
CoVM2: Molecular Biological Data Integration of SARS-CoV-2 Proteins in a Macro-to-Micro Method



Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Datasets and Network Construction
2.2. Homology Modeling for SARS-CoV-2 Variants
2.3. Quality Assessment of Protein Structures
2.4. Molecular Docking of SARS-CoV-2–Human Protein Pairs
3. Results
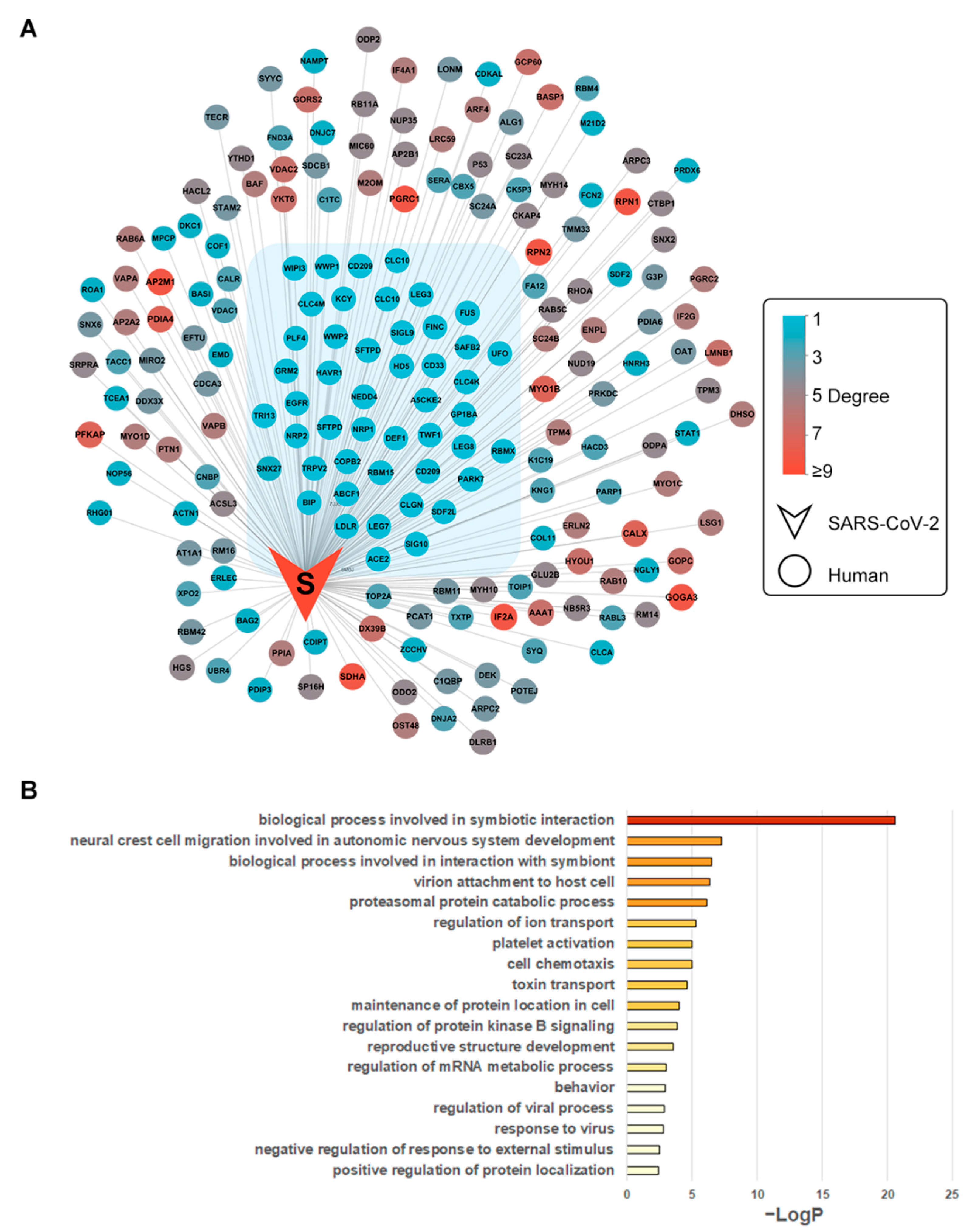
3.1. SARS-CoV-2–Human Protein Interaction Network
3.2. Biological Process Enrichment of Spike Glycoprotein Subnetwork
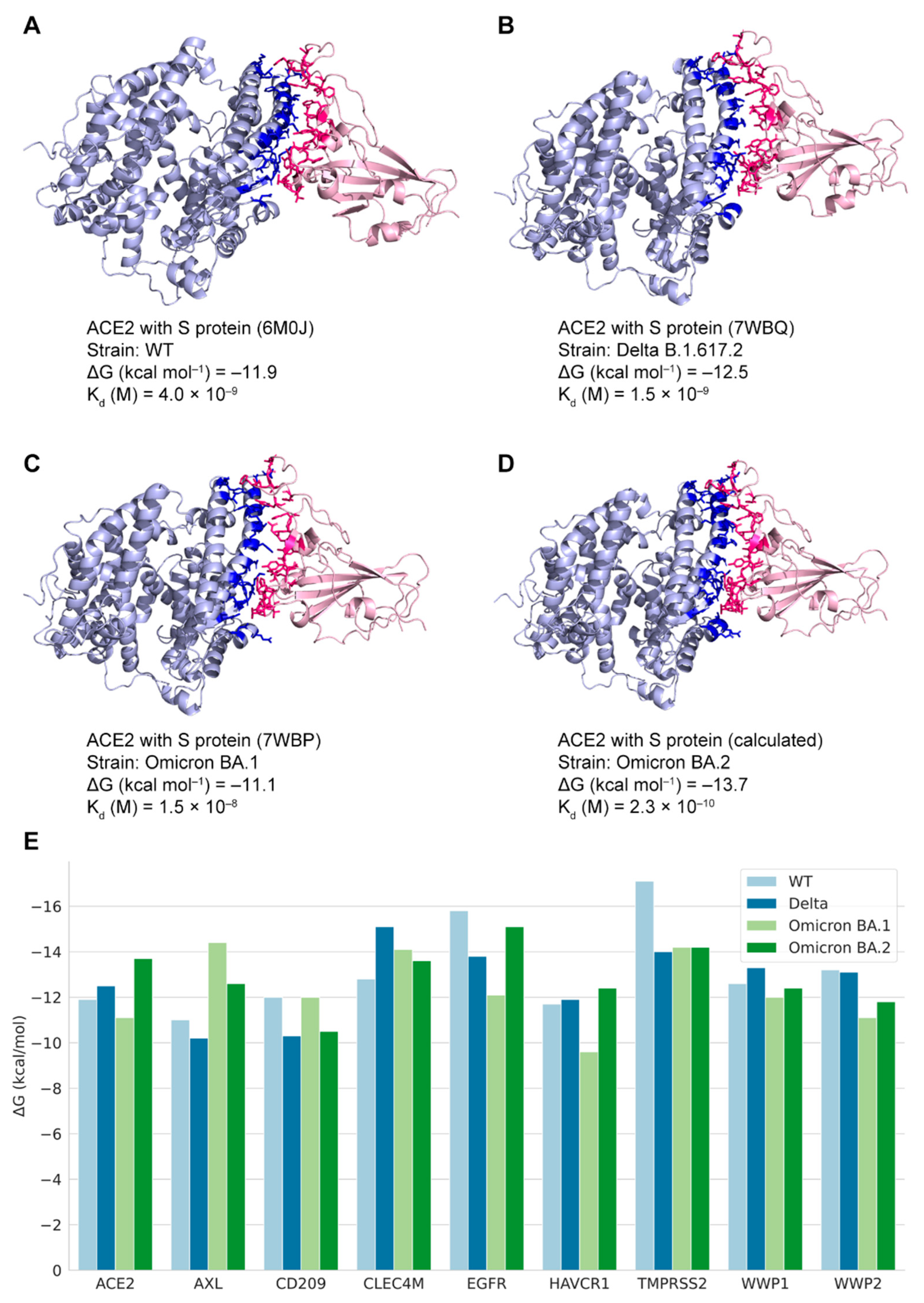
3.3. Crystal Structures of Complexes of Binding Pairs
3.4. The Predicted Docking Structures of Potential S-Protein-Binding Pairs
3.5. The Macro-to-Micro View of the SARS-CoV-2–Human Protein Associations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Khorsand, B.; Savadi, A.; Naghibzadeh, M. SARS-CoV-2-Human Protein-Protein Interaction Network. Inform. Med. Unlocked 2020, 20, 100413. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The Emergence, Genomic Diversity and Global Spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [PubMed]
- Singh, M.; Bansal, V.; Feschotte, C. A Single-Cell RNA Expression Map of Human Coronavirus Entry Factors. Cell Rep. 2020, 32. [Google Scholar] [CrossRef]
- Baggen, J.; Vanstreels, E.; Jansen, S.; Daelemans, D. Cellular Host Factors for SARS-CoV-2 Infection. Nat. Microbiol. 2021, 6, 1219–1232. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL Is a Candidate Receptor for SARS-CoV-2 That Promotes Infection of Pulmonary and Bronchial Epithelial Cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 Is a Host Factor for SARS-CoV-2 Infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Sacco, R.; Stukalov, A.; van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M.; et al. Interlaboratory Reproducibility of Large-Scale Human Protein-Complex Analysis by Standardized AP-MS. Nat. Methods 2013, 10, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhao, C.; Gu, F.; He, Z. A Two-Step Framework for Inferring Direct Protein-Protein Interaction Network from AP-MS Data. BMC Syst. Biol. 2017, 11, 82. [Google Scholar] [CrossRef]
- Liu, X.; Huuskonen, S.; Laitinen, T.; Redchuk, T.; Bogacheva, M.; Salokas, K.; Pöhner, I.; Öhman, T.; Tonduru, A.K.; Hassinen, A.; et al. SARS-CoV-2–Host Proteome Interactions for Antiviral Drug Discovery. Mol. Syst. Biol. 2021, 17, e10396. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A Promiscuous Biotin Ligase Fusion Protein Identifies Proximal and Interacting Proteins in Mammalian Cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N. The MIntAct Project—IntAct as a Common Curation Platform for 11 Molecular Interaction Databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef]
- Guirimand, T.; Delmotte, S.; Navratil, V. VirHostNet 2.0: Surfing on the Web of Virus/Host Molecular Interactions Data. Nucleic Acids Res. 2015, 43, D583–D587. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Ahsan, M.A.; Liu, Y.; Feng, C.; Hofestädt, R.; Chen, M. OverCOVID: An Integrative Web Portal for SARS-CoV-2 Bioinformatics Resources. J. Integr. Bioinforma. 2021, 18, 9–17. [Google Scholar] [CrossRef]
- Satyam, R.; Yousef, M.; Qazi, S.; Bhat, A.M.; Raza, K. COVIDium: A COVID-19 Resource Compendium. Database J. Biol. Databases Curation 2021, 2021, baab057. [Google Scholar] [CrossRef]
- Wierbowski, S.D.; Liang, S.; Liu, Y.; Chen, Y.; Gupta, S.; Andre, N.M.; Lipkin, S.M.; Whittaker, G.R.; Yu, H. A 3D Structural SARS-CoV-2–Human Interactome to Explore Genetic and Drug Perturbations. Nat. Methods 2021, 18, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Tiwari, S.; Gomes, L.G.R.; Pinto, C.H.R.; Andrade, B.S.; Ahmad, S.; Aljabali, A.A.A.; Alzahrani, K.J.; Banjer, H.J.; Hassan, S.S.; et al. SARS-CoV-2 Variants Show a Gradual Declining Pathogenicity and pro-Inflammatory Cytokine Spur, an Increasing Antigenic and Antiinflammatory Cytokine Induction, and Rising Structural Protein Instability. bioRxiv 2022, 2022. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, A.; Keskin, O.; Nussinov, R. Topological Properties of Protein Interaction Networks from a Structural Perspective. Biochem. Soc. Trans. 2008, 36, 1398. [Google Scholar] [CrossRef] [PubMed]
- Vakser, I.A. Challenges in Protein Docking. Curr. Opin. Struct. Biol. 2020, 64, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor Binding and Complex Structures of Human ACE2 to Spike RBD from Omicron and Delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Sumitha, A.; Devi, P.B.; Hari, S.; Dhanasekaran, R. COVID-19-In Silico Structure Prediction and Molecular Docking Studies with Doxycycline and Quinine. Biomed. Pharmacol. J. 2020, 13, 1185–1193. [Google Scholar] [CrossRef]
- Rai, D.K.; Rieder, E. Homology Modeling and Analysis of Structure Predictions of the Bovine Rhinitis B Virus RNA Dependent RNA Polymerase (RdRp). Int. J. Mol. Sci. 2012, 13, 8998–9013. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Cloete, R. Molecular Modeling of Subtype-Specific Tat Protein Signatures to Predict Tat-TAR Interactions That May Be Involved in HIV-Associated Neurocognitive Disorders. Front. Microbiol. 2022, 13, 866611. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. The Protein Structure Prediction Problem Could Be Solved Using the Current PDB Library. Proc. Natl. Acad. Sci. USA 2005, 102, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A Web Server for Protein–Protein and Protein–DNA/RNA Docking Based on a Hybrid Strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein–Protein Complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) Is a Receptor for Severe Acute Respiratory Syndrome Coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef]
- Chan, V.S.F.; Chan, K.Y.K.; Chen, Y.; Poon, L.L.M.; Cheung, A.N.Y.; Zheng, B.; Chan, K.-H.; Mak, W.; Ngan, H.Y.S.; Xu, X.; et al. Homozygous L-SIGN (CLEC4M) Plays a Protective Role in SARS Coronavirus Infection. Nat. Genet. 2006, 38, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and Inhibition of the Papain-like Protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural Basis for Translational Shutdown and Immune Evasion by the Nsp1 Protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhu, K.; Qin, B.; Olieric, V.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Orf9b in Complex with Human TOM70 Suggests Unusual Virus-Host Interactions. Nat. Commun. 2021, 12, 2843. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Ye, F.; Guo, Y.; Xia, L.; Zhong, X.; Chi, X.; Zhou, Q. Structural Basis for the Different States of the Spike Protein of SARS-CoV-2 in Complex with ACE2. Cell Res. 2021, 31, 717–719. [Google Scholar] [CrossRef]
- Szeto, C.; Chatzileontiadou, D.S.M.; Nguyen, A.T.; Sloane, H.; Lobos, C.A.; Jayasinghe, D.; Halim, H.; Smith, C.; Riboldi-Tunnicliffe, A.; Grant, E.J.; et al. The Presentation of SARS-CoV-2 Peptides by the Common HLA-A∗02:01 Molecule. iScience 2021, 24, 102096. [Google Scholar] [CrossRef]
- Wu, D.; Kolesnikov, A.; Yin, R.; Guest, J.D.; Gowthaman, R.; Shmelev, A.; Serdyuk, Y.; Dianov, D.V.; Efimov, G.A.; Pierce, B.G.; et al. Structural Assessment of HLA-A2-Restricted SARS-CoV-2 Spike Epitopes Recognized by Public and Private T-Cell Receptors. Nat. Commun. 2022, 13, 19. [Google Scholar] [CrossRef]
- Chaurasia, P.; Nguyen, T.H.O.; Rowntree, L.C.; Juno, J.A.; Wheatley, A.K.; Kent, S.J.; Kedzierska, K.; Rossjohn, J.; Petersen, J. Structural Basis of Biased T Cell Receptor Recognition of an Immunodominant HLA-A2 Epitope of the SARS-CoV-2 Spike Protein. J. Biol. Chem. 2021, 297, 101065. [Google Scholar] [CrossRef]
- Javorsky, A.; Humbert, P.O.; Kvansakul, M. Structural Basis of Coronavirus E Protein Interactions with Human PALS1 PDZ Domain. Commun. Biol. 2021, 4, 724. [Google Scholar] [CrossRef]
- Biswal, M.; Lu, J.; Song, J. SARS-CoV-2 Nucleocapsid Protein Targets a Conserved Surface Groove of the NTF2-like Domain of G3BP1. J. Mol. Biol. 2022, 434, 167516. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural Basis of SARS-CoV-2 Omicron Immune Evasion and Receptor Engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Padhi, A.K.; Rath, S.L.; Tripathi, T. Accelerating COVID-19 Research Using Molecular Dynamics Simulation. J. Phys. Chem. B 2021, 125, 9078–9091. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.Z.H. Mutational Effect of Some Major COVID-19 Variants on Binding of the S Protein to ACE2. Biomolecules 2022, 12, 572. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, K.B.; Balakrishnan, N.; Ganapathiraju, M.K. Interactome of SARS-CoV-2 Modulated Host Proteins With Computationally Predicted PPIs: Insights From Translational Systems Biology Studies. Front. Syst. Biol. 2022, 2. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, L.; Mo, M.; Liu, T.; Wu, C.; Gong, C.; Lu, K.; Gong, L.; Zhu, W.; Xu, Z. SARS-CoV-2 Omicron RBD Shows Weaker Binding Affinity than the Currently Dominant Delta Variant to Human ACE2. Signal Transduct. Target. Ther. 2022, 7, 8. [Google Scholar] [CrossRef]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta Variant of SARS-CoV-2: A Comparative Computational Study of Spike Protein. J. Med. Virol. 2022, 94, 1641–1649. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | UniProt | Spike Binding | Template for Docking | Chain | Note |
|---|---|---|---|---|---|
| WWP2 | O00308 | 4Y07 | A | ||
| NRP1 | O14786 | 7JJC | 7JJC | A | |
| EGFR | P00533 | 1IVO | A | ||
| AXL | P30530 | Modeled structure | Homology template 5VXZ.1.B | ||
| HAVCR1 | Q96D42 | 5DZO | A | ||
| ACE2 | Q9BYF1 | 6M0J, 7WBQ, 7WBP | 6M0J | A | |
| WWP1 | Q9H0M0 | 1ND7 | A | ||
| CLEC4M | Q9H2X3 | 1K9J | B | ||
| CD209 | Q9NNX6 | 1SL4 | A | ||
| TMPRSS2 | O15393 | 7MEQ | A |
| Model | Percentage in Allowed Region | Z-Score | RMSD | GMQE | Sequence Identity |
|---|---|---|---|---|---|
| Spike Omicron BA.2 | 87.50% | −5.59 | 0.154 | 0.75 | 97.76% |
| Spike Omicron BA.1 | 84.10% | −5.81 | |||
| Spike Delta | 83.90% | −6.04 | |||
| Spike WT | 89.30% | −5.87 | |||
| AXL | 95.10% | −4.49 | 0.168 | 0.93 | 95.10% |
| WWP2 | 92.30% | −7.68 | |||
| ACE2 | 93.30% | −12.87 | |||
| EGFR | 66.10% | −8.46 | |||
| HAVR1 | 89.10% | −4.3 | |||
| WWP1 | 85.20% | −8.32 | |||
| CLC4M | 89.40% | −6.74 | |||
| CD209 | 91.40% | −4.94 | |||
| TMPS2 | 87.90% | −7.11 | |||
| NRP1 | 87.60% | −5.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Hu, X.; Hu, Y.; Zhou, J.; Chen, M. CoVM2: Molecular Biological Data Integration of SARS-CoV-2 Proteins in a Macro-to-Micro Method. Biomolecules 2022, 12, 1067. https://doi.org/10.3390/biom12081067
Chen H, Hu X, Hu Y, Zhou J, Chen M. CoVM2: Molecular Biological Data Integration of SARS-CoV-2 Proteins in a Macro-to-Micro Method. Biomolecules. 2022; 12(8):1067. https://doi.org/10.3390/biom12081067
Chicago/Turabian StyleChen, Hongjun, Xiaotian Hu, Yanshi Hu, Jiawen Zhou, and Ming Chen. 2022. "CoVM2: Molecular Biological Data Integration of SARS-CoV-2 Proteins in a Macro-to-Micro Method" Biomolecules 12, no. 8: 1067. https://doi.org/10.3390/biom12081067
APA StyleChen, H., Hu, X., Hu, Y., Zhou, J., & Chen, M. (2022). CoVM2: Molecular Biological Data Integration of SARS-CoV-2 Proteins in a Macro-to-Micro Method. Biomolecules, 12(8), 1067. https://doi.org/10.3390/biom12081067