

The Origin of Discrepancies between Predictions and Annotations in Intrinsically Disordered Proteins


Abstract
:1. Introduction
2. Materials and Methods
2.1. Disorder Dataset
2.2. Proteomes
2.3. Prediction Methods
2.4. Evolutionary Conservation
3. Results
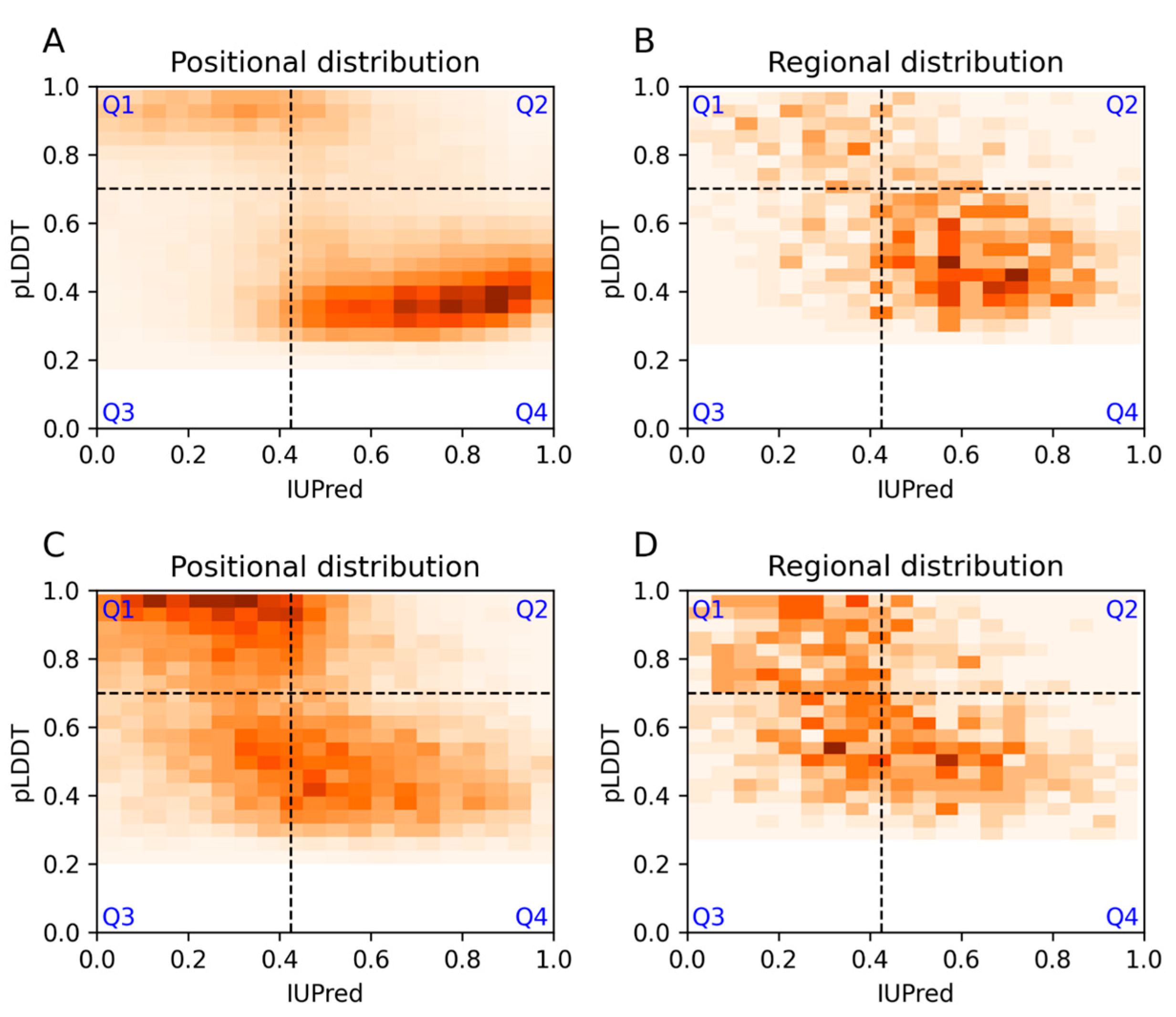
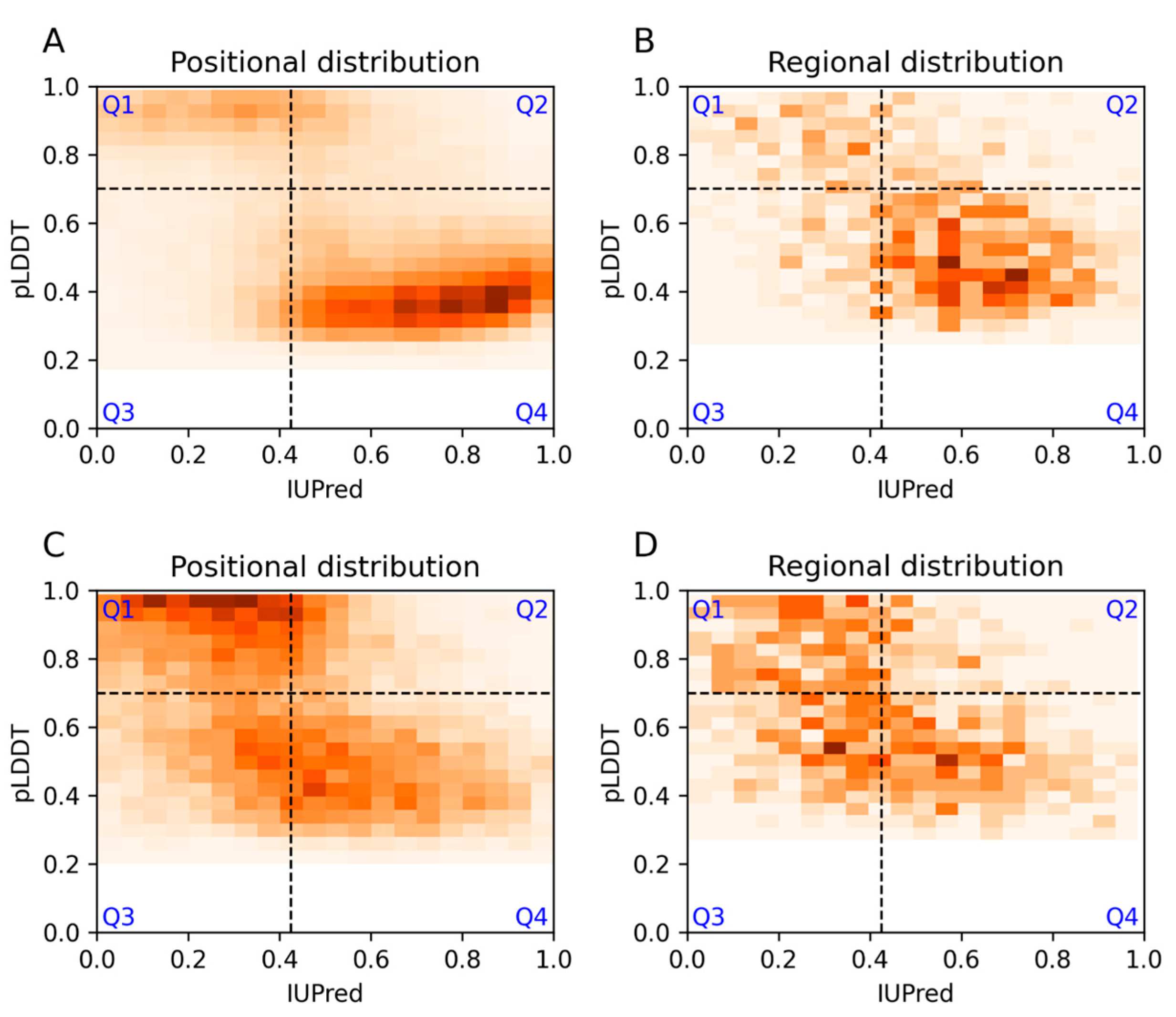
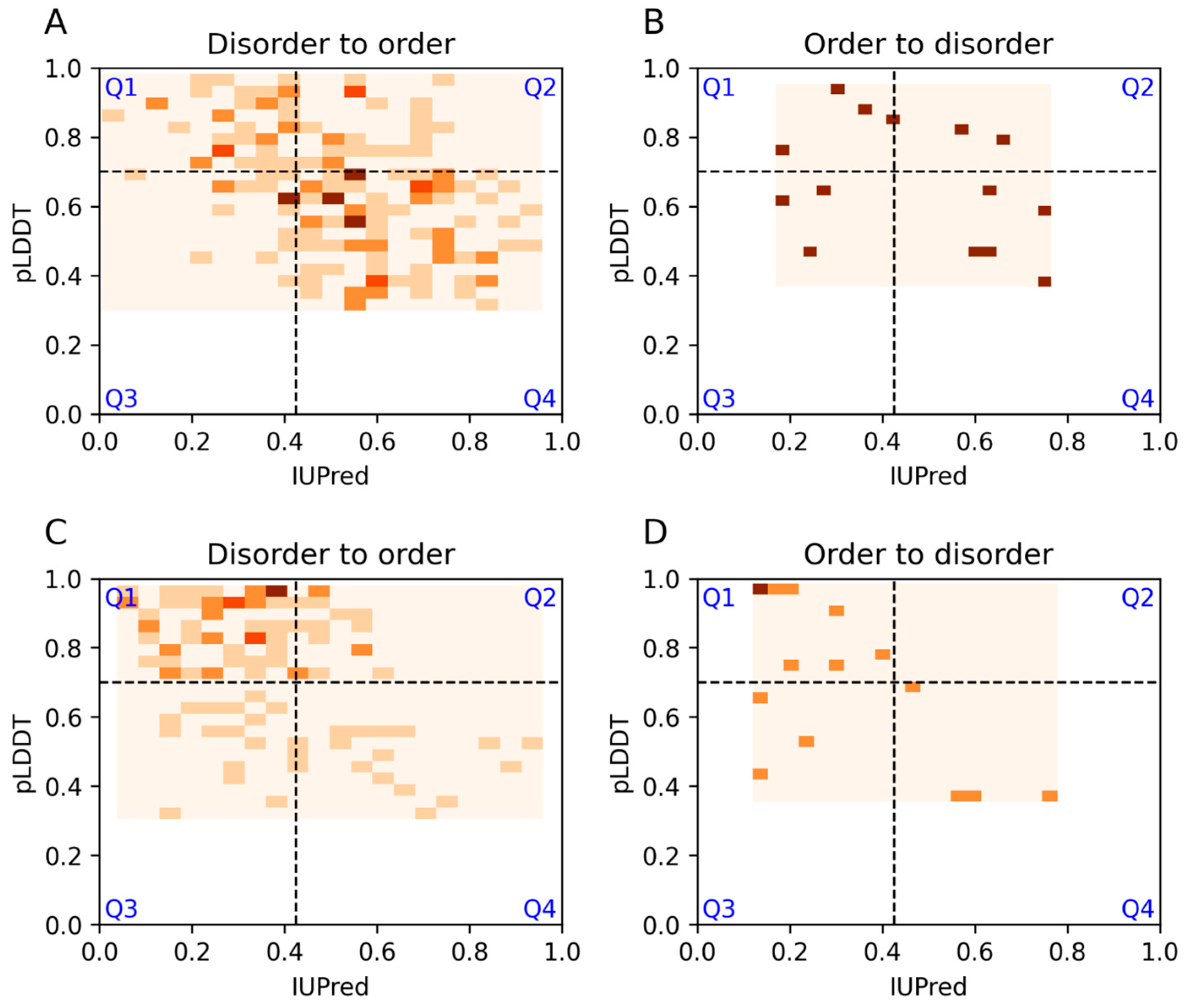
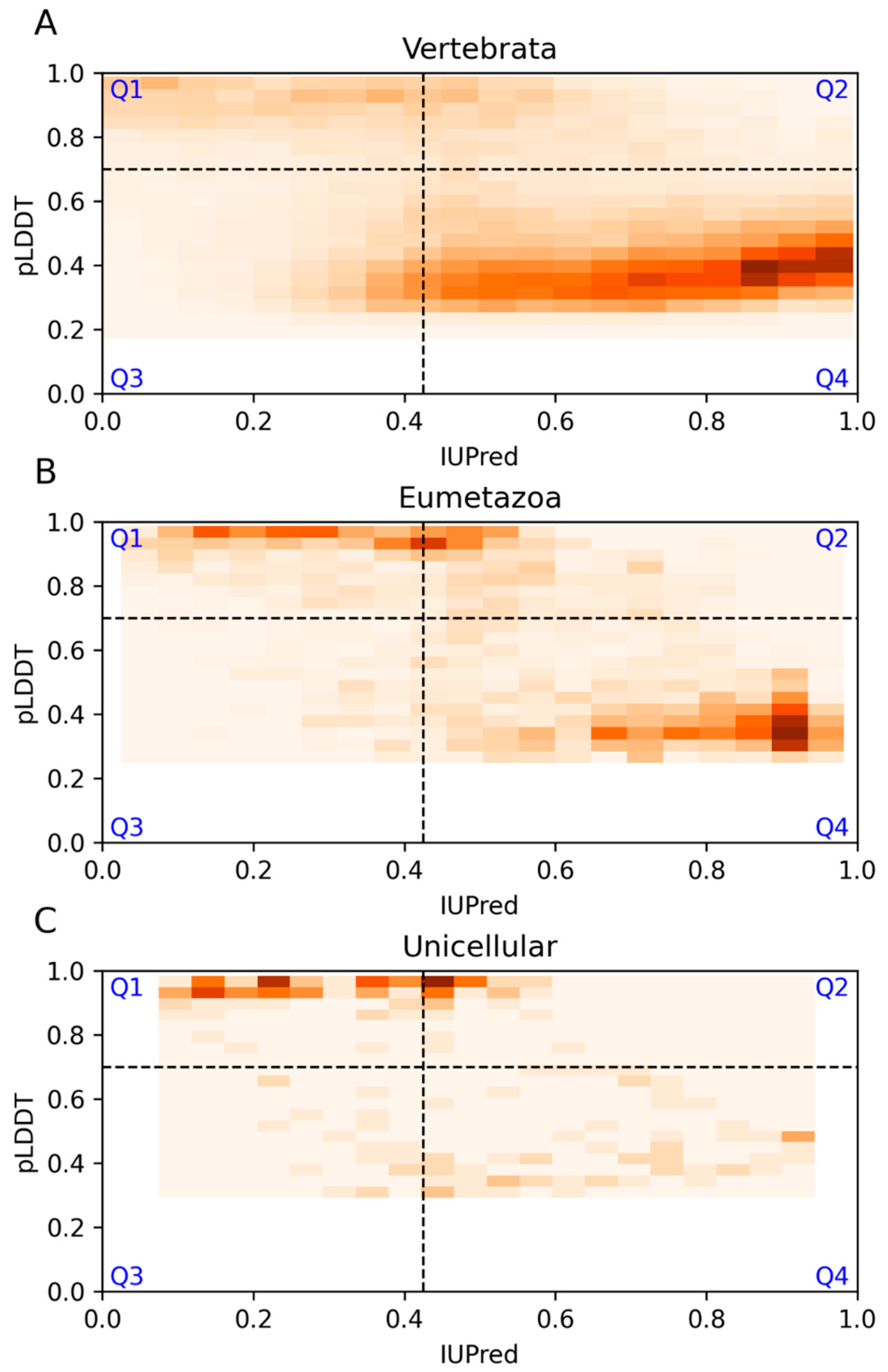
3.1. Agreement and Disagreement between Disorder Predictions
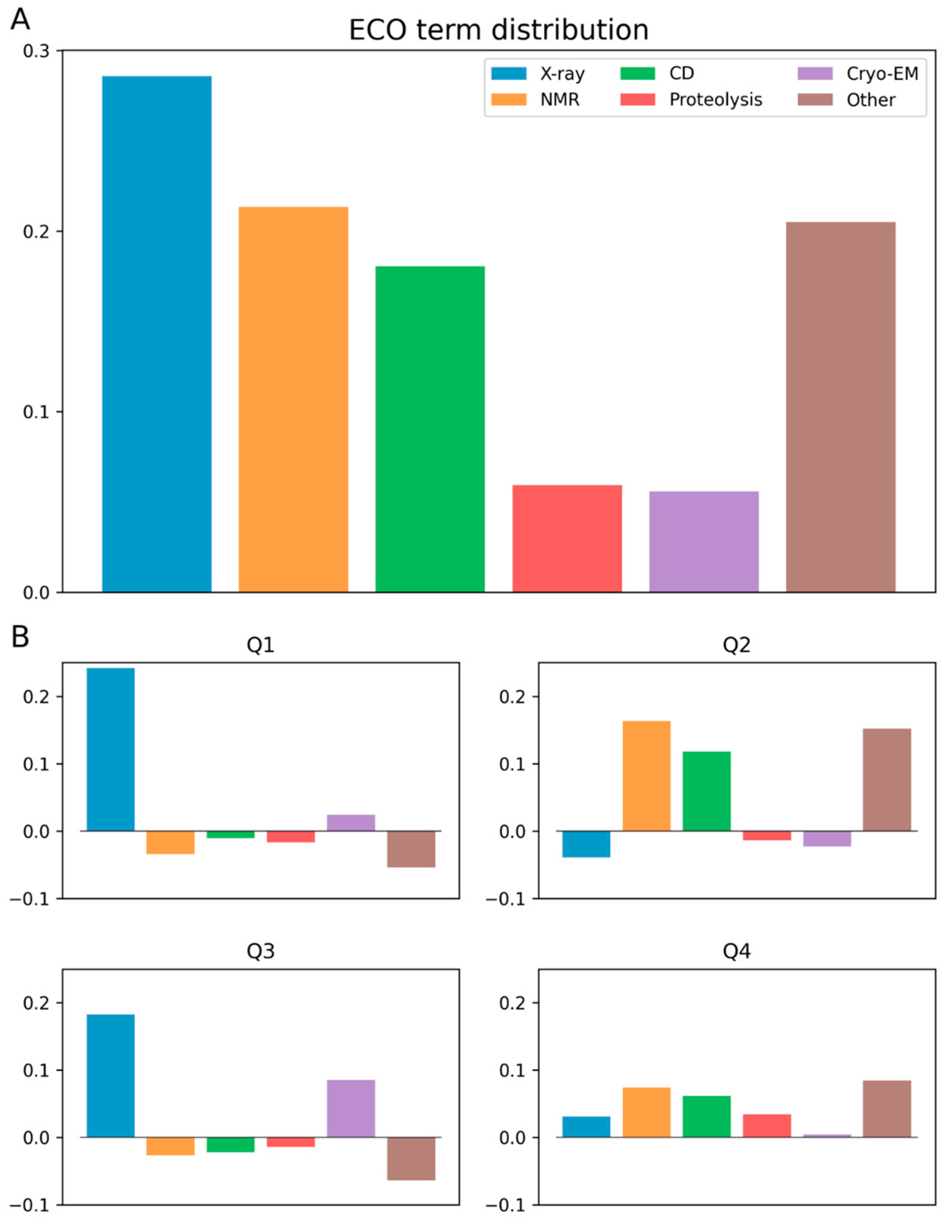
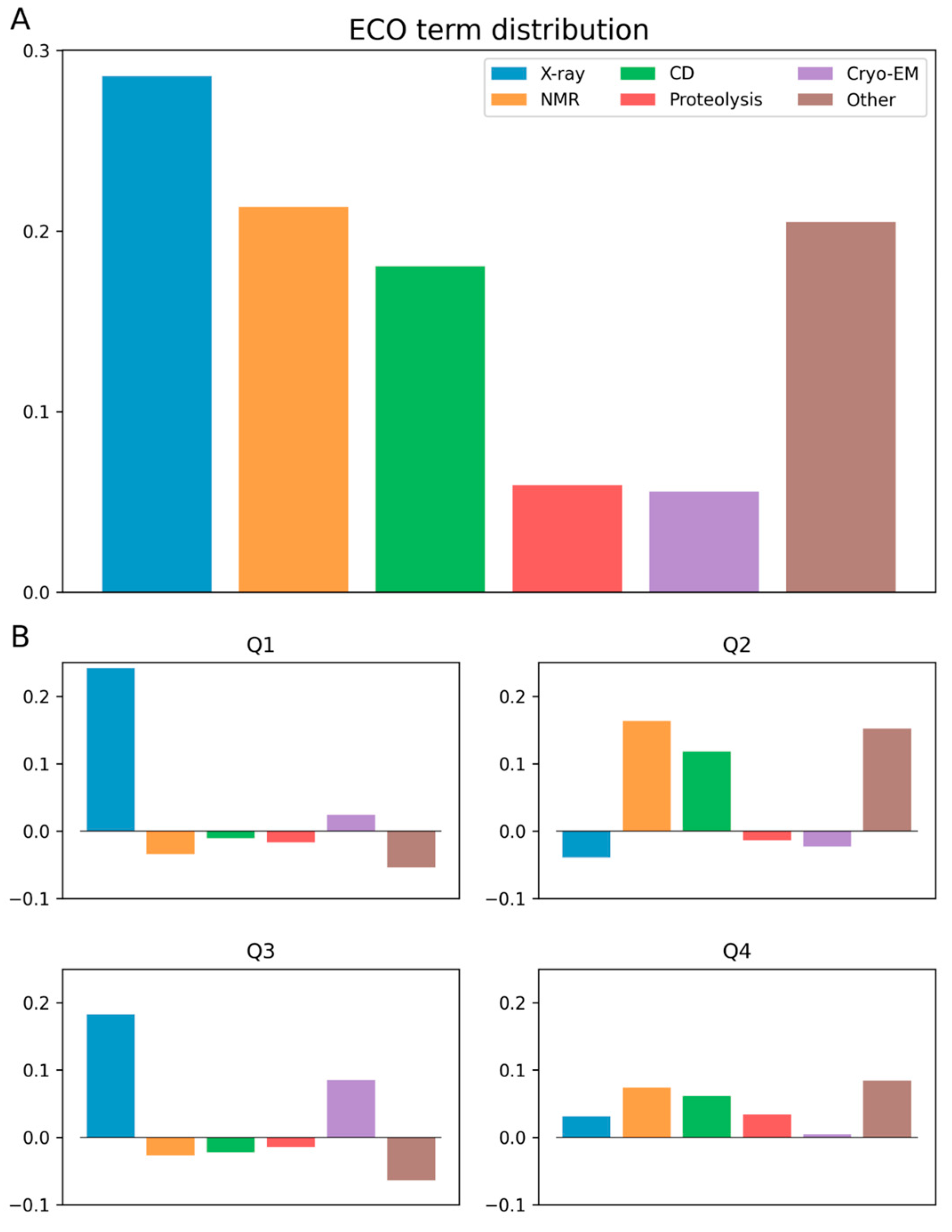
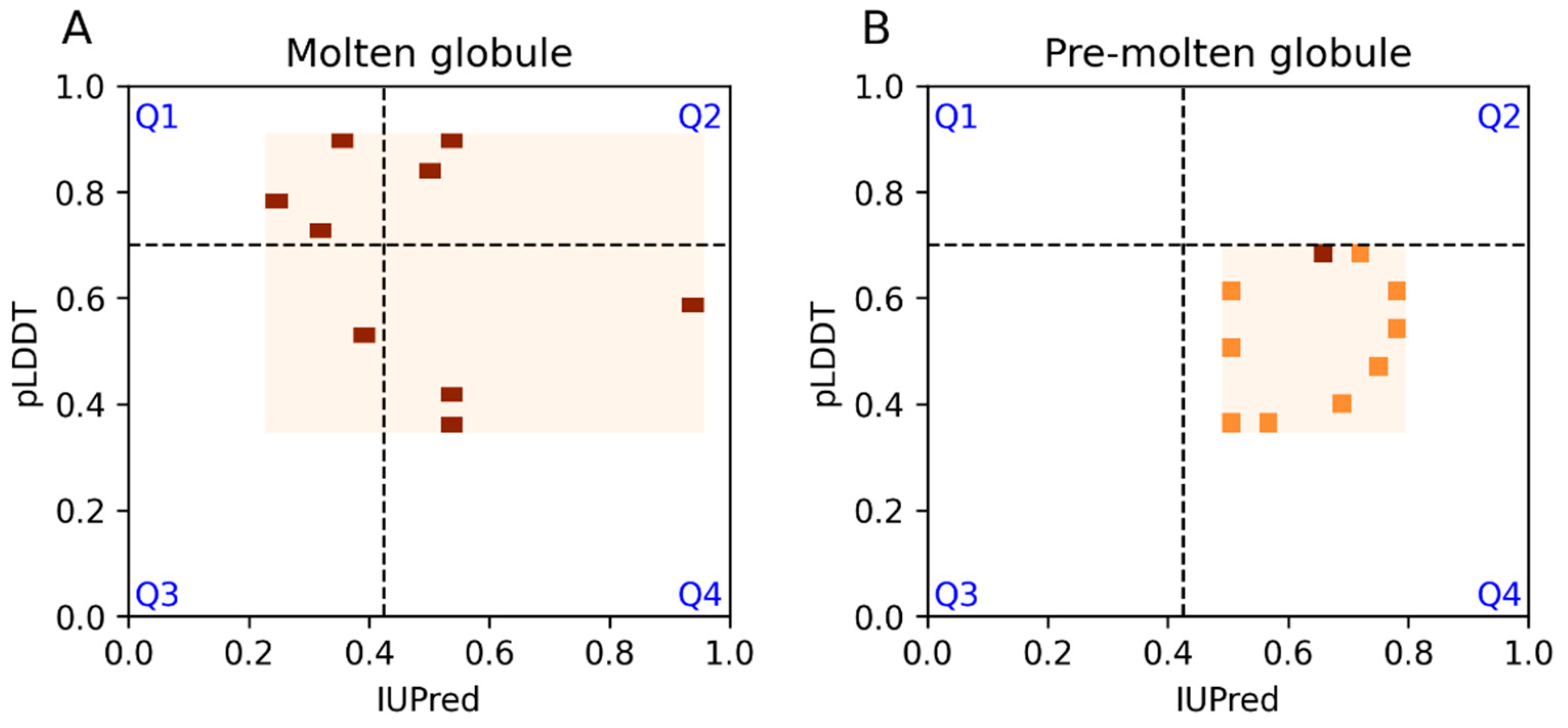
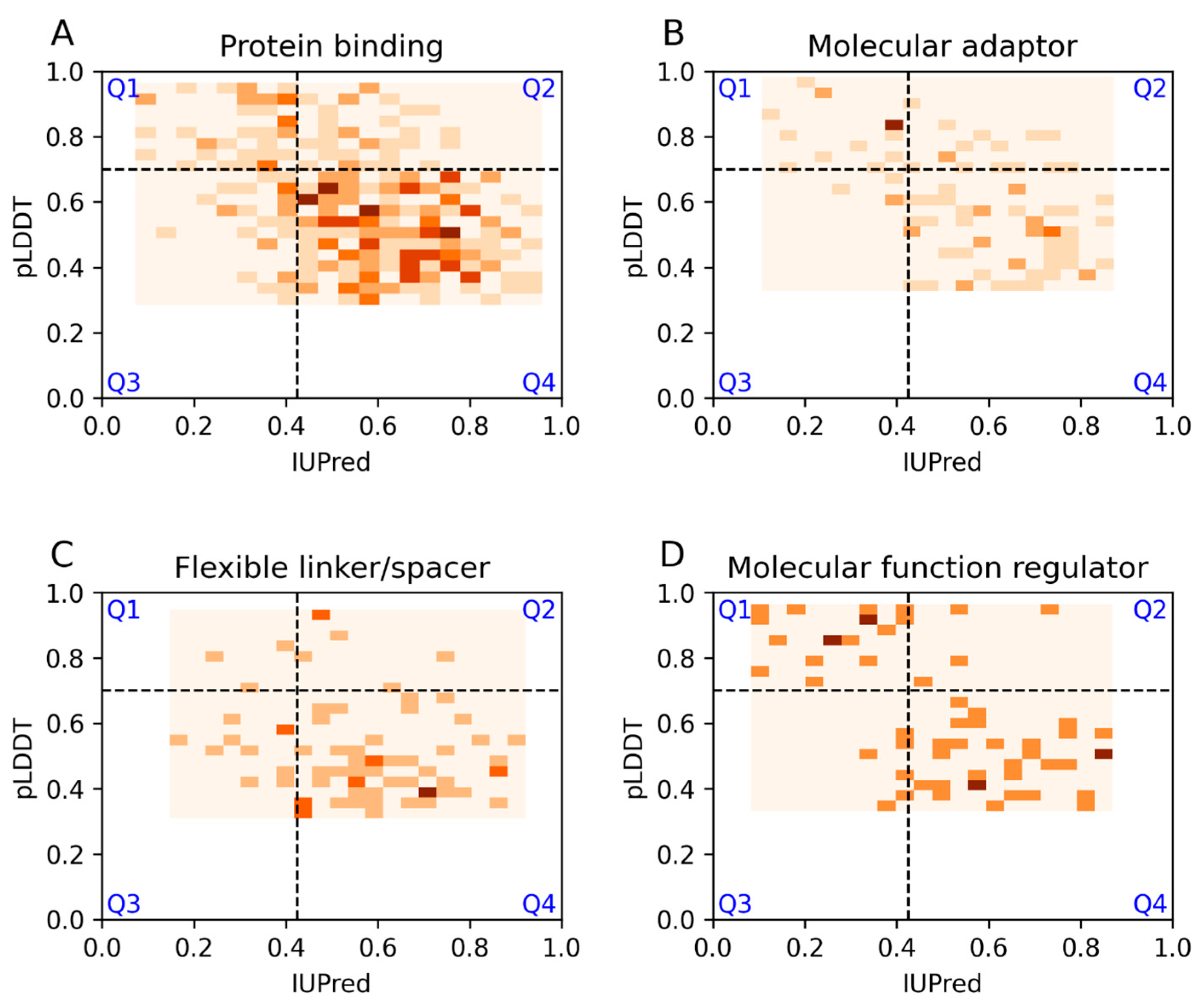
3.2. Ontology Terms
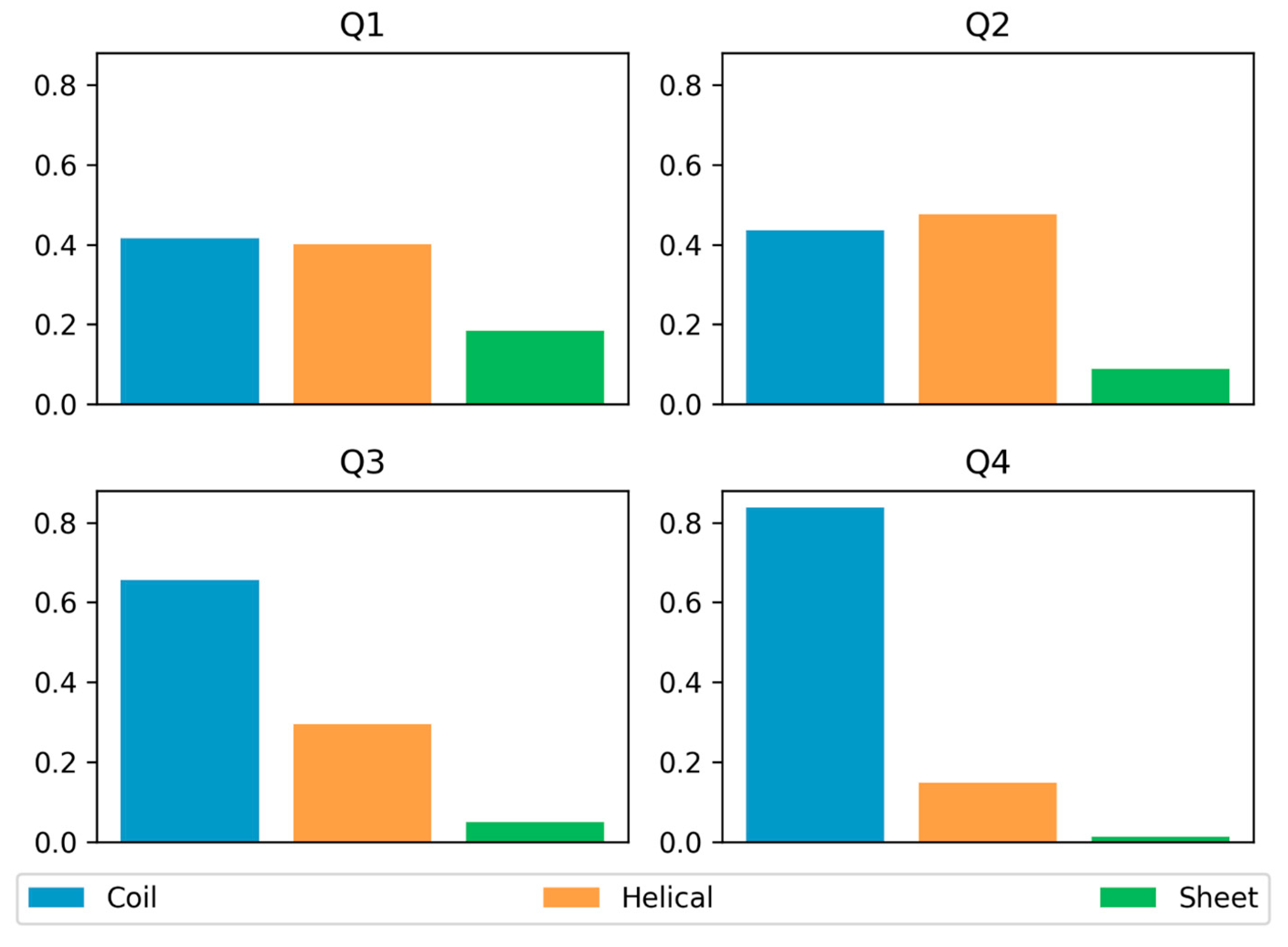
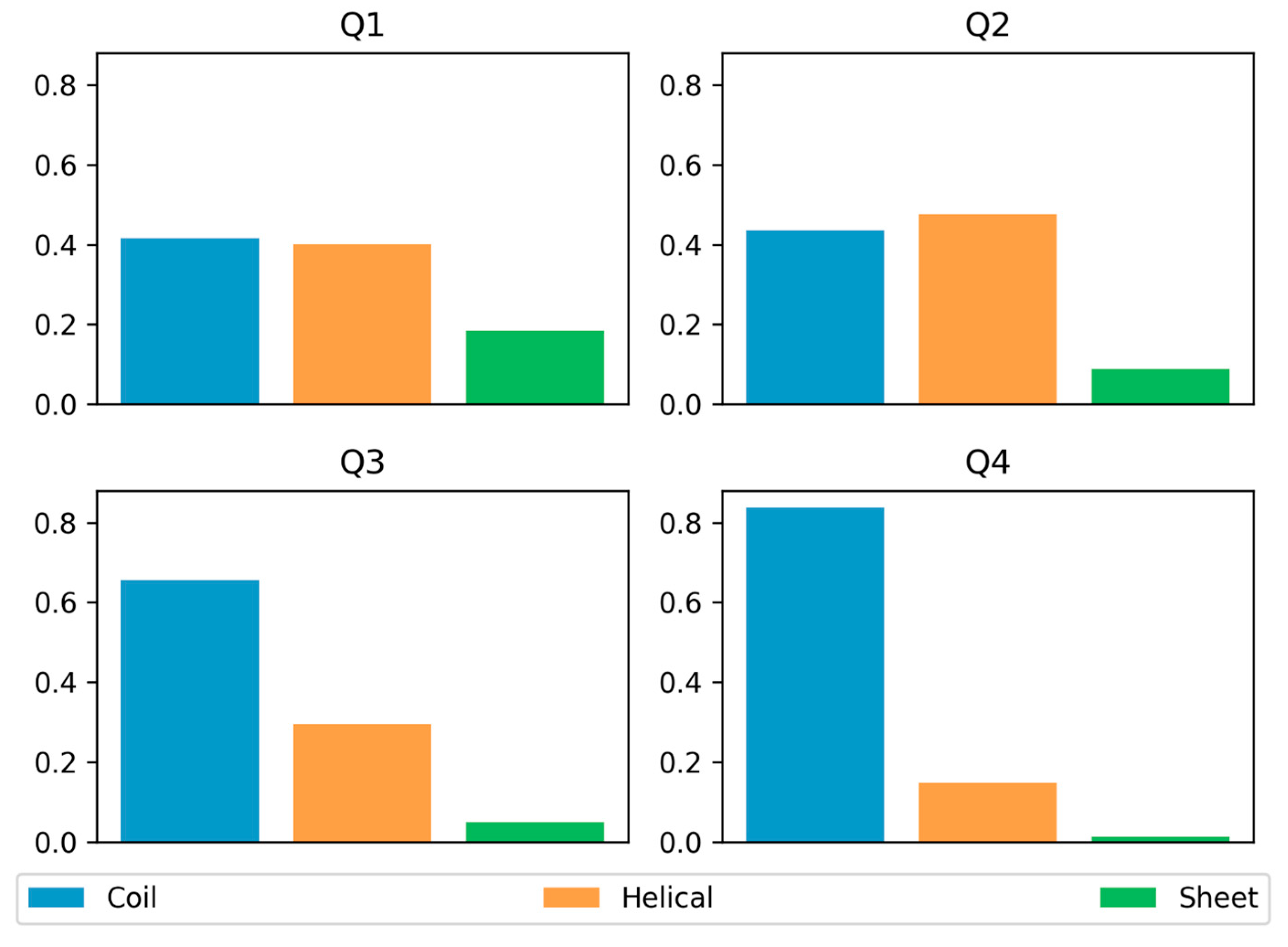
3.3. Secondary Structure Preferences
3.4. Sequence Conservation
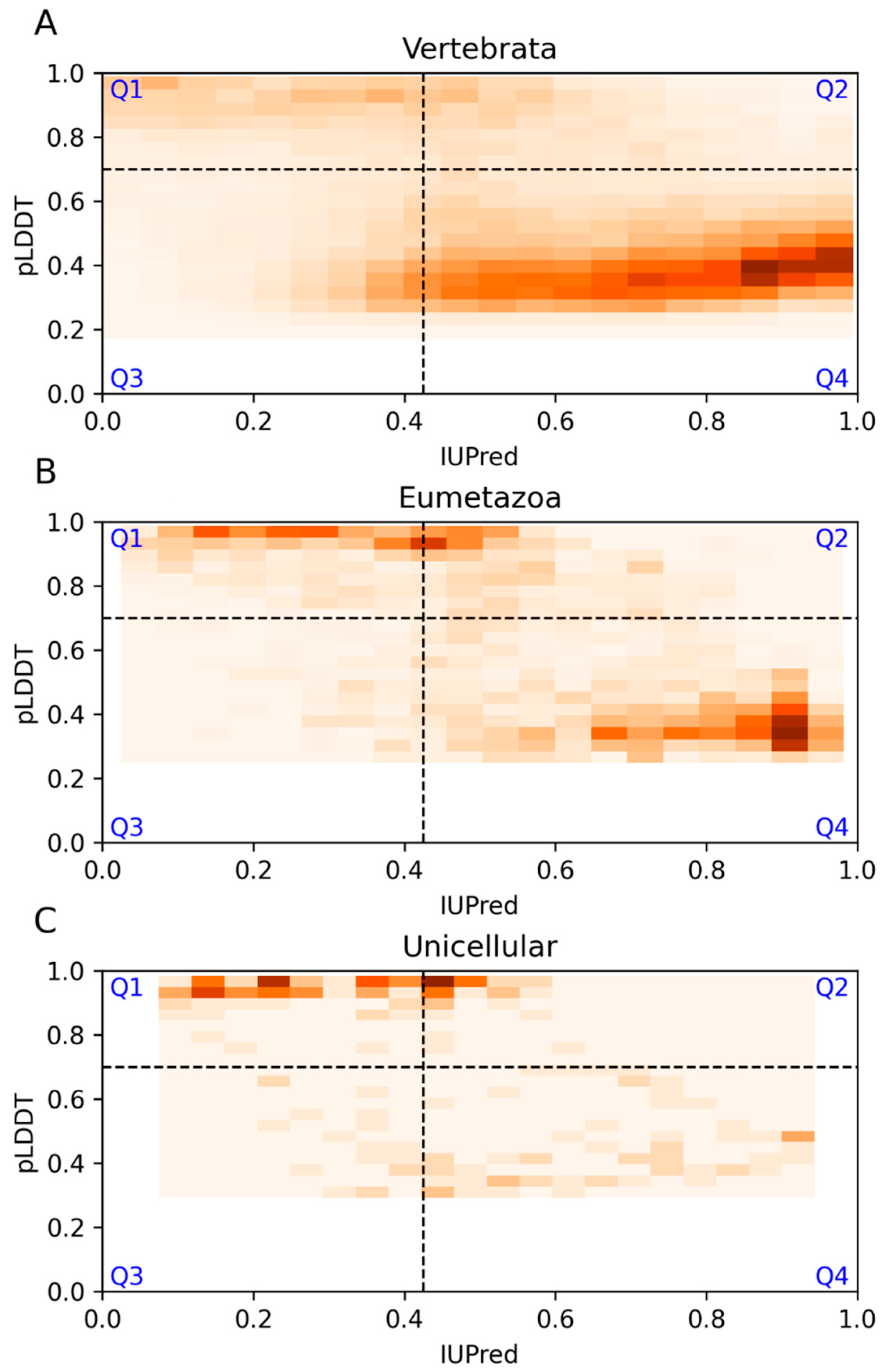
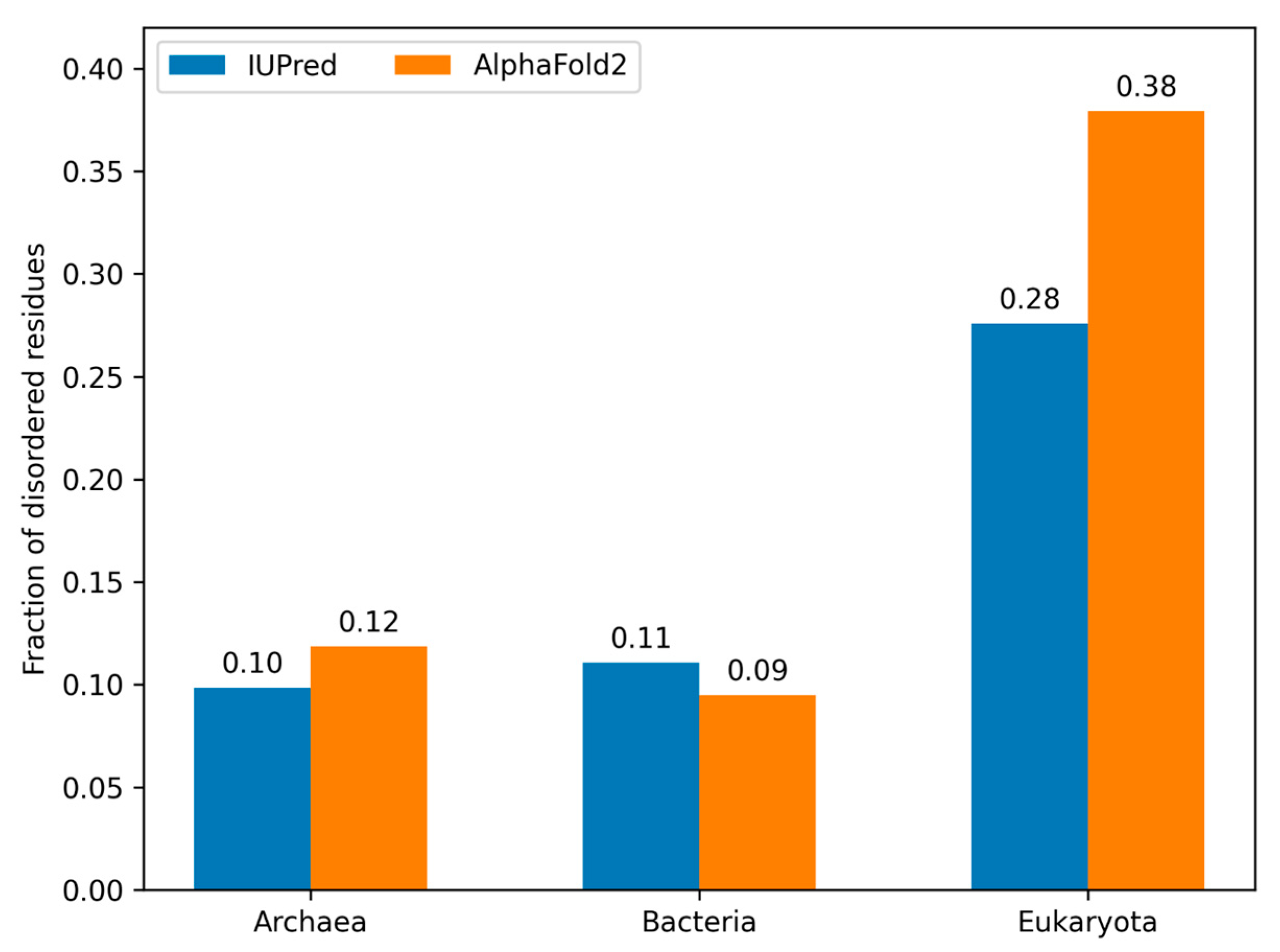
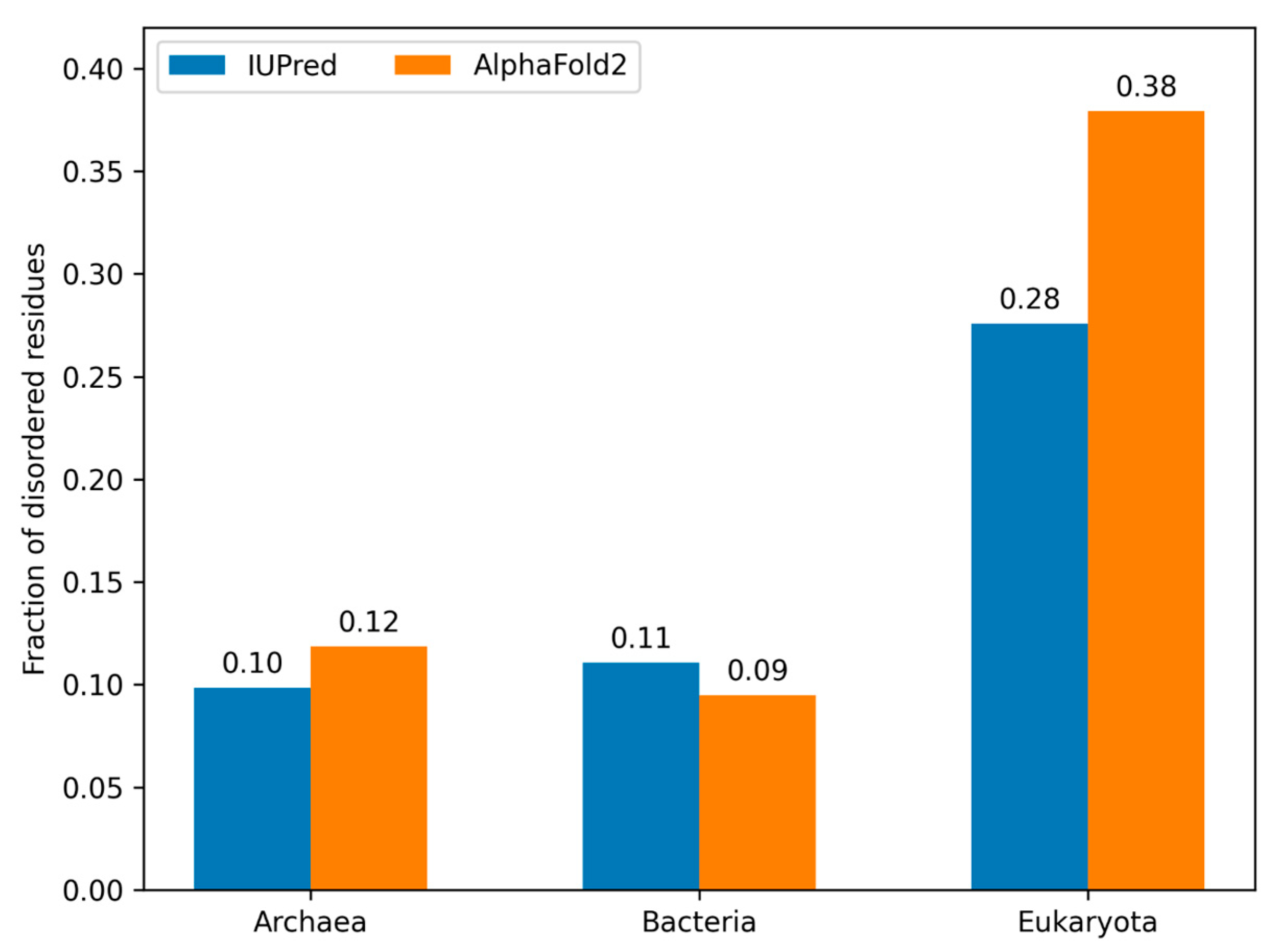
3.5. Model Proteomes
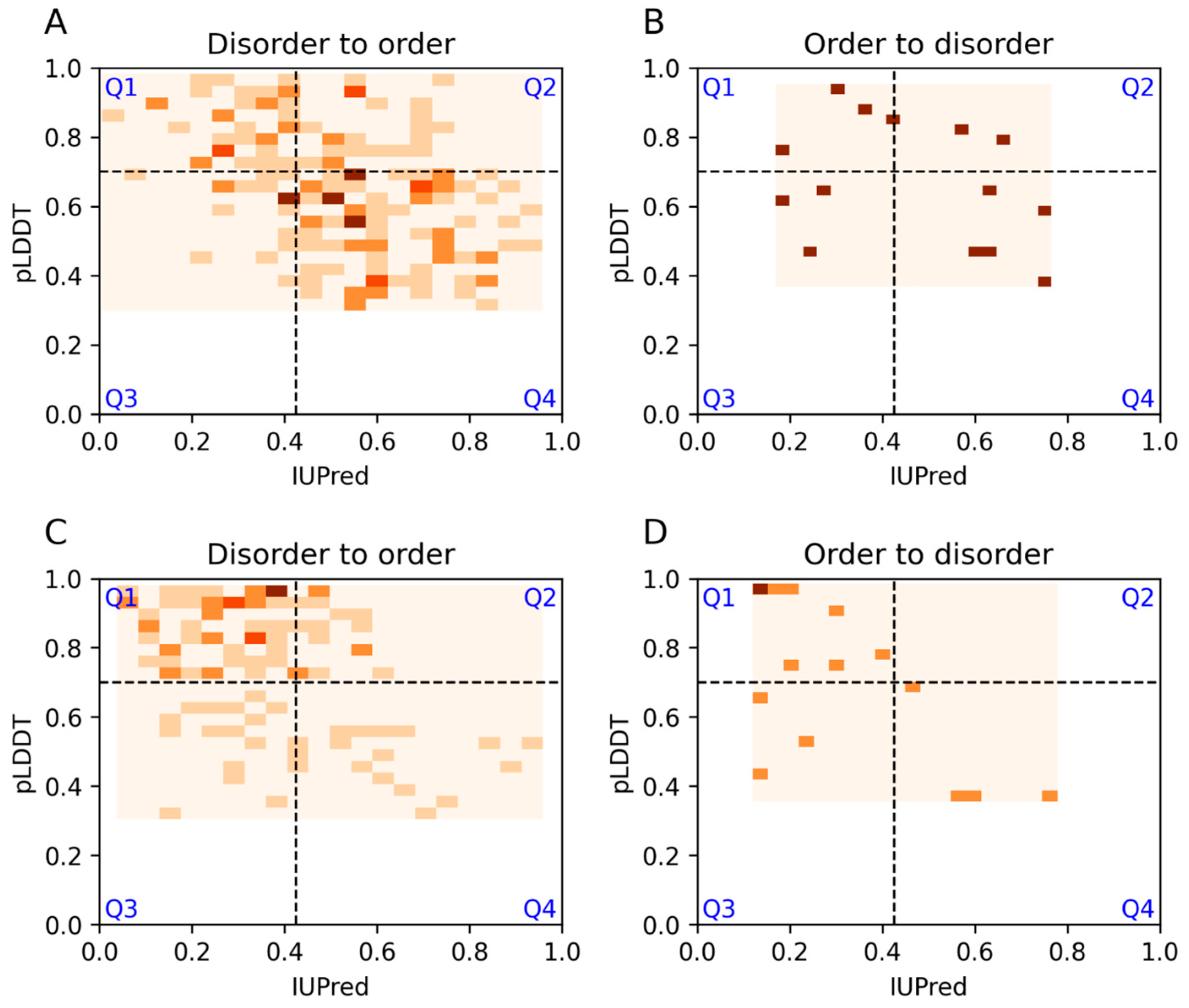
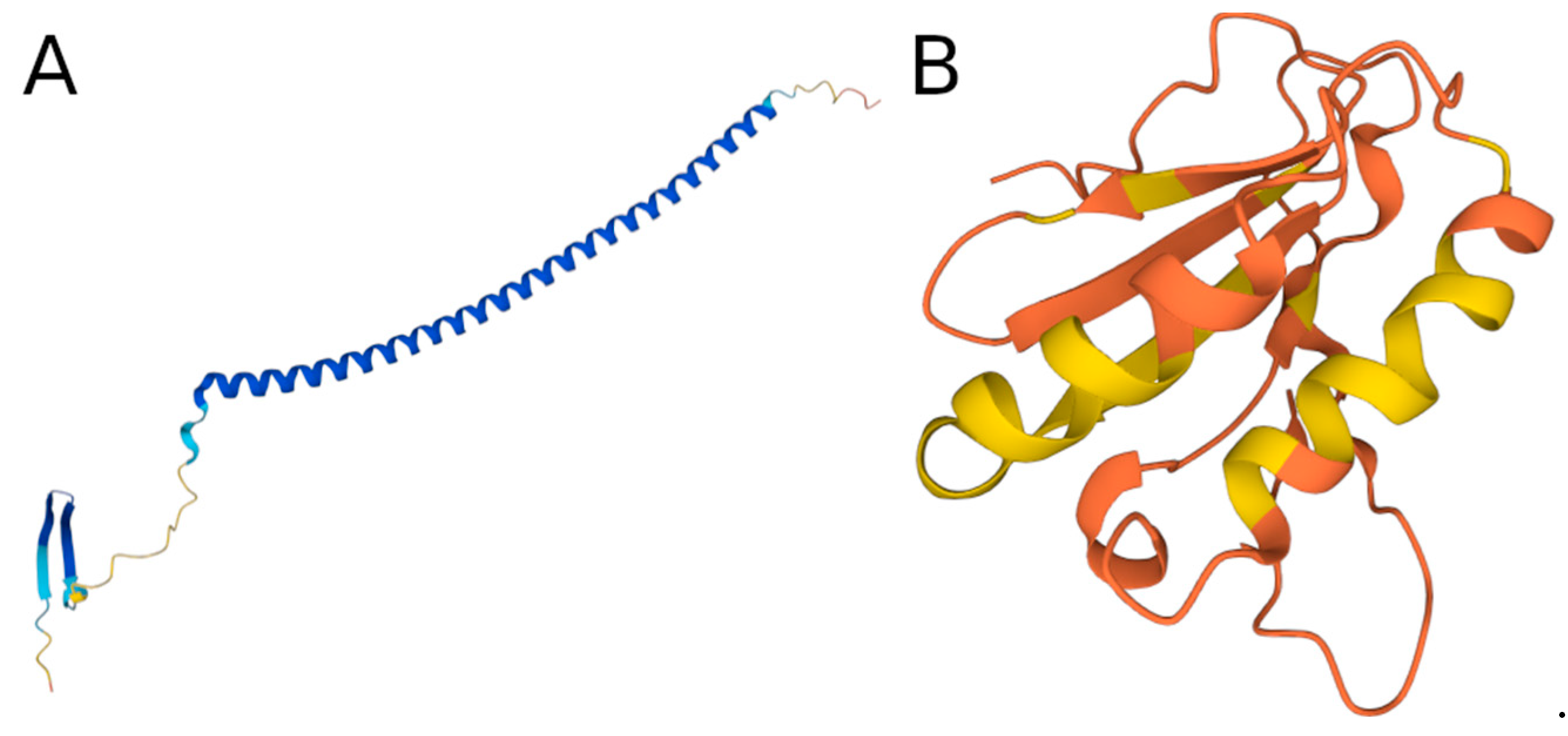
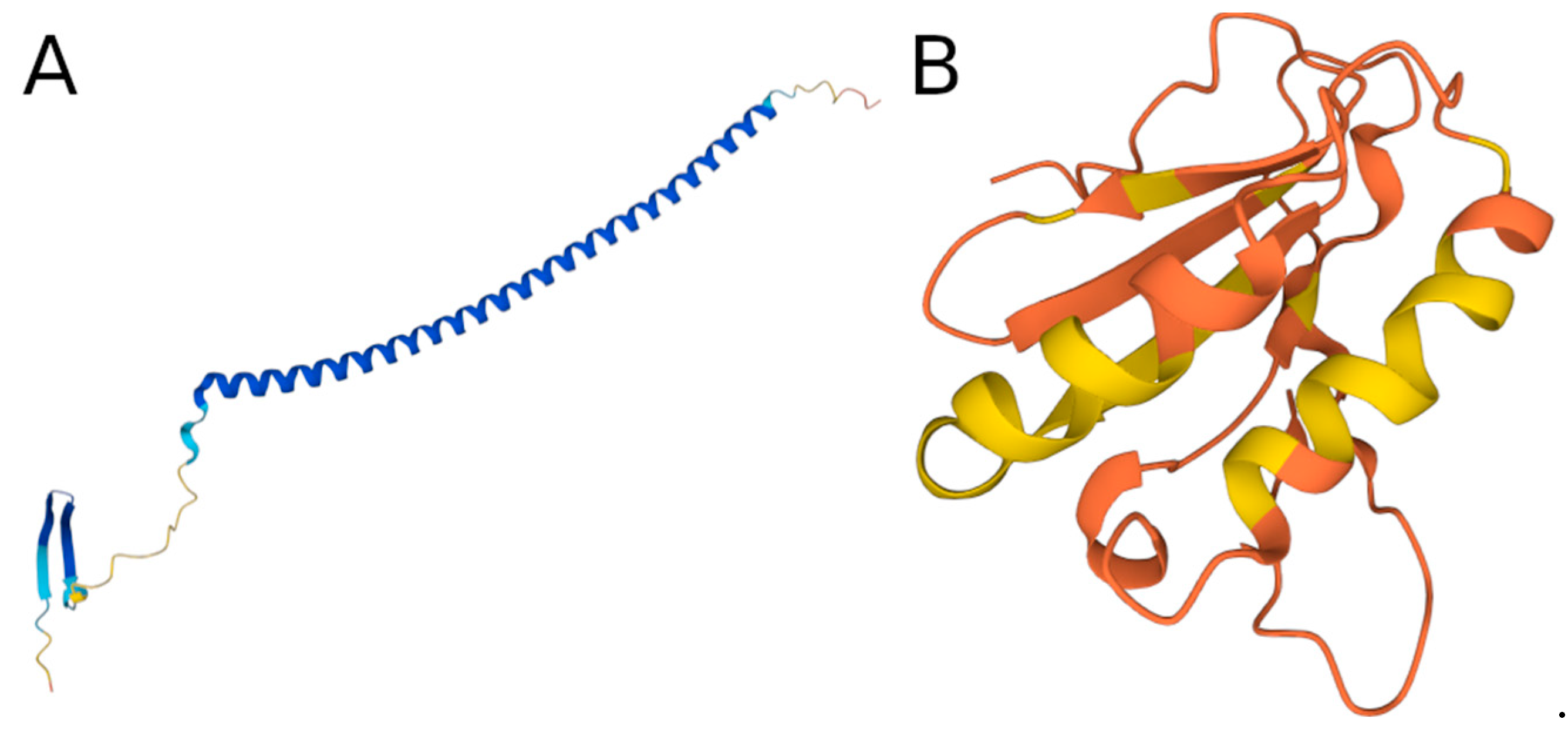
3.6. Examples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically Disordered Protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Natively Unfolded Proteins: A Point Where Biology Waits for Physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The Most Important Thing Is the Tail: Multitudinous Functionalities of Intrinsically Disordered Protein Termini. FEBS Lett. 2013, 587, 1891–1901. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic Disorder and Protein Function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the Structural Biology of Protein-Protein Interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef]
- Uversky, V.N. The Protein Disorder Cycle. Biophys. Rev. 2021, 13, 1155–1162. [Google Scholar] [CrossRef]
- Rubinstein, A.; Lyubchenko, Y.L.; Sherman, S. Dynamic Properties of pH-Dependent Structural Organization of the Amyloidogenic Beta-Protein (1-40). Prion 2009, 3, 31–43. [Google Scholar] [CrossRef]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. Intrinsically Disordered Proteins Play Diverse Roles in Cell Signaling. Cell Commun. Signal. 2022, 20, 20. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, X.; Wu, X.; Zhang, M. Formation of Biological Condensates via Phase Separation: Characteristics, Analytical Methods, and Physiological Implications. J. Biol. Chem. 2019, 294, 14823–14835. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Zardecki, C.; Dutta, S.; Goodsell, D.S.; Lowe, R.; Voigt, M.; Burley, S.K. PDB-101: Educational Resources Supporting Molecular Explorations through Biology and Medicine. Protein Sci. 2022, 31, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Hatos, A.; Palopoli, N.; Quaglia, F.; Salladini, E.; Van Roey, K.; Arthanari, H.; Dosztányi, Z.; Felli, I.C.; Fischer, P.D.; et al. Minimum Information Guidelines for Experiments Structurally Characterizing Intrinsically Disordered Protein Regions. Nat. Methods 2023, 20, 1291–1303. [Google Scholar] [CrossRef]
- Quaglia, F.; Mészáros, B.; Salladini, E.; Hatos, A.; Pancsa, R.; Chemes, L.B.; Pajkos, M.; Lazar, T.; Peña-Díaz, S.; Santos, J.; et al. DisProt in 2022: Improved Quality and Accessibility of Protein Intrinsic Disorder Annotation. Nucleic Acids Res. 2022, 50, D480–D487. [Google Scholar] [CrossRef]
- Galea, C.A.; Wang, Y.; Sivakolundu, S.G.; Kriwacki, R.W. Regulation of Cell Division by Intrinsically Unstructured Proteins: Intrinsic Flexibility, Modularity, and Signaling Conduits. Biochemistry 2008, 47, 7598–7609. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; CAID Predictors; DisProt Curators; Tosatto, S.C.E. Critical Assessment of Protein Intrinsic Disorder Prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Liu, B. A Comprehensive Review and Comparison of Existing Computational Methods for Intrinsically Disordered Protein and Region Prediction. Brief. Bioinform. 2019, 20, 330–346. [Google Scholar] [CrossRef]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein Disorder and the Evolution of Molecular Recognition: Theory, Predictions and Observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Zhao, B.; Kurgan, L. Deep Learning in Prediction of Intrinsic Disorder in Proteins. Comput. Struct. Biotechnol. J. 2022, 20, 1286–1294. [Google Scholar] [CrossRef]
- Erdős, G.; Pajkos, M.; Dosztányi, Z. IUPred3: Prediction of Protein Disorder Enhanced with Unambiguous Experimental Annotation and Visualization of Evolutionary Conservation. Nucleic Acids Res. 2021, 49, W297–W303. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Choy, W.-Y.; Karttunen, M. AlphaFold2: A Role for Disordered Protein/Region Prediction? Int. J. Mol. Sci. 2022, 23, 4591. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Monzon, A.M.; Tosatto, S.C.E. Intrinsic Protein Disorder and Conditional Folding in AlphaFoldDB. Protein Sci. 2022, 31, e4466. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Nevers, Y.; Jones, T.E.M.; Jyothi, D.; Yates, B.; Ferret, M.; Portell-Silva, L.; Codo, L.; Cosentino, S.; Marcet-Houben, M.; Vlasova, A.; et al. The Quest for Orthologs Orthology Benchmark Service in 2022. Nucleic Acids Res. 2022, 50, W623–W632. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Martínez-Pérez, E.; Pajkos, M.; Tosatto, S.C.E.; Gibson, T.J.; Dosztanyi, Z.; Marino-Buslje, C. Pipeline for Transferring Annotations between Proteins beyond Globular Domains. Protein Sci. 2023, 32, e4655. [Google Scholar] [CrossRef]
- Chakravarty, D.; Porter, L.L. AlphaFold2 Fails to Predict Protein Fold Switching. Protein Sci. 2022, 31, e4353. [Google Scholar] [CrossRef]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-Dependent Prediction of Protein Intrinsic Disorder. BMC Bioinformatics 2006, 7, 208. [Google Scholar] [CrossRef]
- Nadendla, S.; Jackson, R.; Munro, J.; Quaglia, F.; Mészáros, B.; Olley, D.; Hobbs, E.T.; Goralski, S.M.; Chibucos, M.; Mungall, C.J.; et al. ECO: The Evidence and Conclusion Ontology, an Update for 2022. Nucleic Acids Res. 2022, 50, D1515–D1521. [Google Scholar] [CrossRef] [PubMed]
- Monzon, A.M.; Necci, M.; Quaglia, F.; Walsh, I.; Zanotti, G.; Piovesan, D.; Tosatto, S.C.E. Experimentally Determined Long Intrinsically Disordered Protein Regions Are Now Abundant in the Protein Data Bank. Int. J. Mol. Sci. 2020, 21, 4496. [Google Scholar] [CrossRef]
- Sun, Q.; Zhu, X.; Qi, J.; An, W.; Lan, P.; Tan, D.; Chen, R.; Wang, B.; Zheng, S.; Zhang, C.; et al. Molecular Architecture of the 90S Small Subunit Pre-Ribosome. Elife 2017, 6, e22086. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, M.; Hubbard, T.J.; Proctor, M.; Freund, S.M.; Murzin, A.G. The Solution Structure of the S1 RNA Binding Domain: A Member of an Ancient Nucleic Acid-Binding Fold. Cell 1997, 88, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Nanga, R.P.R.; Brender, J.R.; Vivekanandan, S.; Popovych, N.; Ramamoorthy, A. NMR Structure in a Membrane Environment Reveals Putative Amyloidogenic Regions of the SEVI Precursor Peptide PAP(248–286). J. Am. Chem. Soc. 2009, 131, 17972–17979. [Google Scholar] [CrossRef]
- Ortlund, E.; LaCount, M.W.; Lebioda, L. Crystal Structures of Human Prostatic Acid Phosphatase in Complex with a Phosphate Ion and Alpha-Benzylaminobenzylphosphonic Acid Update the Mechanistic Picture and Offer New Insights into Inhibitor Design. Biochemistry 2003, 42, 383–389. [Google Scholar] [CrossRef]
- Steinmetz, M.O.; Jahnke, W.; Towbin, H.; García-Echeverría, C.; Voshol, H.; Müller, D.; van Oostrum, J. Phosphorylation Disrupts the Central Helix in Op18/stathmin and Suppresses Binding to Tubulin. EMBO Rep. 2001, 2, 505–510. [Google Scholar] [CrossRef]
- Vucetic, S.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Flavors of Protein Disorder. Proteins 2003, 52, 573–584. [Google Scholar] [CrossRef]
- Del Conte, A.; Bouhraoua, A.; Mehdiabadi, M.; Clementel, D.; Monzon, A.M.; CAID predictors; Tosatto, S.C.E.; Piovesan, D. CAID Prediction Portal: A Comprehensive Service for Predicting Intrinsic Disorder and Binding Regions in Proteins. Nucleic Acids Res. 2023, 51, W62–W69. [Google Scholar] [CrossRef]
- Basu, S.; Kihara, D.; Kurgan, L. Computational Prediction of Disordered Binding Regions. Comput. Struct. Biotechnol. J. 2023, 21, 1487–1497. [Google Scholar] [CrossRef]
- Meng, F.; Kurgan, L. DFLpred: High-Throughput Prediction of Disordered Flexible Linker Regions in Protein Sequences. Bioinformatics 2016, 32, i341–i350. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdos, G.; Dosztányi, Z. IUPred2A: Context-Dependent Prediction of Protein Disorder as a Function of Redox State and Protein Binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Long | Short | |||
|---|---|---|---|---|
| Residues | Regions | Residues | Regions | |
| Q1 | 17,444 (15%) | 139 (15%) | 5240 (28%) | 290 (29%) |
| Q2 | 13,290 (12%) | 85 (9%) | 2259 (13%) | 106 (10%) |
| Q3 | 13,043 (12%) | 119 (12%) | 4444 (24%) | 264 (26%) |
| Q4 | 68,401 (61%) | 603 (64%) | 6557 (35%) | 361 (35%) |
| Sum | 112,178 | 946 | 18,500 | 1021 |
| Vertebrata | Eumetazoa | Unicellular | |
|---|---|---|---|
| Q1 | 1150 (13%) | 578 (22%) | 83 (40%) |
| Q2 | 9925 (12%) | 480 (19%) | 41 (20%) |
| Q3 | 10,705 (13%) | 144 (6%) | 18 (9%) |
| Q4 | 52,222 (62%) | 1378 (53%) | 63 (31%) |
| Sum | 74,002 | 2580 | 205 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajkos, M.; Erdős, G.; Dosztányi, Z. The Origin of Discrepancies between Predictions and Annotations in Intrinsically Disordered Proteins. Biomolecules 2023, 13, 1442. https://doi.org/10.3390/biom13101442
Pajkos M, Erdős G, Dosztányi Z. The Origin of Discrepancies between Predictions and Annotations in Intrinsically Disordered Proteins. Biomolecules. 2023; 13(10):1442. https://doi.org/10.3390/biom13101442
Chicago/Turabian StylePajkos, Mátyás, Gábor Erdős, and Zsuzsanna Dosztányi. 2023. "The Origin of Discrepancies between Predictions and Annotations in Intrinsically Disordered Proteins" Biomolecules 13, no. 10: 1442. https://doi.org/10.3390/biom13101442
APA StylePajkos, M., Erdős, G., & Dosztányi, Z. (2023). The Origin of Discrepancies between Predictions and Annotations in Intrinsically Disordered Proteins. Biomolecules, 13(10), 1442. https://doi.org/10.3390/biom13101442