Abstract
For the past several decades, the prevalence of obesity and type 2 diabetes (T2D) has continued to rise on a global level. The risk contributing to this pandemic implicates both genetic and environmental factors, which are functionally integrated by epigenetic mechanisms. While these conditions are accompanied by major abnormalities in fuel metabolism, evidence indicates that altered immune cell functions also play an important role in shaping of obesity and T2D phenotypes. Interestingly, these events have been shown to be determined by epigenetic mechanisms. Consistently, recent epigenome-wide association studies have demonstrated that immune cells from obese and T2D individuals feature specific epigenetic profiles when compared to those from healthy subjects. In this work, we have reviewed recent literature reporting epigenetic changes affecting the immune cell phenotype and function in obesity and T2D. We will further discuss therapeutic strategies targeting epigenetic marks for treating obesity and T2D-associated inflammation.
1. Introduction
The adoption of unhealthy lifestyles has led to an unprecedented prevalence of obesity and obesity-related disorders, including T2D [1]. This challenge cannot be addressed without a deeper understanding of the molecular mechanisms underlying these disorders. Along with well-investigated metabolic abnormalities, there is now accumulating evidence that the immune system also plays a major mechanistic role in the development of obesity and T2D [2].
The traditional view that immune system functions are confined to fighting pathogens has been challenged during the past decades as it became clear that the immune system plays major roles in metabolism [3]. Inflammation, for instance, is a classical immune-mediated reaction which has established as a defensive reaction against environmental insults. However, when uncontrolled, inflammation can become a chronic condition, triggering a cascade of events leading to metabolic dysfunction and contributing to a variety of metabolic disorders [4]. Indeed, chronic low-grade inflammation has been proposed as a key step in the pathogenesis of obesity-induced insulin resistance (IR) and T2D [5].
Initial evidence supporting the involvement of inflammation in obesity and T2D came from the finding that the pro-inflammatory molecule tumor necrosis factor-alpha (TNF-α) features increased expression in adipose tissue (AT) from obese rodents and humans with amelioration of IR after neutralization of this potent cytokine [6,7]. It is now well-recognized that excess fat mass in obese individuals is characterized by significant changes in the abundance and profile of infiltrating immune cells [8]. These changes include activation of pro-inflammatory macrophages and other immune cells that produce and secrete inflammatory cytokines and chemokines [2]. AT macrophages (ATM) from lean humans and animals are usually of the M2 type, or alternatively activated, which play a role in the attenuation and resolution of inflammation by secreting high levels of anti-inflammatory Interleukin (IL)-10 [9,10]. Conversely, the AT of obese individuals harbors a larger number of M1, or classically activated, macrophages which secrete several pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α [11,12]. These alterations precede and contribute to the development of systemic inflammation and IR, which in turn increases the risk of T2D [2]. In line with these findings, pro-inflammatory TNF-α levels have been found to be higher in T2D-prone Aγ mice than in wild-type mice [13]. As a further example, IL-6 levels, which are elevated in obese individuals and in patients with T2D, predict the development of these disorders [14,15]. Finally, IL-1β, is upregulated in islets of individuals with T2D. This master cytokine controls and is able to recruit several other cytokines, thereby implementing a broad inflammatory response in these conditions [16,17,18]. Overall, these studies indicate that components of the immune system may have specific roles in the etiology and progression of obesity and T2D. This, however, leads to the important question of what triggers the inflammatory events through the progression towards obesity and T2D.
The interactions between environmental and genetic factors appear to play a pivotal role in the onset and progression of obesity and T2D. Lifestyle factors, such as physical activity, glucose, and a high-fat diet, can modulate gene expression through epigenetic modifications [1]. Epigenetic processes control the expression of different genes, including genes involved in metabolism and the regulation of inflammatory pathways [19]. Recent studies have reported distinct epigenetic signatures in immune cells from obese individuals as well as in patients with T2D [3,20]. Thus, dysregulation of epigenetic mechanisms may represent an underlying cause of chronic inflammation in obesity and T2D. In the following section of this review, we will report on the current evidence supporting the important role of epigenetic dysregulation in determining chronic low-grade inflammation in obesity and T2D.
2. Epigenetics of Immune System and Its Role in the Development of Obesity- and T2D-Associated Inflammation
The immune system is one of the most complex networks in the body. Such complexity implicates an intricate circuit of different cell types which are closely interconnected and whose functions are based upon finely-tuned epigenetic regulation. Initial events leading to epigenetic control of immune cells occur at the time of hematopoietic stem cells (HSCs) lineage commitment. HSCs give rise to common lymphoid and myeloid progenitors that are the precursors, respectively, of all lymphoid (B, T, and NK cells) and myeloid cells (monocytes, neutrophils, eosinophils, basophils and mast cells). Importantly, all of these cells differ in their epigenetic programing. Convincing evidence indeed indicates that DNA methylation, chromatin remodeling by histone tail modifications, and non-coding RNA expression, in particular miRNA, represent the most commonly epigenetic events coordinating the main features of the immune system, including cell differentiation, identity and function [21]. Moreover, since altered immune functions and onset of chronic inflammation represent early pathophysiological mechanisms accompanying the development of obesity and T2D, where the epigenome is also profoundly altered, it is possible that epigenetic abnormalities impacting on immune cells relay environmental cues in obesity and T2D (Figure 1).
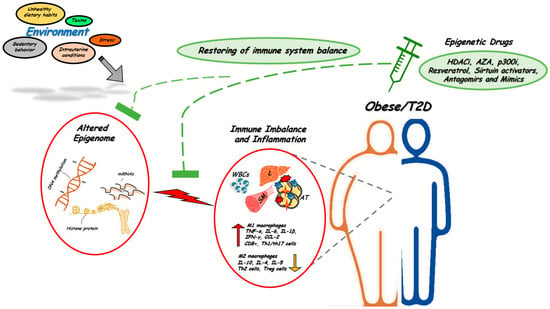
Figure 1.
Alterations in the epigenetic signature of immune system in obesity and T2D. Adverse environmental factors could affect the epigenetic signature of immune cells to generate or aggravate tissue pro-inflammatory states and induce obesity and T2D (red circles). Targeting epigenetic mechanisms offer novel opportunities for treatment and restoring obesity- and T2D-associated inflammation (green circles). Several epigenetic drugs, including HDACi and Antagomirs or Mimics are currently being tested in vivo in human and animal models of metabolic disorders. WBCs, white blood cells; L, liver; AT, adipose tissue; SM, skeletal muscle. ↑: up-regulated expression, ↓: down-regulated expression.
2.1. DNA Methylation
DNA methylation consists in the addition of methyl groups to DNA. The cytosine residues occurring in cytosine–phosphate–guanine dinucleotides (CpG site) are the main targets for DNA methylation. Hypermethylation of CpG-rich regions (i.e., CpG islands) at gene promoters is commonly associated with gene silencing, while their hypomethylation often results in transcriptional activation. Methylation events are controlled by specific enzymes known as DNA methyltransferases (DNMTs, such as DNMT1, DNMT3A, and DNMT3B). DNA methylation is reversible. Also, the enzymes of the ten-eleven translocation (TET) methyl-cytosine dioxygenase family, namely TET1, TET2 and TET3, catalyze the active demethylation process [1].
2.1.1. DNA Methylation and Regulation of Immunity
Changes in the DNA methylation status are of major relevance to the differentiation of HSCs. Genome-wide DNA methylation profiles of differentiating blood cells have revealed that myelopoiesis and lymphopoiesis can be segregated based on cell Methylome, with lymphopoiesis very much depending on the acquisition of DNA methylation marks, and myelopoiesis depending much more on loss of methylation marks [22]. Consistently, the combined loss of Dnmt3a and Dnmt3b in mouse HSCs impairs their differentiation capacity, along with a profound demethylation of genes normally restricted to stem cells (HSC multipotency genes [23,24]). Most studies have shown a remarkable dynamic plasticity in methylation during human monocyte-macrophage differentiation and subsequent activation, where both the gain and loss of methylation are biologically relevant [25,26,27]. In addition, macrophage polarization is actively controlled by DNA methylation. Indeed, a complete DNMT3b knockdown promotes macrophage polarization to the alternatively activated M2 phenotype (anti-inflammatory), together with a decreased expression of inflammatory genes such as TNFα, IL-1β, and an altered chemotactic capacity [28].
B cell development, which is responsible for antibody-mediated immunity, also falls under the control of DNA methylation. From their early differentiation in the bone marrow to their maturation in the spleen, human B cells undergo extensive changes in their DNA Methylome, in up to 30% of all measured CpG sites. Early differentiation stages are accompanied by a gradual, widespread demethylation of the enhancer regions containing binding sequences of key B cell transcription factors (TFs). At variance, late differentiation shows a large-scale demethylation of heterochromatin and methylation gain at polycomb-repressed regions that do not impact on B cell specific/functional genes [29]. Furthermore, several genes involved in proliferation, migration, and nuclear factor-κB (NF-κB) signaling in B cells show an inverse correlation between promoter DNA methylation and gene expression [30]. A strict control of the DNA Methylome is also required for the specification of the T cell subtype as helper (CD4+) or cytotoxic (CD8+) from thymocytes. In particular, a dynamic gain and loss of methylation at the Cd4 locus are essential to maintain CD4 silencing in CD8+ cytotoxic cells, and to enable CD4 stable expression in T helper (CD4+) cells, respectively [31]. Concomitantly, the Cd8b gene, which encodes for the CD8 co-receptor, undergoes promoter hypomethylation, showing an inverse correlation with the expression in thymocytes and lymph node CD8+ T cells [32].
In addition, DNA methylation is involved in the transcriptional regulation of the C-C motif chemokine receptor type 5 (CCR5) gene, which is the primary chemokine receptor utilized by HIV to infect the leukocytes. In fact, monocytes and CD4+ T cells expressing high levels of CCR5 feature an absence of methylation at the Ccr5 promoter region [33]. Similarly, changes in the methylation status at the T-box expressed in T cells (Tbet), Il-4, interferon γ (Ifnγ), and Forkhead box P3 (Foxp3) gene loci are essential for the polarization of CD4+ T cells towards the Th1, Th2, Th17, or to regulatory T cell (Treg) lineages [34,35,36]. The transcriptional regulation of some cytokine genes is also regulated by the active demethylation by TET2. Specifically, the expression of Ifnγ, Il-4, and Il-17 is impaired in TET2−/− mice [37]. Treg, expressing the FOXP3 TF, play a pivotal role in the maintenance of self-tolerance within the immune system, preventing inflammatory excess and autoimmunity. Importantly, DNMT1 actively participates with FOXP3 to differentiate Treg. A loss of this enzyme in mouse T cells impairs methylation maintenance, disrupting the Treg transcriptional program and causing lethal immunity [38,39,40].
2.1.2. DNA Methylation and the Development of Inflammation in Obesity and T2D
In obese humans, global DNA methylation levels in AT have been associated with an increased expression of specific pro-inflammatory genes, including the MCP-1 (monocyte chemoattractant protein-1) gene [41]. The chemokine product of this gene promotes macrophage infiltration into the AT and is directly implicated in the development of IR in obesity [42]. A study by Liu et al. has reported that the methylation level at the MCP-1 promoter decreases in peripheral blood mononuclear cells (PBMCs) from T2D patients. This epigenetic change is associated with serum MCP-1, blood glucose, and triglyceride levels, which may lead to a sustained increase of MCP-1 gene expression and contribute to the development of T2D-associated inflammation [43]. Also, methylation analysis in PBMCs of T2 diabetics showed decreased levels of DNA methylation at the IL-1β promoter, which was accompanied by an increased gene expression. This gene may represent an epigenetic marker of chronic inflammation and T2D development, as methylation at the IL-1β locus significantly correlates with fasting plasma glucose and glycated hemoglobin levels in T2D subjects [44,45].
IL-β1 mRNA production and its accumulation in the adipose tissue are also dependent on a glucose-induced activation of the TXNIP (Thioredoxin Interacting Protein) gene, which is a regulator of peripheral glucose metabolism in humans [46,47]. Interestingly, decreased skeletal muscle and blood TXNIP DNA methylation and increased skeletal muscle TXNIP expression have been reported both in subjects with increased risk of T2D and in individuals with overt disease [48]. Thus, an altered TXNIP methylation seems to represent a pathogenic mechanism involved in the induction of the inflammatory response accompanying T2D.
In another study, obese women were reported to exhibit higher plasma TNF-α levels accompanied by lower methylation at its promoter, linking an altered DNA methylation to systemic inflammation in obesity [49]. Similarly, Korean women with an excessive body weight were shown to feature aberrant methylation at the IL-6 gene promoter in blood cells [50]. Likewise, the MENA (Methyl Epigenome Network Association) project has reported a strong association between altered DNA methylation in PBMCs and waist circumference in specific methylation sites of several inflammatory genes, including GRIK3 (Glutamate ionotropic Receptor Kainate type subunit 3), ZC3H12D (Zinc Finger CCCH-Type Containing 12D) and TREM-1 (Triggering Receptor Expressed on Myeloid Cells 1) [51]. At variance, Simar et al. have reported no correlation between global DNA methylation in PBMCs (consisting of both lymphocytes and monocytes) and obesity or T2D [52]. However, they found that both obese and T2 diabetic individuals are characterized by an increased global DNA methylation in B and NK cells when compared to lean subjects, and this altered methylation profile is gene- and cytosine-specific.
The DNMTs also contribute to altered immune-cell function in metabolic diseases. For example, DNMT3B expression promotes M1 macrophage polarization and AT inflammation by increasing DNA methylation at the PPARγ1 promoter, leading to PPARγ1 downregulation. PPARγ1 has anti-inflammatory properties and favors the transcription of M2 macrophage-associated genes. In addition, DNMT3B is upregulated in in vitro isolated ATM treated with saturated fatty acids, whose plasma levels are increased in obesity. Deletion of this enzyme in macrophages not only enhances M2 macrophage polarization but also restores insulin sensitivity in adipocytes [28,53]. Myeloid DNMT1 inhibition leads to a decreased macrophage PPARγ1 promoter DNA methylation, which, in turn, promotes an increased ATM alternative activation and decreased inflammation in obese animals [54]. DNMT1 is also responsible for the hypermethylation of the adiponectin promoter in obese mice and humans, and causes decreased expression of this anti-inflammatory cytokine, worsening of IR and obesity-associated inflammation [55].
Taken together, these findings indicate that obesity and T2D are intimately linked to a remodeling of the DNA Methylome both in WBCs and in specific immune cell sub-populations, contributing to the altered immune function reported in these metabolic disorders. In the future, changes in the Methylome of immune cells might be used for the prediction of individual disease risk and as targets for treatment.
2.2. Histone Modifications
In the eukaryotes, the basic unit of chromatin is the nucleosome, which is a dynamic structure of 147 base pairs of DNA wrapped around an octamer of core histone proteins (two copies of each histone H2A, H2B, H3, and H4). Each histone has an N-terminal tail, that protrudes from the nucleosome, which can be decorated with various post-translational modifications (PTMs), including acetylation, methylation, phosphorylation and ubiquitination. These PTMs, which are positioned or removed by specialized modifying enzymes, either loosen (enabling access for transcriptional machinery) or lock (preventing access) the packing of the DNA around the histones, thus regulating gene expression [56].
2.2.1. Histone Modifications in Immunity
Parallel to changes in the DNA methylation pattern, histone modifications also represent key regulators of genome structure and gene expression which affect the immune response [57]. Upon activation, naïve CD4+ T cells can differentiate into a variety of T cell subsets, including Th1 or Th2 lineages. CD4+ T cell lineage commitment correlates with dynamic changes in histone PTM deposition at the gene loci associated with directing subset specific CD4+ T cell effector function [58]. For instance, this is the case for the expression of Ifnγ in the CD4+ T cell. The Ifnγ locus shows a strong enrichment of the repressive mark H3K27me3 (tri-methylation of histone H3 at lysine 27) upon Th2 differentiation and in naïve CD4+ T cells, while the same locus features increased levels of the permissive histone mark H3K4me2 during differentiation of Th1 cells [59]. A similar scenario also occurs in directing the naïve CD8+ T cell fate. In particular, the deacetylated histone H3, which is a marker of open chromatin, increases as naive CD8+ T cells develop into memory T cells. Interestingly, this modification has been proposed as a marker for assessing memory T cell functionality, since memory T cells with defective rapid recall ability have less deacetylated histone H3 than their fully functional counterparts [57].
The development and activation of different B-cell subsets are also associated with the acquisition of a distinct pattern of histone marks [60]. For example, analysis of histone lysine methylation in resting cells and activated cells has shown that methylation of histone H3 at lysines 4 (H3K4), 9 (H3K9), 27 (H3K27) and histone H4 at lysine 20 is markedly reduced in quiescent B lymphocytes as compared with activated B cells [61]. The potential role of histone changes in controlling B cell function has been further documented by the ability of histone deacetylase inhibitors (HDACi) to affect B cell proliferation, survival and differentiation in a lupus-prone mouse model [62].
Histone change events are also associated with the determination of macrophage fate. For instance, the enzyme SMYD-3 (SET and MYN domain), an H3K4 methyltransferase, has been shown to positively regulate M2 polarization. Its activity was increased in human macrophages upon exposure to M-CSF (macrophage colony stimulating factor) in combination with IL-4 and IL-13, while it was decreased upon exposure to M1 stimulation [63]. Another typical example is represented by JMJD3 (Jumonji Domain Containing Protein D3), a H3K27 demethylase which has been recognized to control M2 macrophage markers. JMJD3 promotes di- and tri-demethylation of H3K27, leading to an activation of Arg1 (arginase) and Irf-4 (interferon regulatory factor 4), among other M2 markers [64]. Histone acetylation can further influence macrophage polarization. HDAC3, for example, promotes M1 macrophage polarization by acting as an epigenomic brake in macrophage alternative activation [65], while HDAC2, recruited by Tet2, represses transcription of IL-6 via histone deacetylation, promoting a resolution of the inflammation [66]. Moreover, HDAC9 deletion results in downregulation of inflammation-related genes and in polarization of macrophages towards the M2-like phenotype through a strong enrichment of acetylated H3 at ABCA1 (ATP binding cassette subfamily A member 1), ABCG1 (ATP binding cassette subfamily G member 1), and PPARγ (Peroxisome proliferator-activated receptor gamma) macrophage promoters [67]. Macrophage trafficking is also under the control of histone marks. Indeed, H3K9ac and H3K4me3 regulate the expression of the CCL2 and CCL3 chemokines (C-C motif chemokine ligand) and their receptors [68].
Thus, the activation of immune cells is dependent on a complex network of transcriptional regulators whose loci are characterized by histone marks which dynamically switch from a repressive to a permissive state in order to enable a prompt response to external stimuli. Histone modifying agents (e.g., HDACi) are currently under extensive investigation for their potential efficacy in inflammatory disorders.
2.2.2. Histone Marks in Obesity and T2D
A number of changes in histone PTMs with impact on inflammatory pathways have been reported in obesity and T2D [69]. Culturing monocytes in the presence of high glucose levels leads to increased H3 hyperacetylation at the promoters of NF-κB-dependent inflammatory genes, mimicking findings in T2D patients. Consistently, increased levels of histone acetylation at the promoter of TNF-α and COX-2 (Cyclooxygenase-2) have been observed upon treatment of monocytes with HDACi [70]. The histone methyltransferase SET7/9 (SET domain containing 7) is an essential co-activator of NF-κB and mediates methylation at H3K4. In human monocytes, SET7/9 enhances the recruitment of p65, which acts as a trigger for the induction of expression of NF-κB-related genes. In monocytes exposed to TNF-α, SET7/9 deletion not only inhibits inflammatory genes, including TNF-α, MCP-1 and IL-6, but also attenuates diabetes-induced inflammatory processes [71]. Importantly, Panemi et al. have shown that in PBMCs from T2 diabetics, SET7/9 increases methylation of H3K4 at the NF-κB p65 promoter leading to an higher expression of MCP-1, ICAM-1 (Intercellular Adhesion Molecule 1) and COX-2 [72]. Gallagher et al. also reported that, at the IL-2 gene promoter, H3K27me3, a repressive histone methylation mark, is decreased in bone marrow progenitors, which is passed to wounded macrophages in a murine model of glucose intolerance. These epigenetically pre-programming generates poised macrophages in peripheral tissues and negatively impact on wound repair. Decreased methylation at H3K27 is accomplished by Jmjd3. Its inhibition enhances H3K27me and IL-12 levels, providing an innovative, though potential, epigenetic approach for wound healing in T2D [73]. Furthermore, in human monocytes, hyperglycemia induces decreases in the histone repressor mark H3K9me3 at IL-6 promoter, accompanied by increased IL-6 expression and exacerbation of inflammation.
Obese individuals exhibit reduced levels of HDAC4 in PBMC and AT, which is negatively correlated with circulating levels of the pro-inflammatory chemokine RANTES/CCL5. Intriguingly, in the same subjects HDAC4 expression was restored after physical exercise, which makes HDAC4 an appealing, though potential, target for mitigating obesity-associated inflammation [74]. In addition, gene expression analyses have revealed a correlation between different HDACs (HDAC2, 4, 5, and 6), adiposity parameters, and obesity-associated inflammatory events in subcutaneous and visceral fat depots of obese women [75]. Of note, dietary-induced and genetic mouse models of obesity display a decreased level of KDM1A (Lysine Demethylase 1A), a histone demethylase, in AT, which correlates with an increased expression of pro-inflammatory genes [76].
It has also been reported that treatment of hepatocytes with free fatty acid upregulates the expression of the Brahma-related gene1 (Brg1), which, in turn, promotes inflammation by increasing permissive histone acetylation at the promoter regions of the IL-1, IL-6 and MCP-1 genes. Depletion of Brg1 attenuates the release of pro-inflammatory mediators in the liver and significantly ameliorates obesity-related hepatic inflammation [77]. In vitro lipopolysaccharide (LPS)-treated hepatocytes display an increased histone acetylation of H3K9/K18Ac at the inflammatory TNF-α and Ccl2 gene loci [78]. In addition, the H3 deacetylase SIRT (Sirtuin)-1 exhibits a decreased expression in the AT of obese patients, resulting in enhanced macrophage recruitment through chemoattractant and cytokine secretion [79]. On the same line, obese children with IR show downregulation of both SIRT1 and 2 in PBMCs [80,81], while administration of hypocaloric diets in obese individuals results in SIRT1 and 2 upregulation [82]. The crucial role of histone modifying enzymes (e.g., HDACs) and their inhibitors (e.g., HDACi) in the regulation of inflammatory events makes them ideally suited targets for the treatment of inflammatory diseases, such as obesity and T2D.
2.3. microRNAs
MiRNAs are single-stranded RNAs containing 20–24 bases. They primarily act as post-transcriptional repressors by targeting mRNA 3′-untranslated regions and stimulating their degradation and translation repression [83,84]. In mammals, miRNAs are predicted to directly control up to 60% of all protein-coding genes. In addition, a specific miRNA is able to regulate more than one target mRNA even within the same signaling pathway, thereby creating different steps of gene regulation [85].
2.3.1. Role of microRNAs in the Immune Response
Emerging evidence indicates that miRNAs play very important roles in immune cells, including HSC differentiation and maturation [86]. For example, the combined deletion of miR-23a and miR-23b leads to an improper hematopoietic progenitor cell production and differentiation in mice [87]. Other miRNAs which are responsible for regulating hematopoietic lineage differentiation include miR-181, miR-223, and miR-142 [88]. Furthermore, Shangqin Guo et al. [89] have reported an essential role of Dicer (the enzyme responsible for miRNA biogenesis [90]) in HSC maintenance and have identified miR-125a as a positive regulator of the HSC regeneration of hematopoiesis, at least in part, by reducing the pro-apoptotic protein Bak1 (Bcl2 antagonist/killer 1 [89]).
MiRNAs finely tune the differentiation and activation of B cells. The expression of miR-1246 in normal B cells regulates cell responsiveness by upregulating the expression of several molecules that allow B cells to interact with T cells and provide an effective immune response [91]. MiR-16 and miRNA-let-7c modulate the ability of CD4 T cells to discriminate between activating and energizing stimuli by targeting the mTOR (mechanistic target of rapamycin kinase) components Mtor and Rictor (RPTOR independent companion of mTOR complex 2 [92]). Also, Dicer−/− CD4+ T cells favor Th1 cell differentiation via an increased expression of the Th1-specific transcription factor Tbx21 (T-box transcription factor 21) and production of IFNγ [93]. At variance, miR-29a and miR-29b negatively regulate Th1 differentiation by repressing Tbet and Eomes (Eomesodermin), two transcription factors known to induce Th1 lineage and IFNγ production [93]. Several studies have further identified differentially expressed miRNAs that provide M1 and M2-polarized macrophages signatures [94]. In particular, M1-polarized macrophages are feature a high expression of miR-155, miR-9, miR-146a, and miR-19, whereas higher levels of miR-26a-2-3p and let-7c identify M2-polarized macrophages [95]. MiR-127 [96] and miR-125b [97] also induce M1 polarization by targeting Bcl6 (B cell leukemia/lymphoma 6) and IRF4 (Interferon regulatory factor 4), respectively, with a consequent increase in the expression of pro-inflammatory cytokines.
The NF-κB family of transcription factors coordinates the expression of an array of genes involved in the inflammatory responses (e.g., TNF-α, IL-1β, and IL-6). Interestingly, a specific subset of miRNAs, including miR-146a, miR-155, and miR-9, are directly controlled by this inflammatory signalling pathway, and in several cases these miRNAs act as a feedback control system of the NF-κB-dependent immune response by regulating its key members [94]. Thus, the intricate miRNA network represents a pivotal player which actively participates in the modulation of immune cell function and inflammatory response.
2.3.2. MicroRNAs and the Development of Inflammation in Metabolic Disorders
A growing body of evidence supports the hypothesis that specific miRNAs regulate obesity- and T2D-associated inflammation. Mir-320, involved in the modulation of insulin signaling, is increased in the AT from obese individuals and exhibits a positive correlation with elevated levels of several inflammatory markers, including TNF-α, NF-κB, and IL-6. Interestingly, adipocyte knockdown of mir-320 induces downregulation of these inflammatory factors, suggesting that selectively inhibiting mir-320 represents a strategy to ameliorate inflammation response in obesity settings [98]. Likewise, in obese patients, a reduced expression of mir-126 and mir-193b is associated with upregulation of the chemokine CCL2, while their overexpression attenuates CCL2 production in human monocytes, thus affecting immune cell recruitment and AT inflammation [99]. By using an integrated computational strategy, Zhang et al. have identified 23 active miRNA-TF gene regulatory pathways which are significantly associated with obesity-related inflammation. Among these, mir-193b is of particular interest since its expression strongly correlates with signaling through the TNF-α pathway [100]. Several exosomal miRNAs are also differentially expressed in subcutaneous and visceral AT from patients with obesity. Notably, these miRNAs preferentially target genes involved in key inflammatory processes. It is proposed that these miRNAs and their target genes represent novel therapeutic targets for metabolic disease management [101]. Besides, the expression of adipocyte-secreted mir-34a has been reported to increase progressively with the development of dietary obesity, whereas its adipose-selective ablation leads to macrophage polarization into the anti-inflammatory M2 phenotype, and protects mice from inflammation and IR caused by dietary stress [102].
It has also been reported that several anti-inflammatory miRNAs, such as mir-1934, mir-532-5p and mir-146a are downregulated in overweight and obesity, which may mechanistically contribute to the chronic inflammatory state associated with these disorders [103,104,105,106]. T2D patients are associated with decreased PBMC expression of mir-146a, which is correlated with IR, poor glycemic control, and expression of several pro-inflammatory genes with increased plasma levels of TNF-α and IL-6 [107]. Analysis of plasma miRNA expression profiles in patients with T2D has revealed decreased levels of a set of miRNAs involved in inflammatory pathways [108]. For example, mir-15a shows a downregulation under hyperglycemic conditions, while its overexpression attenuates the pro-inflammatory signaling of IL-1β, TNF-α and NF-κB [109]. Anti-inflammatory function role has also been proposed for mir-20b and mir-29b, as these miRNAs target proteins responsible for the differentiation of the Th17 and Th1 T-helper cells, which produce pro-inflammatory cytokines [110,111]. Interestingly, deregulation of mir-29b along with that of mir-15a precedes T2D onset [108].
Mir-103 and mir-143 are implicated in adipogenesis dysfunction and IR, and feature increased expression in PBMCs from murine models of pre-diabetes and T2D [112,113]. Increased expression of several members of the mir-200 family is associated with T2D-related vascular inflammation. In particular, mir-200b, mir-200c, and mir-492 induced overexpression of COX-2 and MCP-1 in vascular smooth muscle cells (VSMCs) from diabetic mice by repressing the Zeb1 (Zinc Finger E-Box Binding Homeobox 1), which negatively regulates inflammatory genes [114]. Additionally, mir-504 was also found to be increased in VSMCs from diabetic mice and shown to reduce the expression of two target genes, namely Grb10 (Growth factor receptor bound protein 10) and Egr2 (Early growth response 2), whose downregulation facilities the expression of a set of inflammatory genes in mice [115].
These studies underline the involvement of specific miRNAs in the development of obesity- and T2D-related inflammation. Some of these miRNAs may not only become promising tools for treatment of obesity and T2D, but may also be used as biomarkers for the early detection and staging of metabolic disorders. The use of miRNAs as diagnostic and therapeutic tools in clinical settings holds several advantages, including their stability and easy detectability in body fluids. Nonetheless, factors such as a lack of standardized measurements and/or an absence of a disease-specific miRNA database make this attractive goal still challenging.
2.4. Adipocyte Hypertrophy: A Paradigm of Epigenetic Derangement in Chronic Inflammation
Obesity and a family history of T2D are major predisposing factors for the development of T2D [116]. In both of these conditions an inappropriate expansion of subcutaneous adipose cells, has been reported [117]. Key characteristics of this abnormality, in addition to enlarged adipocytes, is the impaired adipocyte differentiation, presence of senescent fat cells, fibrosis and, most importantly, evidence of inflammatory state [118]. Notably, hypertrophic obesity has long been known to be an independent predictor of T2D [119,120]. Research by our group has recently identified epigenetic processes as key mechanisms in the development of these abnormalities. In these studies, we have also shown that adipocyte hypertrophy in first-degree relatives of T2 diabetics (FDR) and in obese subjects is accompanied by epigenetic alterations that affect genes responsible for the development of a pro-inflammatory state (Table 1 and Table 2). This makes of adipocyte hypertrophy a paradigm of epigenetic-associated inflammatory disease.

Table 1.
Epigenetic alterations affecting inflammatory genes/pathways in human hypertrophic obesity models.
We have recently reported that the Methylome profile of subcutaneous adipocyte precursor cells (APCs) from individuals who are FDR is associated with extensive hypomethylation events affecting major molecular pathways involved in the transduction signals controlling inflammation processes [117]. The strongest hypomethylation signal was identified at the PTPRD (Protein Tyrosine Phosphatase Receptor Type D) gene, which marks hypertrophic obesity in FDR. This methylation pattern was also replicated in obese individuals. PTPRD belongs to the protein tyrosine phosphatase (PTP) superfamily and has been identified as a T2D susceptibility gene [126,127]. Importantly, PTPs have been associated with multiple immune-related disorders in both animal models and in human diseases [128]. Even more recently, Spinelli et al. have reported that in healthy FDR subjects, the senescence-related ZMAT3 (Zing Finger Matrin-Type 3) gene exhibited a decreased methylation, which was responsible for its upregulation [121]. ZMAT3 regulates p53 which in turn acts as a molecular link between pathways involved in inflammation and IR in the AT [13,129]. Of note, APCs from these FDR subjects displayed an enhanced secretion of several pro-inflammatory factors, such as IL-6, MCP-1, RANTES, and IL-8, which further appeared to be connected with ZMAT3 hypo-methylation [121].
Table 2.
Epigenetic alterations affecting inflammatory genes/pathways in mouse hypertrophic obesity models.
Table 2.
Epigenetic alterations affecting inflammatory genes/pathways in mouse hypertrophic obesity models.
| Study Model | Epigenetic Marks | Position | Processes | Medical Condition | Species | Ref |
|---|---|---|---|---|---|---|
| AT from Diet-induced Obesity model | DNA Hyper-methylation | Hoxa5 | Hox Gene Family, Adipogenesis, AT Macrophage Genes | Obesity, Impaired Glucose Metabolism, AT Inflammation | Mouse | [130] |
| AT from Diet-induced Obesity model | - | Hoxa5 | ER Stress Signalling pathways, M2 Macrophage Polarization | Obesity, Impaired Glucose Metabolism, AT Inflammation | Mouse | [131] |
| PMCs from Diet-induced Obesity Model | - | Zfp423 | Nf-κb Inflammatory pathway, ATM accumulation, LPS-induced Inflammation | Obesity, AT Inflammation | Mouse | [132] |
| AT from Diet-induced Obesity and from Obese subjects | DNA Hyper-methylation | Ankrd26 | Adipocyte pro-inflammatory secretion, Il-8, Mcp-1, Rantes/Pro-inflammatory profile in AT | Obesity, Impaired Glucose Metabolism, Adiposity, AT Inflammation | Mouse | [133] |
AT, adipose tissue; APC, adipocyte precursor cells; PBL, peripheral blood leukocytes; FDR, first-degree relatives of type 2 diabetics; ER, endoplasmic reticulum; PMCs, perivascular mesen-chymal cells; ATM, adipose tissue macrophages; LPS, lipopolysaccharides.
Not only loss of DNA methylation, but also the increase in methylation levels at specific genes and genomic regions is biologically relevant in shaping the phenotype of individual conditions. For instance, we have found that, in FDR and obese individuals, hypertrophic obesity is associated with increased methylation levels at the promoter of the HOXA5 (Homeobox A5) gene. In human pre-adipocytes, HOXA5 silencing leads to an impaired adipogenesis along with inappropriate activation of WNT-signaling (Wingless-related integration site) genes, which leads to local and systemic inflammation in AT [122]. Importantly, Hoxa5 reduced inflammatory cytokine secretion and promoted an increased number of M2 macrophages in the AT of high-fat diet mice [131], whereas its methylation-dependent silencing was associated with an elevated expression of macrophage marker genes, including F4/80, Cd68 (Cluster of differentiation 68) and Mcp-1, and a disturbed glucose metabolism [130]. Similarly, we have observed massive hypermethylation at the promoter of the human ZNF423 (Zinc Finger Protein 423) gene, which accounted for the reduced ZNF423 expression observed in hypertrophic obesity [123]. Zfp423 has been identified as a transcriptional break on NF-κB signaling. In perivascular mesenchymal cells Zfp423 suppresses inflammatory signaling and attenuates metabolic inflammation in the AT of diet-induced obesity, while its ablation, facilitating the activation of NF-κB signaling, exacerbates AT macrophage accrual and promotes AT dysfunction [132]. Furthermore, we demonstrated that diet-induced obesity in mice led to a hyper-methylation of the Ankrd26 (Ankyrin repeat domain containing 26) gene promoter, which further contributed to increased secretion of pro-inflammatory factors in AT [133]. This gene has been previously associated with the development of obesity and T2D. Consistently, in the PBL from obese individuals, epigenetic silencing of the ANKRD26 gene by promoter hyper-methylation correlated with increased levels of several inflammatory factors including IL-6, IL-8, and RANTES [124].
In addition to DNA methylation changes, alterations in miRNA expression and histone modifications also contribute to the inflammatory state accompanying hypertrophic AT changes. Analysis of miRNome profile in APC from FDR identified a set of differentially expressed miRNAs featuring a strong enrichment in pro-inflammatory pathways, which may be relevant to the hypertrophic changes of FDR individuals [125]. Several of these miRNAs targeted the Insulin-like Growth Factor 2 (IGF2) gene, whose dysregulation induces inflammatory disease [134]. Finally, in FDR subjects, altered histone marks were identified at genes related to mitochondrial dysfunction (unpublished data), a major cause of adipose tissue inflammation [135].
All together, these findings strengthen the role of epigenetic mechanisms in inflammation-related metabolic disorders, and reveal the existence of different layers of epigenetic dysregulation which determine subcutaneous adipocyte hypertrophy.
3. Epigenetic Changes as Anti-Inflammatory Targets for Treatment of Metabolic Disorders
In the recent past, significant progress has been made in the understanding and treatment of metabolic disorders. However, it appears that traditional medications are not able to prevent the progression of these disorders, indicating that additional therapeutic strategies are needed. In addition, epigenetic changes are often reversible making them a promising therapeutic target [1]. Since the evidence presented in the previous section of this review indicates that aberrant epigenetic variations lead to dysregulation of the inflammatory pathways involved in obesity and T2D, immunomodulatory therapies targeting the epigenetic profiles of obesity and diabetes may offer additional benefits for the treatment of these disorders. Indeed, patients with metabolic diseases receive beneficial effects on the glucose metabolism and IR upon treatment with anti-inflammatory drugs [136,137,138].
Several epigenetic modifiers have been identified, including HDACi, DNMT inhibitors (e.g., azacytidine) and novel miRNA-based drugs (Figure 1, [139]). HDACi have been successfully used in the treatment of several inflammatory disorders such as cancer, immune and infectious diseases [140]. These drugs have been proposed to display beneficial effects on T2D and obesity as well. For instance, the HDACi sodium phenylbutyrate ameliorates IR and the β-cell dysfunction induced by prolonged elevation of free fatty acid levels in obese individuals, by reducing inflammatory events associated to endoplasmic reticulum stress [141]. Lewis et al. have reported that oral administration of HDACi ITF2357 increases β-cell survival and enhances insulin secretion, while reducing the production and/or activities of pro-inflammatory chemokines [142]. Similarly, HDAC3 inhibition improves inflammation, hyperglycemia and insulin secretion in obese diabetic rats [143]. Resveratrol, which features HDACi activity, inhibits pro-inflammatory gene expression, significantly ameliorating glucose control and insulin sensitivity in T2D patients [144]. Resveratrol is also a potent activator of SIRT1, and its supplementation decreases systemic inflammation markers, such as IL-6 and TNF-α, along with increasing insulin sensitivity in obese humans, with no adverse effects [145]. The histone acetyltransferase (HAT) p300 plays an important role in the activation of NF-kB target genes [146]. Thus, p300 targeting may specifically prevent inflammatory events. Curcumin, for instance, has been identified as a specific inhibitor of p300, with several studies showing anti-inflammatory properties and supporting its use in treatment of both IR and T2D [147]. Recently, two other competitive inhibitors of p300, C646, and A-485, have been identified [148,149]. These molecules have been effectively used in cancer-related inflammation [150,151], suggesting that these compounds may also have great therapeutic potential in preventing an altered immune response in metabolic conditions.
The DNA methyltransferases inhibitor 5-azacytidine (AZA) and its derivate 5-aza 2′ deoxycytidine (DAC) have also been proposed as potential candidates for treatment of inflammatory disorders, including obesity and T2D. Both of these drugs prevent pro-inflammatory events by fostering generation of anti-inflammatory Treg cells [152]. Therefore, promoting Treg production by targeting DNA methylation could offer a promising novel treatment option.
However, concerns related to the use of epigenetic drugs in metabolic dysfunction need to be addressed. The low specificity and global action of these agents could potentially compromise host immunity, as described in [153,154]. Moreover, the number of epigenetic changes that must be reversed is uncertain and, given that a single molecule regulates several signaling pathways and has different biological targets, one may also anticipate multiple side effects [155]. Another issue that needs to be addressed is the dynamic nature of epigenetic changes. While making epigenetic targets pharmacologically attractive, their plasticity may also render them too unstable.
Recently, miRNAs have gained increasing interest as research tools and as a potential treatment options of several diseases [156]. Novel miRNA-based therapies have been proposed to represent valuable experimental strategies for the treatment of cancer and infectious diseases [157,158]. They are commonly based on the adoption of antagomirs, which efficiently and specifically silence endogenous miRNAs or RNA small molecules which mimic endogenous miRNA function (i.e., miRNA mimics). Thus, while several issues still need to be addressed (e.g., target specificity and delivery, reduction of off-target effects or long-term safety concerns) miRNA-based therapy has, in the near future, the potential for representing a very appealing strategy for rescuing immune system function in obesity and T2D.
4. Conclusions
The studies reviewed in the present work indicate that aberrant changes in the epigenome represent one of the central underlying mechanisms of the immune function perturbation involved in the onset and progression of common metabolic disorders. The dynamic and reversible nature of epigenetic marks creates both challenges and unique opportunities for the development of strategies for the treatment and prevention of the inflammatory derangement associated with obesity and T2D. Indeed, several classes of epigenetic drugs, including HDAC and p300 inhibitors, SIRT1 activators and anti-miRNA molecules, have already demonstrated promising results in the treatment and management of these disorders. Despite this encouraging scenario, significant investments are required before epigenetic therapies can be adopted in clinical practice.
Author Contributions
Conceptualization, F.Z., G.A.R., L.P. and F.B.; study selection, I.P., A.L. and M.C.; writing—original draft preparation, L.P. and F.B.; writing—review and editing, G.A.R., V.D.R., L.P. and F.B.; supervision, L.P. and F.B. F.B. and L.P. equally contributed to this study as last authors. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Ministero dell’Istruzione, Università e Ricerca Scientifica (grants PRIN 2017-2017CPLH32 and PON “RICERCA E INNOVAZIONE” 2014–2020 E FSC-progetto IDF SHARID-ARS01_01270) and by Regione Campania POR FESR 2014–2020 Objective 1.2. Realization of Technology Platform to fight oncologic diseases (RARE PLAT NET-CUP B63D18000380007, SATIN-CUP B61C17000070007 and COEPICA-CUP B63D18000640007 Projects).
Acknowledgments
The technical help of Domenico Liguoro and Said Maouali is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Parrillo, L.; Spinelli, R.; Nicolo, A.; Longo, M.; Mirra, P.; Raciti, G.A.; Miele, C.; Beguinot, F. Nutritional Factors, DNA Methylation, and Risk of Type 2 Diabetes and Obesity: Perspectives and Challenges. Int. J. Mol. Sci. 2019, 20, 2983. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, S.; Donkin, I.; Versteyhe, S.; Barres, R.; Simar, D. The Emerging Role of Epigenetics in Inflammation and Immunometabolism. Trends Endocrinol. Metab. 2016, 27, 782–795. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, E.; Sauter, N.S.; Boni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): Implication of macrophages resident in the AT. J. Clin. Endocrinol. Metab. 2005, 90, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Boni-Schnetzler, M.; Boller, S.; Debray, S.; Bouzakri, K.; Meier, D.T.; Prazak, R.; Kerr-Conte, J.; Pattou, F.; Ehses, J.A.; Schuit, F.C.; et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 2009, 150, 5218–5229. [Google Scholar] [CrossRef] [PubMed]
- Boni-Schnetzler, M.; Thorne, J.; Parnaud, G.; Marselli, L.; Ehses, J.A.; Kerr-Conte, J.; Pattou, F.; Halban, P.A.; Weir, G.C.; Donath, M.Y. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 2008, 93, 4065–4074. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Akbari, M.; Hassan-Zadeh, V. The inflammatory effect of epigenetic factors and modifications in type 2 diabetes. Inflammopharmacology 2020, 28, 345–362. [Google Scholar] [CrossRef]
- Xu, X.; Su, S.; Barnes, V.A.; De Miguel, C.; Pollock, J.; Ownby, D.; Shi, H.; Zhu, H.; Snieder, H.; Wang, X. A genome-wide methylation study on obesity: Differential variability and differential methylation. Epigenetics 2013, 8, 522–533. [Google Scholar] [CrossRef]
- Calle-Fabregat, C.; Morante-Palacios, O.; Ballestar, E. Understanding the Relevance of DNA Methylation Changes in Immune Differentiation and Disease. Genes 2020, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ehrlich, L.I.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.Q.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.C.; Xi, Y.X.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar] [CrossRef]
- Dekkers, K.F.; Neele, A.E.; Jukema, J.W.; Heijmans, B.T.; de Winther, M.P.J. Human monocyte-to-macrophage differentiation involves highly localized gain and loss of DNA methylation at transcription factor binding sites. Epigenetics Chromatin 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gomez, A.; Li, T.; Kerick, M.; Catala-Moll, F.; Comet, N.R.; Rodriguez-Ubreva, J.; de la Rica, L.; Branco, M.R.; Martin, J.; Ballestar, E. TET2- and TDG-mediated changes are required for the acquisition of distinct histone modifications in divergent terminal differentiation of myeloid cells. Nucleic Acids Res. 2017, 45, 10002–10017. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Garcia-Gomez, A.; Morante-Palacios, O.; Ciudad, L.; Ozkaramehmet, S.; Van Dijck, E.; Rodriguez-Ubreva, J.; Vaquero, A.; Ballestar, E. SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Res. 2020, 48, 665–681. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef]
- Kulis, M.; Merkel, A.; Heath, S.; Queiros, A.C.; Schuyler, R.P.; Castellano, G.; Beekman, R.; Raineri, E.; Esteve, A.; Clot, G.; et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet. 2015, 47, 746–756. [Google Scholar] [CrossRef]
- Shaknovich, R.; Cerchietti, L.; Tsikitas, L.; Kormaksson, M.; De, S.; Figueroa, M.E.; Ballon, G.; Yang, S.N.; Weinhold, N.; Reimers, M.; et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood 2011, 118, 3559–3569. [Google Scholar] [CrossRef]
- Sellars, M.; Huh, J.R.; Day, K.; Issuree, P.D.; Galan, C.; Gobeil, S.; Absher, D.; Green, M.R.; Littman, D.R. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat. Immunol. 2015, 16, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Ellmeier, W.; Haust, L.; Tschismarov, R. Transcriptional control of CD4 and CD8 coreceptor expression during T cell development. Cell Mol. Life Sci. 2013, 70, 4537–4553. [Google Scholar] [CrossRef] [PubMed]
- Wierda, R.J.; Kuipers, H.F.; van Eggermond, M.C.; Benard, A.; van Leeuwen, J.C.; Carluccio, S.; Geutskens, S.B.; Jukema, J.W.; Marquez, V.E.; Quax, P.H.; et al. Epigenetic control of CCR5 transcript levels in immune cells and modulation by small molecules inhibitors. J. Cell Mol. Med. 2012, 16, 1866–1877. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.C.; Hutchins, A.S.; High, F.A.; Lee, H.W.; Sykes, K.J.; Chodosh, L.A.; Reiner, S.L. Hlx is induced by and genetically interacts with T-bet to promote heritable T(H)1 gene induction. Nat. Immunol. 2002, 3, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.D.; Bopp, T.; Schmitt, E.; et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007, 5, e38. [Google Scholar] [CrossRef]
- Lee, D.U.; Agarwal, S.; Rao, A. Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity 2002, 16, 649–660. [Google Scholar] [CrossRef]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Beier, U.H.; Han, R.; Bhatti, T.R.; Akimova, T.; Hancock, W.W. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood 2013, 121, 3631–3639. [Google Scholar] [CrossRef]
- Helmin, K.A.; Morales-Nebreda, L.; Torres Acosta, M.A.; Anekalla, K.R.; Chen, S.Y.; Abdala-Valencia, H.; Politanska, Y.; Cheresh, P.; Akbarpour, M.; Steinert, E.M.; et al. Maintenance DNA methylation is essential for regulatory T cell development and stability of suppressive function. J. Clin. Investig. 2020, 130, 6571–6587. [Google Scholar] [CrossRef]
- Petrus, P.; Bialesova, L.; Checa, A.; Kerr, A.; Naz, S.; Backdahl, J.; Gracia, A.; Toft, S.; Dahlman-Wright, K.; Heden, P.; et al. Adipocyte Expression of SLC19A1 Links DNA Hypermethylation to Adipose Tissue Inflammation and Insulin Resistance. J. Clin. Endocrinol. Metab. 2018, 103, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Chen, L.L.; Deng, X.L.; Song, H.J.; Liao, Y.F.; Zeng, T.S.; Zheng, J.; Li, H.Q. Methylation status of CpG sites in the MCP-1 promoter is correlated to serum MCP-1 in Type 2 diabetes. J. Endocrinol. Investig. 2012, 35, 585–589. [Google Scholar] [CrossRef]
- Roshanzamir, N.; Hassan-Zadeh, V. Methylation of Specific CpG Sites in IL-1beta and IL1R1 Genes is Affected by Hyperglycaemia in Type 2 Diabetic Patients. Immunol. Investig. 2020, 49, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Hassan-Zadeh, V. Hyperglycemia Affects the Expression of Inflammatory Genes in Peripheral Blood Mononuclear Cells of Patients with Type 2 Diabetes. Immunol. Investig. 2018, 47, 654–665. [Google Scholar] [CrossRef]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef]
- Dayeh, T.; Tuomi, T.; Almgren, P.; Perfilyev, A.; Jansson, P.A.; de Mello, V.D.; Pihlajamaki, J.; Vaag, A.; Groop, L.; Nilsson, E.; et al. DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics 2016, 11, 482–488. [Google Scholar] [CrossRef]
- Hermsdorff, H.H.; Mansego, M.L.; Campion, J.; Milagro, F.I.; Zulet, M.A.; Martinez, J.A. TNF-alpha promoter methylation in peripheral white blood cells: Relationship with circulating TNFalpha, truncal fat and n-6 PUFA intake in young women. Cytokine 2013, 64, 265–271. [Google Scholar] [CrossRef]
- Na, Y.K.; Hong, H.S.; Lee, W.K.; Kim, Y.H.; Kim, D.S. Increased methylation of interleukin 6 gene is associated with obesity in Korean women. Mol. Cells 2015, 38, 452–456. [Google Scholar] [CrossRef]
- Arpon, A.; Milagro, F.I.; Ramos-Lopez, O.; Mansego, M.L.; Riezu-Boj, J.I.; Martinez, J.A.; Project, M. Methylome-Wide Association Study in Peripheral White Blood Cells Focusing on Central Obesity and Inflammation. Genes 2019, 10, 444. [Google Scholar] [CrossRef] [PubMed]
- Simar, D.; Versteyhe, S.; Donkin, I.; Liu, J.; Hesson, L.; Nylander, V.; Fossum, A.; Barres, R. DNA methylation is altered in B and NK lymphocytes in obese and type 2 diabetic human. Metabolism 2014, 63, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016, 1, e87748. [Google Scholar] [CrossRef]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Dispirito, J.R.; Shen, H. Histone acetylation at the single-cell level: A marker of memory CD8+ T cell differentiation and functionality. J. Immunol. 2010, 184, 4631–4636. [Google Scholar] [CrossRef]
- Allan, R.S.; Zueva, E.; Cammas, F.; Schreiber, H.A.; Masson, V.; Belz, G.T.; Roche, D.; Maison, C.; Quivy, J.P.; Almouzni, G.; et al. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature 2012, 487, 249–253. [Google Scholar] [CrossRef]
- Schoenborn, J.R.; Dorschner, M.O.; Sekimata, M.; Santer, D.M.; Shnyreva, M.; Fitzpatrick, D.R.; Stamatoyannopoulos, J.A.; Wilson, C.B. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat. Immunol. 2007, 8, 732–742. [Google Scholar] [CrossRef]
- Good-Jacobson, K.L. Regulation of germinal center, B-cell memory, and plasma cell formation by histone modifiers. Front. Immunol. 2014, 5, 596. [Google Scholar] [CrossRef]
- Baxter, J.; Sauer, S.; Peters, A.; John, R.; Williams, R.; Caparros, M.L.; Arney, K.; Otte, A.; Jenuwein, T.; Merkenschlager, M.; et al. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J. 2004, 23, 4462–4472. [Google Scholar] [CrossRef] [PubMed]
- Waibel, M.; Christiansen, A.J.; Hibbs, M.L.; Shortt, J.; Jones, S.A.; Simpson, I.; Light, A.; O’Donnell, K.; Morand, E.F.; Tarlinton, D.M.; et al. Manipulation of B-cell responses with histone deacetylase inhibitors. Nat. Commun. 2015, 6, 6838. [Google Scholar] [CrossRef] [PubMed]
- Kittan, N.A.; Allen, R.M.; Dhaliwal, A.; Cavassani, K.A.; Schaller, M.; Gallagher, K.A.; Carson, W.F.t.; Mukherjee, S.; Grembecka, J.; Cierpicki, T.; et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE 2013, 8, e78045. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef]
- Mullican, S.E.; Gaddis, C.A.; Alenghat, T.; Nair, M.G.; Giacomin, P.R.; Everett, L.J.; Feng, D.; Steger, D.J.; Schug, J.; Artis, D.; et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011, 25, 2480–2488. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Cao, Q.; Rong, S.; Repa, J.J.; St Clair, R.; Parks, J.S.; Mishra, N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1871–1879. [Google Scholar] [CrossRef]
- Kiguchi, N.; Kobayashi, Y.; Saika, F.; Kishioka, S. Epigenetic upregulation of CCL2 and CCL3 via histone modifications in infiltrating macrophages after peripheral nerve injury. Cytokine 2013, 64, 666–672. [Google Scholar] [CrossRef]
- Ding, Q.; Gao, Z.; Chen, K.; Zhang, Q.; Hu, S.; Zhao, L. Inflammation-Related Epigenetic Modification: The Bridge Between Immune and Metabolism in Type 2 Diabetes. Front. Immunol. 2022, 13, 883410. [Google Scholar] [CrossRef]
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 2004, 279, 18091–18097. [Google Scholar] [CrossRef]
- Li, Y.; Reddy, M.A.; Miao, F.; Shanmugam, N.; Yee, J.K.; Hawkins, D.; Ren, B.; Natarajan, R. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J. Biol. Chem. 2008, 283, 26771–26781. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.A.; Joshi, A.; Carson, W.F.; Schaller, M.; Allen, R.; Mukerjee, S.; Kittan, N.; Feldman, E.L.; Henke, P.K.; Hogaboam, C.; et al. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes 2015, 64, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Abu-Farha, M.; Tiss, A.; Abubaker, J.; Khadir, A.; Al-Ghimlas, F.; Al-Khairi, I.; Baturcam, E.; Cherian, P.; Elkum, N.; Hammad, M.; et al. Proteomics analysis of human obesity reveals the epigenetic factor HDAC4 as a potential target for obesity. PLoS ONE 2013, 8, e75342. [Google Scholar] [CrossRef]
- Shanaki, M.; Omidifar, A.; Shabani, P.; Toolabi, K. Association between HDACs and pro-inflammatory cytokine gene expressions in obesity. Arch. Physiol. Biochem. 2020, 1–7. [Google Scholar] [CrossRef]
- Hanzu, F.A.; Musri, M.M.; Sanchez-Herrero, A.; Claret, M.; Esteban, Y.; Kaliman, P.; Gomis, R.; Parrizas, M. Histone demethylase KDM1A represses inflammatory gene expression in preadipocytes. Obesity 2013, 21, E616–E625. [Google Scholar] [CrossRef]
- Tian, W.; Xu, H.; Fang, F.; Chen, Q.; Xu, Y.; Shen, A. Brahma-related gene 1 bridges epigenetic regulation of proinflammatory cytokine production to steatohepatitis in mice. Hepatology 2013, 58, 576–588. [Google Scholar] [CrossRef]
- Mikula, M.; Majewska, A.; Ledwon, J.K.; Dzwonek, A.; Ostrowski, J. Obesity increases histone H3 lysine 9 and 18 acetylation at Tnfa and Ccl2 genes in mouse liver. Int. J. Mol. Med. 2014, 34, 1647–1654. [Google Scholar] [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef]
- Arab Sadeghabadi, Z.; Nourbakhsh, M.; Pasalar, P.; Emamgholipour, S.; Golestani, A.; Larijani, B.; Razzaghy-Azar, M. Reduced gene expression of sirtuins and active AMPK levels in children and adolescents with obesity and insulin resistance. Obes. Res. Clin. Pract. 2018, 12, 167–173. [Google Scholar] [CrossRef]
- Zhou, S.; Tang, X.; Chen, H.Z. Sirtuins and Insulin Resistance. Front. Endocrinol. 2018, 9, 748. [Google Scholar] [CrossRef] [PubMed]
- Crujeiras, A.B.; Parra, D.; Goyenechea, E.; Martinez, J.A. Sirtuin gene expression in human mononuclear cells is modulated by caloric restriction. Eur. J. Clin. Investig. 2008, 38, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell. Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef]
- Chen, C.Z.; Schaffert, S.; Fragoso, R.; Loh, C. Regulation of immune responses and tolerance: The microRNA perspective. Immunol. Rev. 2013, 253, 112–128. [Google Scholar] [CrossRef]
- Kurkewich, J.L.; Boucher, A.; Klopfenstein, N.; Baskar, R.; Kapur, R.; Dahl, R. The mirn23a and mirn23b microrna clusters are necessary for proper hematopoietic progenitor cell production and differentiation. Exp. Hematol. 2018, 59, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Lu, J.; Schlanger, R.; Zhang, H.; Wang, J.Y.; Fox, M.C.; Purton, L.E.; Fleming, H.H.; Cobb, B.; Merkenschlager, M.; et al. MicroRNA miR-125a controls hematopoietic stem cell number. Proc. Natl. Acad. Sci. USA 2010, 107, 14229–14234. [Google Scholar] [CrossRef]
- Song, M.S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef]
- Luo, S.; Liu, Y.; Liang, G.; Zhao, M.; Wu, H.; Liang, Y.; Qiu, X.; Tan, Y.; Dai, Y.; Yung, S.; et al. The role of microRNA-1246 in the regulation of B cell activation and the pathogenesis of systemic lupus erythematosus. Clin. Epigenet. 2015, 7, 24. [Google Scholar] [CrossRef]
- Marcais, A.; Blevins, R.; Graumann, J.; Feytout, A.; Dharmalingam, G.; Carroll, T.; Amado, I.F.; Bruno, L.; Lee, K.; Walzer, T.; et al. microRNA-mediated regulation of mTOR complex components facilitates discrimination between activation and anergy in CD4 T cells. J. Exp. Med. 2014, 211, 2281–2295. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.F.; Thomas, M.F.; Hu, J.K.; Yang, Z.; Babiarz, J.E.; Allen, C.D.; Matloubian, M.; Blelloch, R.; Ansel, K.M. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity 2011, 35, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Curtale, G.; Rubino, M.; Locati, M. MicroRNAs as Molecular Switches in Macrophage Activation. Front. Immunol. 2019, 10, 799. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; McCurdy, S.; Huang, S.; Zhu, X.; Peplowska, K.; Tiirikainen, M.; Boisvert, W.A.; Garmire, L.X. Time Series miRNA-mRNA integrated analysis reveals critical miRNAs and targets in macrophage polarization. Sci. Rep. 2016, 6, 37446. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kang, Y.; Zhang, H.; Zhao, D.; Xia, J.; Lu, Z.; Wang, H.; Xu, F.; Shi, L. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J. Immunol. 2015, 194, 1239–1251. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; So, A.Y.; Sinha, N.; Gibson, W.S.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b potentiates macrophage activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef]
- Liu, L.; Li, X. Downregulation of miR-320 Alleviates Endoplasmic Reticulum Stress and Inflammatory Response in 3T3-L1 Adipocytes. Exp. Clin. Endocrinol. Diabetes 2021, 129, 131–137. [Google Scholar] [CrossRef]
- Arner, E.; Mejhert, N.; Kulyte, A.; Balwierz, P.J.; Pachkov, M.; Cormont, M.; Lorente-Cebrian, S.; Ehrlund, A.; Laurencikiene, J.; Heden, P.; et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes 2012, 61, 1986–1993. [Google Scholar] [CrossRef]
- Zhang, X.M.; Guo, L.; Chi, M.H.; Sun, H.M.; Chen, X.W. Identification of active miRNA and transcription factor regulatory pathways in human obesity-related inflammation. BMC Bioinform. 2015, 16, 76. [Google Scholar] [CrossRef]
- Yang, Z.; Wei, Z.; Wu, X.; Yang, H. Screening of exosomal miRNAs derived from subcutaneous and visceral adipose tissues: Determination of targets for the treatment of obesity and associated metabolic disorders. Mol. Med. Rep. 2018, 18, 3314–3324. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Hui, X.; Hoo, R.L.C.; Ye, D.; Chan, C.Y.C.; Feng, T.; Wang, Y.; Lam, K.S.L.; Xu, A. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J. Clin. Investig. 2019, 129, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, J.G.; Diehl, K.J.; Bammert, T.D.; Kavlich, P.J.; Lincenberg, G.M.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Influence of Overweight and Obesity on Circulating Inflammation-Related microRNA. Microrna 2018, 7, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Q.; Xiao, X.; Wu, C.; Gao, R.; Peng, C.; Li, D.; Zhang, W.; Du, T.; Wang, Y.; et al. miR-1934, downregulated in obesity, protects against low-grade inflammation in adipocytes. Mol. Cell. Endocrinol. 2016, 428, 109–117. [Google Scholar] [CrossRef]
- Ge, Q.; Gerard, J.; Noel, L.; Scroyen, I.; Brichard, S.M. MicroRNAs regulated by adiponectin as novel targets for controlling adipose tissue inflammation. Endocrinology 2012, 153, 5285–5296. [Google Scholar] [CrossRef]
- Runtsch, M.C.; Nelson, M.C.; Lee, S.H.; Voth, W.; Alexander, M.; Hu, R.; Wallace, J.; Petersen, C.; Panic, V.; Villanueva, C.J.; et al. Anti-inflammatory microRNA-146a protects mice from diet-induced metabolic disease. PLoS Genet. 2019, 15, e1007970. [Google Scholar] [CrossRef]
- Balasubramanyam, M.; Aravind, S.; Gokulakrishnan, K.; Prabu, P.; Sathishkumar, C.; Ranjani, H.; Mohan, V. Impaired miR-146a expression links subclinical inflammation and insulin resistance in Type 2 diabetes. Mol. Cell. Biochem. 2011, 351, 197–205. [Google Scholar] [CrossRef]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef]
- Ye, E.A.; Liu, L.; Jiang, Y.; Jan, J.; Gaddipati, S.; Suvas, S.; Steinle, J.J. miR-15a/16 reduces retinal leukostasis through decreased pro-inflammatory signaling. J. Neuroinflamm. 2016, 13, 305. [Google Scholar] [CrossRef]
- Smith, K.M.; Guerau-de-Arellano, M.; Costinean, S.; Williams, J.L.; Bottoni, A.; Mavrikis Cox, G.; Satoskar, A.R.; Croce, C.M.; Racke, M.K.; Lovett-Racke, A.E.; et al. miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J. Immunol. 2012, 189, 1567–1576. [Google Scholar] [CrossRef]
- Zhu, E.; Wang, X.; Zheng, B.; Wang, Q.; Hao, J.; Chen, S.; Zhao, Q.; Zhao, L.; Wu, Z.; Yin, Z. miR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORgammat and STAT3. J. Immunol. 2014, 192, 5599–5609. [Google Scholar] [CrossRef] [PubMed]
- Vatandoost, N.; Amini, M.; Iraj, B.; Momenzadeh, S.; Salehi, R. Dysregulated miR-103 and miR-143 expression in peripheral blood mononuclear cells from induced prediabetes and type 2 diabetes rats. Gene 2015, 572, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Lim, B.; Lodish, H.F. MicroRNAs induced during adipogenesis that accelerate fat cell development are downregulated in obesity. Diabetes 2009, 58, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Jin, W.; Villeneuve, L.; Wang, M.; Lanting, L.; Todorov, I.; Kato, M.; Natarajan, R. Pro-inflammatory role of microrna-200 in vascular smooth muscle cells from diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Das, S.; Zhuo, C.; Jin, W.; Wang, M.; Lanting, L.; Natarajan, R. Regulation of Vascular Smooth Muscle Cell Dysfunction Under Diabetic Conditions by miR-504. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 864–873. [Google Scholar] [CrossRef]
- InterAct, C.; Scott, R.A.; Langenberg, C.; Sharp, S.J.; Franks, P.W.; Rolandsson, O.; Drogan, D.; van der Schouw, Y.T.; Ekelund, U.; Kerrison, N.D.; et al. The link between family history and risk of type 2 diabetes is not explained by anthropometric, lifestyle or genetic risk factors: The EPIC-InterAct study. Diabetologia 2013, 56, 60–69. [Google Scholar] [CrossRef]
- Parrillo, L.; Spinelli, R.; Longo, M.; Desiderio, A.; Mirra, P.; Nigro, C.; Fiory, F.; Hedjazifar, S.; Mutarelli, M.; Carissimo, A.; et al. Altered PTPRD DNA methylation associates with restricted adipogenesis in healthy first-degree relatives of Type 2 diabetes subjects. Epigenomics 2020, 12, 873–888. [Google Scholar] [CrossRef]
- Henninger, A.M.; Eliasson, B.; Jenndahl, L.E.; Hammarstedt, A. Adipocyte hypertrophy, inflammation and fibrosis characterize subcutaneous adipose tissue of healthy, non-obese subjects predisposed to type 2 diabetes. PLoS ONE 2014, 9, e105262. [Google Scholar] [CrossRef]
- Weyer, C.; Foley, J.E.; Bogardus, C.; Tataranni, P.A.; Pratley, R.E. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia 2000, 43, 1498–1506. [Google Scholar] [CrossRef]
- Lonn, M.; Mehlig, K.; Bengtsson, C.; Lissner, L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 2010, 24, 326–331. [Google Scholar] [CrossRef]
- Spinelli, R.; Florese, P.; Parrillo, L.; Zatterale, F.; Longo, M.; D’Esposito, V.; Desiderio, A.; Nerstedt, A.; Gustafson, B.; Formisano, P.; et al. ZMAT3 hypomethylation contributes to early senescence of preadipocytes from healthy first-degree relatives of type 2 diabetics. Aging Cell 2022. [Google Scholar] [CrossRef] [PubMed]
- Parrillo, L.; Spinelli, R.; Costanzo, M.; Florese, P.; Cabaro, S.; Desiderio, A.; Prevenzano, I.; Raciti, G.A.; Smith, U.; Miele, C.; et al. Epigenetic Dysregulation of the Homeobox A5 (HOXA5) Gene Associates with Subcutaneous Adipocyte Hypertrophy in Human Obesity. Cells 2022, 11, 728. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Raciti, G.A.; Zatterale, F.; Parrillo, L.; Desiderio, A.; Spinelli, R.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Nigro, C.; et al. Epigenetic modifications of the Zfp/ZNF423 gene control murine adipogenic commitment and are dysregulated in human hypertrophic obesity. Diabetologia 2018, 61, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, A.; Longo, M.; Parrillo, L.; Campitelli, M.; Cacace, G.; De Simone, S.; Spinelli, R.; Zatterale, F.; Cabaro, S.; Dolce, P.; et al. Epigenetic silencing of the ANKRD26 gene correlates to the pro-inflammatory profile and increased cardio-metabolic risk factors in human obesity. Clin. Epigenetics 2019, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Mirra, P.; Desiderio, A.; Spinelli, R.; Nigro, C.; Longo, M.; Parrillo, L.; D’Esposito, V.; Carissimo, A.; Hedjazifar, S.; Smith, U.; et al. Adipocyte precursor cells from first degree relatives of type 2 diabetic patients feature changes in hsa-mir-23a-5p, -193a-5p, and -193b-5p and insulin-like growth factor 2 expression. FASEB J. 2021, 35, e21357. [Google Scholar] [CrossRef]
- Tsai, F.J.; Yang, C.F.; Chen, C.C.; Chuang, L.M.; Lu, C.H.; Chang, C.T.; Wang, T.Y.; Chen, R.H.; Shiu, C.F.; Liu, Y.M.; et al. A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS Genet. 2010, 6, e1000847. [Google Scholar] [CrossRef]
- Pike, K.A.; Tremblay, M.L. Protein Tyrosine Phosphatases: Regulators of CD4 T Cells in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2504. [Google Scholar] [CrossRef]
- Hale, A.J.; Ter Steege, E.; den Hertog, J. Recent advances in understanding the role of protein-tyrosine phosphatases in development and disease. Dev. Biol. 2017, 428, 283–292. [Google Scholar] [CrossRef]
- Vilborg, A.; Glahder, J.A.; Wilhelm, M.T.; Bersani, C.; Corcoran, M.; Mahmoudi, S.; Rosenstierne, M.; Grander, D.; Farnebo, M.; Norrild, B.; et al. The p53 target Wig-1 regulates p53 mRNA stability through an AU-rich element. Proc. Natl. Acad. Sci. USA 2009, 106, 15756–15761. [Google Scholar] [CrossRef]
- Parrillo, L.; Costa, V.; Raciti, G.A.; Longo, M.; Spinelli, R.; Esposito, R.; Nigro, C.; Vastolo, V.; Desiderio, A.; Zatterale, F.; et al. Hoxa5 undergoes dynamic DNA methylation and transcriptional repression in the adipose tissue of mice exposed to high-fat diet. Int. J. Obes. 2016, 40, 929–937. [Google Scholar] [CrossRef]
- Cao, W.; Zhang, T.; Feng, R.; Xia, T.; Huang, H.; Liu, C.; Sun, C. Hoxa5 alleviates obesity-induced chronic inflammation by reducing ER stress and promoting M2 macrophage polarization in mouse adipose tissue. J. Cell. Mol. Med. 2019, 23, 7029–7042. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.; Shao, M.; Zhang, Q.; Hepler, C.; Paschoal, V.A.; Barnes, S.D.; Vishvanath, L.; An, Y.A.; Jia, L.; Malladi, V.S.; et al. Perivascular mesenchymal cells control adipose-tissue macrophage accrual in obesity. Nat. Metab. 2020, 2, 1332–1349. [Google Scholar] [CrossRef] [PubMed]
- Raciti, G.A.; Spinelli, R.; Desiderio, A.; Longo, M.; Parrillo, L.; Nigro, C.; D’Esposito, V.; Mirra, P.; Fiory, F.; Pilone, V.; et al. Specific CpG hyper-methylation leads to Ankrd26 gene down-regulation in white adipose tissue of a mouse model of diet-induced obesity. Sci. Rep. 2017, 7, 43526. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Lin, L.; Li, Q.; Liu, K.; Huang, Y.; Wang, X.; Cao, K.; Chen, X.; Cao, W.; Li, F.; et al. IGF-2 Preprograms Maturing Macrophages to Acquire Oxidative Phosphorylation-Dependent Anti-inflammatory Properties. Cell Metab. 2019, 29, 1363–1375. [Google Scholar] [CrossRef]
- Woo, C.Y.; Jang, J.E.; Lee, S.E.; Koh, E.H.; Lee, K.U. Mitochondrial Dysfunction in Adipocytes as a Primary Cause of Adipose Tissue Inflammation. Diabetes Metab. J. 2019, 43, 247–256. [Google Scholar] [CrossRef]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 2017, 127, 83–93. [Google Scholar] [CrossRef]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef]
- Arguelles, A.O.; Meruvu, S.; Bowman, J.D.; Choudhury, M. Are epigenetic drugs for diabetes and obesity at our door step? Drug Discov. Today 2016, 21, 499–509. [Google Scholar] [CrossRef]
- Stathis, A.; Hotte, S.J.; Chen, E.X.; Hirte, H.W.; Oza, A.M.; Moretto, P.; Webster, S.; Laughlin, A.; Stayner, L.A.; McGill, S.; et al. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin. Cancer Res. 2009, 27, 3528. [Google Scholar] [CrossRef]
- Xiao, C.; Giacca, A.; Lewis, G.F. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 2011, 60, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.C.; Blaabjerg, L.; Storling, J.; Ronn, S.G.; Mascagni, P.; Dinarello, C.A.; Mandrup-Poulsen, T. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet beta cells in vivo and in vitro. Mol. Med. 2011, 17, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Lundh, M.; Galbo, T.; Poulsen, S.S.; Mandrup-Poulsen, T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes. Metab. 2015, 17, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhou, R.; Wang, B.; Mi, M.T. Effect of resveratrol on glucose control and insulin sensitivity: A meta-analysis of 11 randomized controlled trials. Am. J. Clin. Nutr. 2014, 99, 1510–1519. [Google Scholar] [CrossRef]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef]
- Bhatt, D.; Ghosh, S. Regulation of the NF-kappaB-Mediated Transcription of Inflammatory Genes. Front. Immunol. 2014, 5, 71. [Google Scholar] [CrossRef]
- Pivari, F.; Mingione, A.; Brasacchio, C.; Soldati, L. Curcumin and Type 2 Diabetes Mellitus: Prevention and Treatment. Nutrients 2019, 11, 1837. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef]
- Wang, R.; He, Y.; Robinson, V.; Yang, Z.; Hessler, P.; Lasko, L.M.; Lu, X.; Bhathena, A.; Lai, A.; Uziel, T.; et al. Targeting Lineage-specific MITF Pathway in Human Melanoma Cell Lines by A-485, the Selective Small-molecule Inhibitor of p300/CBP. Mol. Cancer Ther. 2018, 17, 2543–2550. [Google Scholar] [CrossRef]
- Gu, M.L.; Wang, Y.M.; Zhou, X.X.; Yao, H.P.; Zheng, S.; Xiang, Z.; Ji, F. An inhibitor of the acetyltransferases CBP/p300 exerts antineoplastic effects on gastrointestinal stromal tumor cells. Oncol. Rep. 2016, 36, 2763–2770. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Landman, S.; van der Horst, C.; van Erp, P.E.J.; Joosten, I.; de Vries, R.; Koenen, H. Immune responses to azacytidine in animal models of inflammatory disorders: A systematic review. J. Transl. Med. 2021, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Nam, K.H.; Kim, J.; Baek, M.W.; Park, J.E.; Park, H.Y.; Kwon, H.J.; Kwon, O.S.; Kim, D.Y.; Oh, G.T. Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2404–2409. [Google Scholar] [CrossRef] [PubMed]
- Roger, T.; Lugrin, J.; Le Roy, D.; Goy, G.; Mombelli, M.; Koessler, T.; Ding, X.C.; Chanson, A.L.; Reymond, M.K.; Miconnet, I.; et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011, 117, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.U.; Rehman, M.S.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an Alternative Epigenetic Modulator: Mechanism of Action and Potential Effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2021, 28, 127–138. [Google Scholar] [CrossRef]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).