
Mitochondrial Dysfunction in Rheumatoid Arthritis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
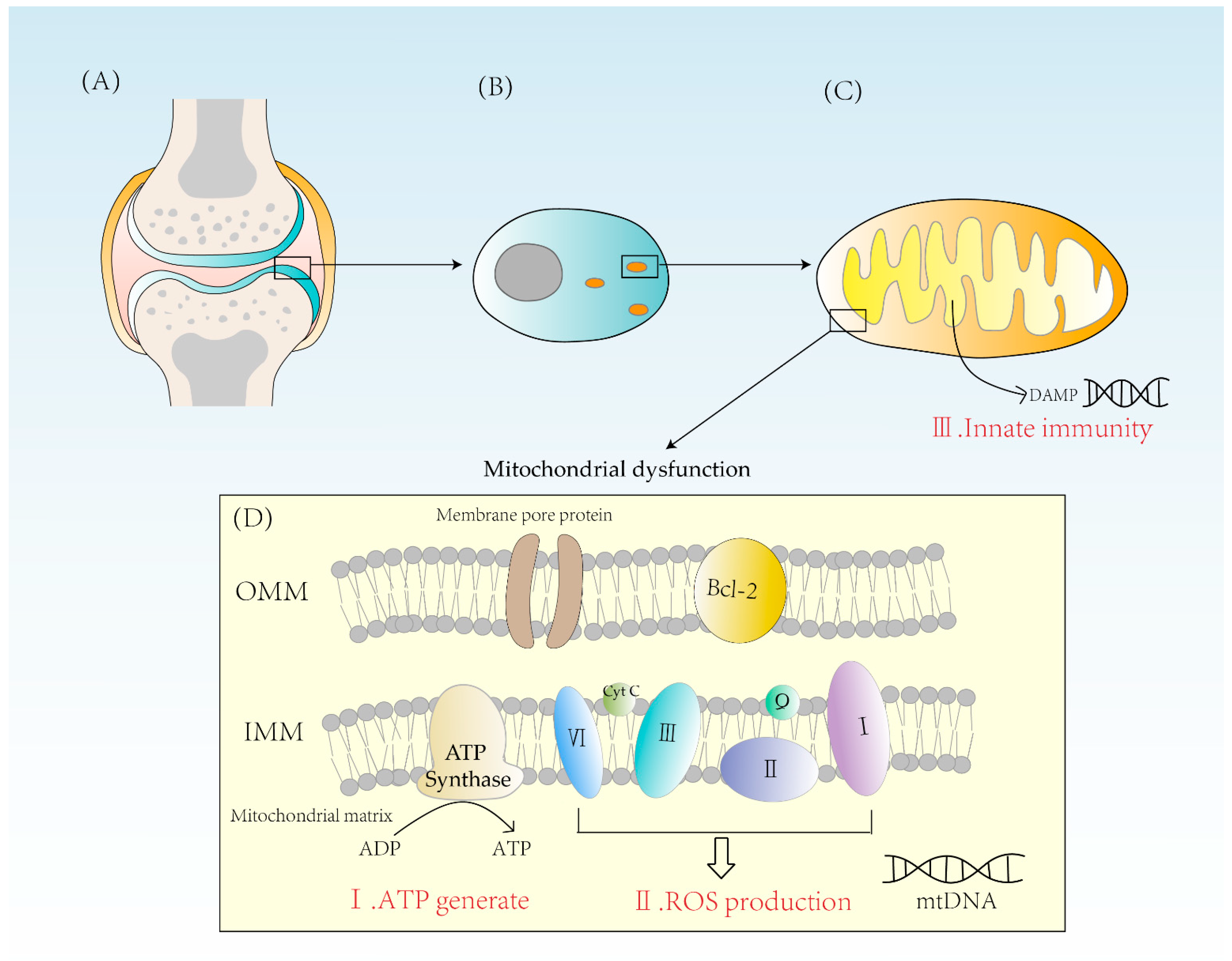
2. Three Ways That Mitochondrial Dysfunction Leads to RA
2.1. Abnormal Energy Metabolism
2.2. Excess ROS Production
2.3. Activation of Innate Immunity
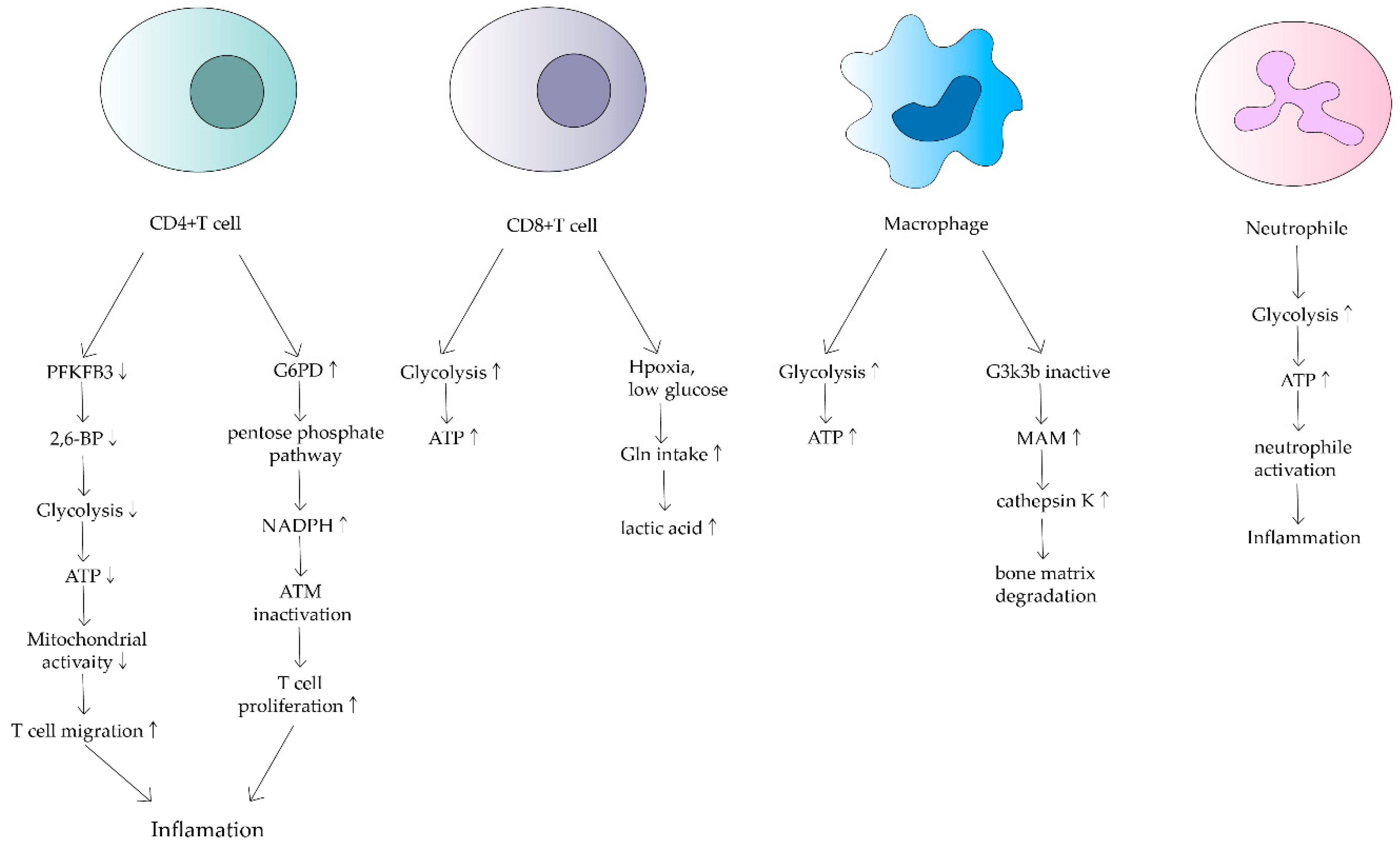
3. Effects of Mitochondrial Dysfunction on Immune Cells in RA
3.1. T Cell
3.2. Macrophage
3.3. Neutrophil
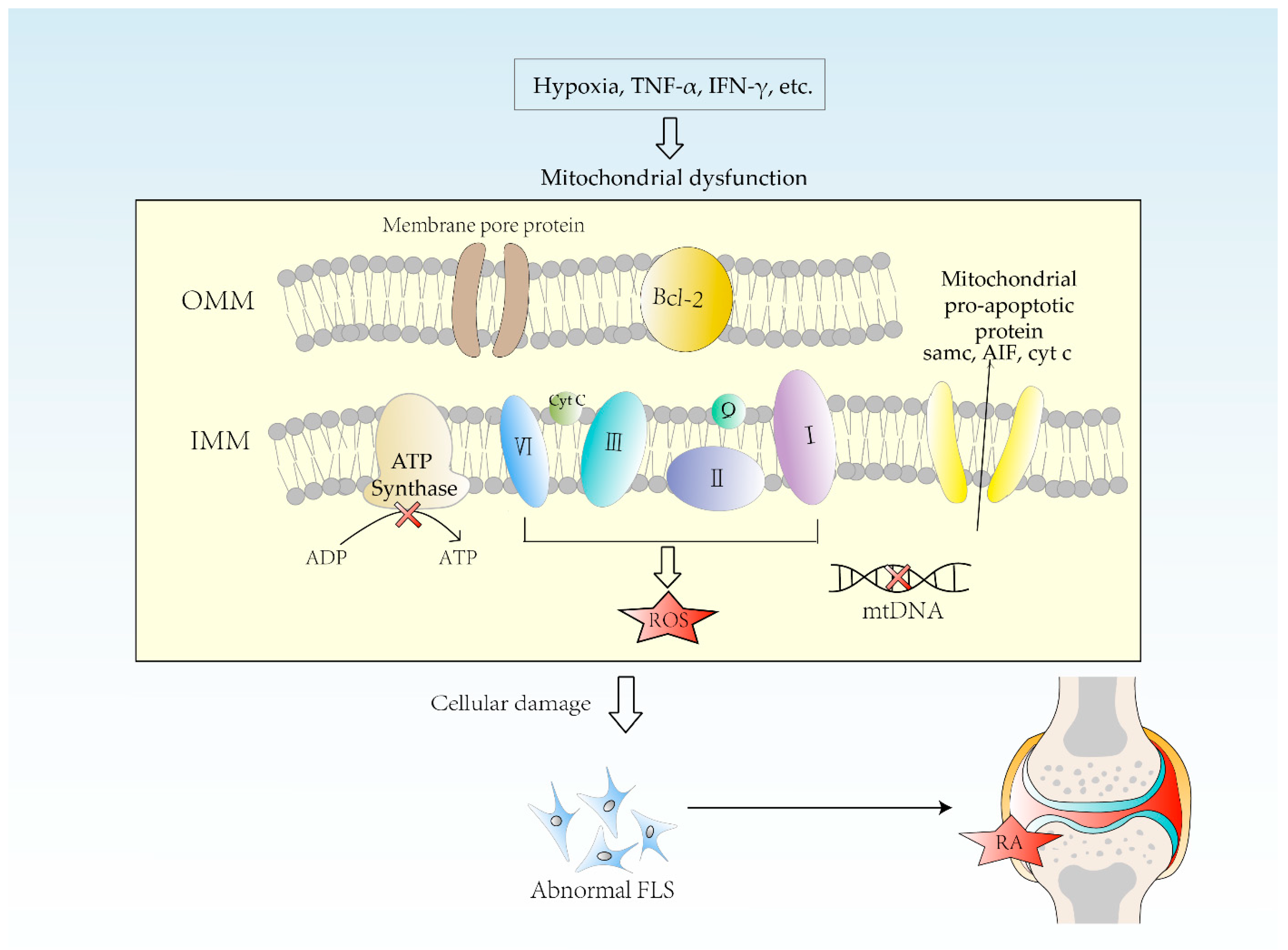
4. Etiology of Mitochondrial Dysfunction in RA
4.1. Hypoxia
4.2. mtDNA Mutation
4.3. Oxidative Stress
5. Impact of Mitochondrial Dysfunction on RA
5.1. Chondrocyte Autophagy
5.2. Immune and Pro-Inflammatory Responses
5.3. Apoptosis Pathway Disorders
6. Drugs Associated with Mitochondria in RA Treatment
6.1. Conventional Synthetic Anti-Rheumatic Drugs (csDMARDs)
6.1.1. Methotrexate
6.1.2. Leflunomide
6.1.3. Sulfasalazine
6.2. Biological Agents DMARDs (bDMARDs)
6.3. Targeted Synthetic DMARDs (tsDMARDs)
7. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clayton, S.A.; MacDonald, L.; Kurowska-Stolarska, M.; Clark, A.R. Mitochondria as Key Players in the Pathogenesis and Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 673916. [Google Scholar] [CrossRef] [PubMed]
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Duan, M.; Liu, Y.; Wang, C.; Xie, J. Role of Mitochondria in Physiology of Chondrocytes and Diseases of Osteoarthritis and Rheumatoid Arthritis. Cartilage 2021, 13 (Suppl. 2), 1102S–1121S. [Google Scholar] [CrossRef] [PubMed]
- Breda, C.N.S.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.A. Rheumatoid Arthritis. Ann. Intern. Med. 2019, 170, ITC1–ITC16. [Google Scholar] [CrossRef]
- Deane, K.D.; Holers, V.M. Rheumatoid Arthritis Pathogenesis, Prediction, and Prevention: An Emerging Paradigm Shift. Arthritis Rheumatol. 2021, 73, 181–193. [Google Scholar] [CrossRef]
- Petrelli, F.; Mariani, F.M.; Alunno, A.; Puxeddu, I. Pathogenesis of rheumatoid arthritis: One year in review 2022. Clin. Exp. Rheumatol. 2022, 40, 475–482. [Google Scholar] [CrossRef]
- Van Delft, M.A.M.; Huizinga, T.W.J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef]
- Almutairi, K.; Nossent, J.; Preen, D.; Keen, H.; Inderjeeth, C. The global prevalence of rheumatoid arthritis: A meta-analysis based on a systematic review. Rheumatol. Int. 2021, 41, 863–877. [Google Scholar] [CrossRef]
- Bingham, C.O.; Butanis, A.L.; Orbai, A.M.; Jones, M.; Ruffing, V.; Lyddiatt, A.; Schrandt, M.S.; Bykerk, V.P.; Cook, K.F.; Bartlett, S.J. Patients and clinicians define symptom levels and meaningful change for PROMIS pain interference and fatigue in RA using bookmarking. Rheumatology 2021, 60, 4306–4314. [Google Scholar] [CrossRef]
- Li, M.; Luo, X.; Long, X.; Jiang, P.; Jiang, Q.; Guo, H.; Chen, Z. Potential role of mitochondria in synoviocytes. Clin. Rheumatol. 2021, 40, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J. Oxidative Stress and Mitochondrial Dysfunction in Human Diseases: Pathophysiology, Predictive Biomarkers, Therapeutic. Biomolecules 2020, 10, 1558. [Google Scholar] [CrossRef] [PubMed]
- Phull, A.R.; Nasir, B.; Haq, I.U.; Kim, S.J. Oxidative stress, consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem. Biol. Interact. 2018, 281, 121–136. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074.e8. [Google Scholar] [CrossRef] [PubMed]
- Telang, S.; Clem, B.F.; Klarer, A.C.; Clem, A.L.; Trent, J.O.; Bucala, R.; Chesney, J. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. J. Transl. Med. 2012, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Fujii, H.; Mohan, S.V.; Goronzy, J.J.; Weyand, C.M. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med. 2013, 210, 2119–2134. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shen, Y.; Oishi, H.; Matteson, E.L.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 331ra38. [Google Scholar] [CrossRef]
- Wu, B.; Qiu, J.; Zhao, T.V.; Wang, Y.; Maeda, T.; Goronzy, I.N.; Akiyama, M.; Ohtsuki, S.; Jin, K.; Tian, L.; et al. Succinyl-CoA Ligase Deficiency in Pro-Inflammatory and Tissue-Invasive T Cells. Cell Metab. 2020, 32, 967–980.e5. [Google Scholar] [CrossRef]
- Mellado, M.; Martínez-Muñoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodríguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef]
- Souto-Carneiro, M.M.; Klika, K.D.; Abreu, M.T.; Meyer, A.P.; Saffrich, R.; Sandhoff, R.; Jennemann, R.; Kraus, F.V.; Tykocinski, L.; Eckstein, V.; et al. Effect of Increased Lactate Dehydrogenase A Activity and Aerobic Glycolysis on the Proinflammatory Profile of Autoimmune CD8+ T Cells in Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 2050–2064. [Google Scholar] [CrossRef]
- Udalova, I.A.; Mantovani, A.; Feldmann, M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 472–485. [Google Scholar] [CrossRef]
- Zeisbrich, M.; Yanes, R.E.; Zhang, H.; Watanabe, R.; Li, Y.; Brosig, L.; Hong, J.; Wallis, B.B.; Giacomini, J.C.; Assimes, T.L.; et al. Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann. Rheum. Dis. 2018, 77, 1053–1062. [Google Scholar] [CrossRef]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef]
- Hao, L.; Zhu, G.; Lu, Y.; Wang, M.; Jules, J.; Zhou, X.; Chen, W. Deficiency of cathepsin K prevents inflammation and bone erosion in rheumatoid arthritis and periodontitis and reveals its shared osteoimmune role. FEBS Lett. 2015, 589, 1331–1339. [Google Scholar] [CrossRef]
- Sun, P.; Liu, Y.; Deng, X.; Yu, C.; Dai, N.; Yuan, X.; Chen, L.; Yu, S.; Si, W.; Wang, X.; et al. An inhibitor of cathepsin K, icariin suppresses cartilage and bone degradation in mice of collagen-induced arthritis. Phytomedicine 2013, 20, 975–979. [Google Scholar] [CrossRef]
- Injarabian, L.; Devin, A.; Ransac, S.; Marteyn, B.S. Neutrophil Metabolic Shift during their Lifecycle: Impact on their Survival and Activation. Int. J. Mol. Sci. 2019, 21, 287. [Google Scholar] [CrossRef]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Biniecka, M.; Canavan, M.; McGarry, T.; Gao, W.; McCormick, J.; Cregan, S.; Gallagher, L.; Smith, T.; Phelan, J.J.; Ryan, J.; et al. Dysregulated bioenergetics: A key regulator of joint inflammation. Ann. Rheum. Dis. 2016, 75, 2192–2200. [Google Scholar] [CrossRef]
- Vega, R.B.; Horton, J.L.; Kelly, D.P. Maintaining ancient organelles: Mitochondrial biogenesis and maturation. Circ. Res. 2015, 116, 1820–1834. [Google Scholar] [CrossRef]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [Green Version]
- Harty, L.C.; Biniecka, M.; O’Sullivan, J.; Fox, E.; Mulhall, K.; Veale, D.J.; Fearon, U. Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann. Rheum. Dis. 2012, 71, 582–588. [Google Scholar] [CrossRef]
- Biniecka, M.; Fox, E.; Gao, W.; Ng, C.T.; Veale, D.J.; Fearon, U.; O’Sullivan, J. Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis Rheum. 2011, 63, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, S.; Hosomichi, K.; Okudaira, Y.; Nakaoka, H.; Suzuki, Y.; Kuwana, M.; Sato, S.; Kaneko, Y.; Homma, Y.; Oka, A.; et al. Aggregation of rare/low-frequency variants of the mitochondria respiratory chain-related proteins in rheumatoid arthritis patients. J. Hum. Genet. 2015, 60, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Hitchon, C.A.; El-Gabalawy, H.S. Oxidation in rheumatoid arthritis. Arthritis Res. Ther. 2004, 6, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Y.; Gao, Y.; Qi, D.; Zhao, L.; Zhao, L.; Liu, C.; Tao, T.; Zhou, C.; Sun, X.; et al. NR1D1 modulates synovial inflammation and bone destruction in rheumatoid arthritis. Cell Death Dis. 2020, 11, 129. [Google Scholar] [CrossRef]
- Pulkki, K.J.; Eerola, E.T.; Saario, R.M.; Toivanen, A.; Vuorio, E.I. Activated monocytes induce arthritis-associated changes in mitochondria of cultured synovial fibroblasts. Scand. J. Rheumatol. 1988, 17, 131–141. [Google Scholar] [CrossRef]
- Hollander, J.M.; Zeng, L. The Emerging Role of Glucose Metabolism in Cartilage Development. Curr. Osteoporos. Rep. 2019, 17, 59–69. [Google Scholar] [CrossRef]
- Olofsson, P.; Holmberg, J.; Tordsson, J.; Lu, S.; Akerström, B.; Holmdahl, R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat. Genet. 2003, 33, 25–32. [Google Scholar] [CrossRef]
- Clancy, R.M.; Rediske, J.; Tang, X.; Nijher, N.; Frenkel, S.; Philips, M.; Abramson, S.B. Outside-in signaling in the chondrocyte. Nitric oxide disrupts fibronectin-induced assembly of a subplasmalemmal actin/rho A/focal adhesion kinase signaling complex. J. Clin. Investig. 1997, 100, 1789–1796. [Google Scholar] [CrossRef]
- Wang, S.; Deng, Z.; Ma, Y.; Jin, J.; Qi, F.; Li, S.; Liu, C.; Lyu, F.J.; Zheng, Q. The Role of Autophagy and Mitophagy in Bone Metabolic Disorders. Int. J. Biol. Sci. 2020, 16, 2675–2691. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Caramés, B.; Martín, M.A.; Cillero-Pastor, B.; Lires-Dean, M.; Fuentes-Boquete, I.; Arenas, J.; Blanco, F.J. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthr. Cartil. 2006, 14, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.F.; Feng, L. Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Zhang, J.; Li, Y.; Chen, Y.X.; Wu, X.M.; Li, X.; Zhang, X.F.; Ma, L.Z.; Yang, Y.Z.; Zheng, K.M.; et al. Advanced Oxidation Protein Products Induce G1/G0-Phase Arrest in Ovarian Granulosa Cells via the ROS-JNK/p38 MAPK-p21 Pathway in Premature Ovarian Insufficiency. Oxidative Med. Cell. Longev. 2021, 2021, 6634718. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhong, Z.M.; Zhu, S.Y.; Liao, C.R.; Pan, Y.; Zeng, J.H.; Zheng, S.; Ding, R.T.; Lin, Q.S.; Ye, Q.; et al. Advanced oxidation protein products induce chondrocyte apoptosis via receptor for advanced glycation end products-mediated, redox-dependent intrinsic apoptosis pathway. Apoptosis 2016, 21, 36–50. [Google Scholar] [CrossRef]
- Barrera, M.J.; Aguilera, S.; Castro, I.; Carvajal, P.; Jara, D.; Molina, C.; González, S.; González, M.J. Dysfunctional mitochondria as critical players in the inflammation of autoimmune diseases: Potential role in Sjögren’s syndrome. Autoimmun. Rev. 2021, 20, 102867. [Google Scholar] [CrossRef]
- Henrotin, Y.; Kurz, B.; Aigner, T. Oxygen and reactive oxygen species in cartilage degradation: Friends or foes? Osteoarthr. Cartil. 2005, 13, 643–654. [Google Scholar] [CrossRef]
- Qiu, J.; Wu, B.; Goodman, S.B.; Berry, G.J.; Goronzy, J.J.; Weyand, C.M. Metabolic Control of Autoimmunity and Tissue Inflammation in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 652771. [Google Scholar] [CrossRef]
- Tu, J.; Hong, W.; Zhang, P.; Wang, X.; Körner, H.; Wei, W. Ontology and Function of Fibroblast-like and Macrophage-like Synoviocytes: How Do They Talk to Each Other and Can They Be Targeted for Rheumatoid Arthritis Therapy? Front. Immunol. 2018, 9, 1467. [Google Scholar] [CrossRef]
- Kim, E.K.; Kwon, J.E.; Lee, S.Y.; Lee, E.J.; Kim, D.S.; Moon, S.J.; Lee, J.; Kwok, S.K.; Park, S.H.; Cho, M.L. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 1998, 3, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Kurowska, M.; Rudnicka, W.; Kontny, E.; Janicka, I.; Chorazy, M.; Kowalczewski, J.; Ziółkowska, M.; Ferrari-Lacraz, S.; Strom, T.B.; Maśliński, W. Fibroblast-like synoviocytes from rheumatoid arthritis patients express functional IL-15 receptor complex: Endogenous IL-15 in autocrine fashion enhances cell proliferation and expression of Bcl-x(L) and Bcl-2. J. Immunol. 2002, 169, 1760–1767. [Google Scholar] [CrossRef]
- Kim, S.K.; Park, K.Y.; Yoon, W.C.; Park, S.H.; Park, K.K.; Yoo, D.H.; Choe, J.Y. Melittin enhances apoptosis through suppression of IL-6/sIL-6R complex-induced NF-κB and STAT3 activation and Bcl-2 expression for human fibroblast-like synoviocytes in rheumatoid arthritis. Jt. Bone Spine 2011, 78, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Al-Azab, M.; Qaed, E.; Ouyang, X.; Elkhider, A.; Walana, W.; Li, H.; Li, W.; Tang, Y.; Adlat, S.; Wei, J.; et al. TL1A/TNFR2-mediated mitochondrial dysfunction of fibroblast-like synoviocytes increases inflammatory response in patients with rheumatoid arthritis via reactive oxygen species generation. FEBS J. 2020, 287, 3088–3104. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Bedoui, Y.; Guillot, X.; Sélambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023. [Google Scholar] [CrossRef]
- Herman, S.; Zurgil, N.; Deutsch, M. Low dose methotrexate induces apoptosis with reactive oxygen species involvement in T lymphocytic cell lines to a greater extent than in monocytic lines. Inflamm. Res. 2005, 54, 273–280. [Google Scholar] [CrossRef]
- Huang, C.; Hsu, P.; Hung, Y.; Liao, Y.; Liu, C.; Hour, C.; Kao, M.; Tsay, G.J.; Hung, H.; Liu, G.Y. Ornithine decarboxylase prevents methotrexate-induced apoptosis by reducing intracellular reactive oxygen species production. Apoptosis 2005, 10, 895–907. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, S.H.; Lee, S.W.; Lee, S.H.; Son, M.K.; Choi, Y.H.; Chung, W.T.; Yoo, Y.H. Synoviocyte apoptosis may differentiate responder and non-responder patients to methotrexate treatment in rheumatoid arthritis. Arch. Pharm. Res. 2014, 37, 1286–1294. [Google Scholar] [CrossRef]
- Heidari, R.; Ahmadi, A.; Mohammadi, H.; Ommati, M.M.; Azarpira, N.; Niknahad, H. Mitochondrial dysfunction and oxidative stress are involved in the mechanism of methotrexate-induced renal injury and electrolytes imbalance. Biomed. Pharmacother. 2018, 107, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Al Maruf, A.; O’Brien, P.J.; Naserzadeh, P.; Fathian, R.; Salimi, A.; Pourahmad, J. Methotrexate induced mitochondrial injury and cytochrome c release in rat liver hepatocytes. Drug Chem. Toxicol. 2018, 41, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Hemshekhar, M.; Thushara, R.M.; Sundaram, M.S.; NaveenKumar, S.K.; Naveen, S.; Devaraja, S.; Somyajit, K.; West, R.; Basappa; et al. Methotrexate Promotes Platelet Apoptosis via JNK-Mediated Mitochondrial Damage: Alleviation by N-Acetylcysteine and N-Acetylcysteine Amide. PLoS ONE 2015, 10, e0127558. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.; Keeling, S.O.; Katz, S.J.; Maksymowych, W.P.; Eurich, D.T.; Hall, J.J. Clinical effectiveness and safety of leflunomide in inflammatory arthritis: A report from the RAPPORT database with supporting patient survey. Clin. Rheumatol. 2017, 36, 1471–1478. [Google Scholar] [CrossRef]
- Miret-Casals, L.; Sebastián, D.; Brea, J.; Rico-Leo, E.M.; Palacín, M.; Fernández-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278.e4. [Google Scholar] [CrossRef]
- Klotz, L.; Eschborn, M.; Lindner, M.; Liebmann, M.; Herold, M.; Janoschka, C.; Torres Garrido, B.; Schulte-Mecklenbeck, A.; Gross, C.C.; Breuer, J.; et al. Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity-dependent effects. Sci. Transl. Med. 2019, 11, eaao5563. [Google Scholar] [CrossRef]
- Fang, J.; Uchiumi, T.; Yagi, M.; Matsumoto, S.; Amamoto, R.; Takazaki, S.; Yamaza, H.; Nonaka, K.; Kang, D. Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Biosci. Rep. 2013, 33, e00021. [Google Scholar] [CrossRef]
- Xuan, J.; Ren, Z.; Qing, T.; Couch, L.; Shi, L.; Tolleson, W.H.; Guo, L. Mitochondrial dysfunction induced by leflunomide and its active metabolite. Toxicology 2018, 396–397, 33–45. [Google Scholar] [CrossRef]
- Liptay, S.; Fulda, S.; Schanbacher, M.; Bourteele, S.; Ferri, K.F.; Kroemer, G.; Adler, G.; Debatin, K.M.; Schmid, R.M. Molecular mechanisms of sulfasalazine-induced T-cell apoptosis. Br. J. Pharmacol. 2002, 137, 608–620. [Google Scholar] [CrossRef]
- Niknahad, H.; Heidari, R.; Mohammadzadeh, R.; Ommati, M.M.; Khodaei, F.; Azarpira, N.; Abdoli, N.; Zarei, M.; Asadi, B.; Rasti, M.; et al. Sulfasalazine induces mitochondrial dysfunction and renal injury. Ren. Fail. 2017, 39, 745–753. [Google Scholar] [CrossRef] [Green Version]
- Jie, L.; Du, H.; Huang, Q.; Wei, S.; Huang, R.; Sun, W. Tanshinone IIA induces apoptosis in fibroblast-like synoviocytes in rheumatoid arthritis via blockade of the cell cycle in the G2/M phase and a mitochondrial pathway. Biol. Pharm. Bull. 2014, 37, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, G.; Zhang, Y.; Zhang, J.; Cao, W.; Chen, X. Effect of lentivirus-mediated overexpression or silencing of MnSOD on apoptosis of resveratrol-treated fibroblast-like synoviocytes in rheumatoid arthritis. Eur. J. Pharmacol. 2019, 844, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Costa, N.T.; Iriyoda, T.M.V.; Alfieri, D.F.; Simão, A.N.C.; Dichi, I. Influence of disease-modifying antirheumatic drugs on oxidative and nitrosative stress in patients with rheumatoid arthritis. Inflammopharmacology 2018, 26, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Biniecka, M.; Kennedy, A.; Ng, C.T.; Chang, T.C.; Balogh, E.; Fox, E.; Veale, D.J.; Fearon, U.; O’Sullivan, J.N. Successful tumour necrosis factor (TNF) blocking therapy suppresses oxidative stress and hypoxia-induced mitochondrial mutagenesis in inflammatory arthritis. Arthritis Res. Ther. 2011, 13, R121. [Google Scholar] [CrossRef] [PubMed]
- Meugnier, E.; Coury, F.; Tebib, J.; Ferraro-Peyret, C.; Rome, S.; Bienvenu, J.; Vidal, H.; Sibilia, J.; Fabien, N. Gene expression profiling in peripheral blood cells of patients with rheumatoid arthritis in response to anti-TNF-alpha treatments. Physiol. Genom. 2011, 43, 365–371. [Google Scholar] [CrossRef]
- Jamilloux, Y.; El Jammal, T.; Vuitton, L.; Gerfaud-Valentin, M.; Kerever, S.; Sève, P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 2019, 18, 102390. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- McGarry, T.; Orr, C.; Wade, S.; Biniecka, M.; Wade, S.; Gallagher, L.; Low, C.; Veale, D.J.; Fearon, U. JAK/STAT Blockade Alters Synovial Bioenergetics, Mitochondrial Function, and Proinflammatory Mediators in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1959–1970. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, C.; Wang, J.; Hong, F.; Yang, S. Mitochondrial Dysfunction in Rheumatoid Arthritis. Biomolecules 2022, 12, 1216. https://doi.org/10.3390/biom12091216
Ma C, Wang J, Hong F, Yang S. Mitochondrial Dysfunction in Rheumatoid Arthritis. Biomolecules. 2022; 12(9):1216. https://doi.org/10.3390/biom12091216
Chicago/Turabian StyleMa, Chen, Jie Wang, Fenfang Hong, and Shulong Yang. 2022. "Mitochondrial Dysfunction in Rheumatoid Arthritis" Biomolecules 12, no. 9: 1216. https://doi.org/10.3390/biom12091216