regional ischemia. The role of cyclic AMP in ventricular fibrillation and the effect of dibutyryl cyclic AMP. Jpn. Circ. J. 1984, 48, 247–252. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Couette, B.; Bourinet, E.; Platzer, J.; Reimer, D.; Striessnig, J.; Nargeot, J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. USA 2003, 100, 5543–5548. [Google Scholar] [CrossRef]
- Toyoda, F.; Ding, W.G.; Matsuura, H. Responses of the sustained inward current to autonomic agonists in guinea-pig sino-atrial node pacemaker cells. Br. J. Pharmacol. 2005, 144, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, F.; Ding, W.G.; Matsuura, H. Heterogeneous functional expression of the sustained inward Na(+) current in guinea pig sinoatrial node cells. Pflug. Arch. Eur. J. Physiol. 2018, 470, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, F.; Mesirca, P.; Dubel, S.; Ding, W.G.; Striessnig, J.; Mangoni, M.E.; Matsuura, H. Ca(V)1.3 L-type Ca(2+) channel contributes to the heartbeat by generating a dihydropyridine-sensitive persistent Na(+) current. Sci. Rep. 2017, 7, 7869. [Google Scholar] [CrossRef] [PubMed]
- Ebert, T.J.; Muzi, M.; Berens, R.; Goff, D.; Kampine, J.P. Sympathetic responses to induction of anesthesia in humans with propofol or etomidate. Anesthesiology 1992, 76, 725–733. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimizu, M.; Mi, X.; Toyoda, F.; Kojima, A.; Ding, W.-G.; Fukushima, Y.; Omatsu-Kanbe, M.; Kitagawa, H.; Matsuura, H. Propofol, an Anesthetic Agent, Inhibits HCN Channels through the Allosteric Modulation of the cAMP-Dependent Gating Mechanism. Biomolecules 2022, 12, 570. https://doi.org/10.3390/biom12040570
Shimizu M, Mi X, Toyoda F, Kojima A, Ding W-G, Fukushima Y, Omatsu-Kanbe M, Kitagawa H, Matsuura H. Propofol, an Anesthetic Agent, Inhibits HCN Channels through the Allosteric Modulation of the cAMP-Dependent Gating Mechanism. Biomolecules. 2022; 12(4):570. https://doi.org/10.3390/biom12040570
Chicago/Turabian StyleShimizu, Morihiro, Xinya Mi, Futoshi Toyoda, Akiko Kojima, Wei-Guang Ding, Yutaka Fukushima, Mariko Omatsu-Kanbe, Hirotoshi Kitagawa, and Hiroshi Matsuura. 2022. "Propofol, an Anesthetic Agent, Inhibits HCN Channels through the Allosteric Modulation of the cAMP-Dependent Gating Mechanism" Biomolecules 12, no. 4: 570. https://doi.org/10.3390/biom12040570
APA StyleShimizu, M., Mi, X., Toyoda, F., Kojima, A., Ding, W.-G., Fukushima, Y., Omatsu-Kanbe, M., Kitagawa, H., & Matsuura, H. (2022). Propofol, an Anesthetic Agent, Inhibits HCN Channels through the Allosteric Modulation of the cAMP-Dependent Gating Mechanism. Biomolecules, 12(4), 570. https://doi.org/10.3390/biom12040570