
ATP12A Proton Pump as an Emerging Therapeutic Target in Cystic Fibrosis and Other Respiratory Diseases

 , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ATP12A Proton Pump Structure and Assembly
3. ATP12A Drives Airway Acidification in CF: Lessons from Animal Models
4. ATP12A Expression, Function, and Modulation in CF and Other Respiratory Diseases
5. ATP12A as a Modifier of Meconium Ileus in CF
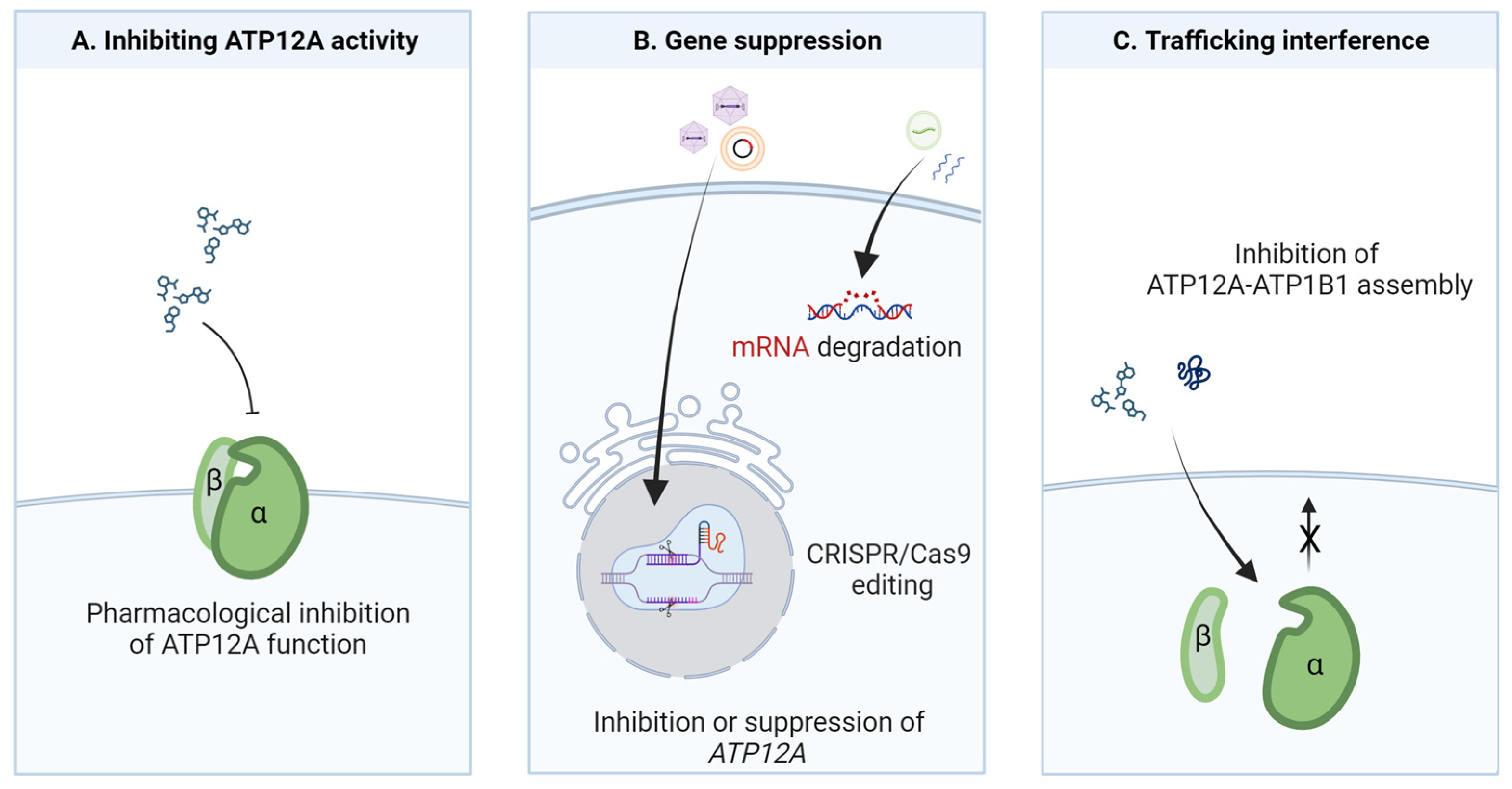
6. Potential Strategies for ATP12A Targeting
Author Contributions
Funding
Conflicts of Interest
References
- Sverdlov, V.E.; Kostina, M.B.; Modyanov, N.N. Genomic Organization of the Human ATP1AL1 Gene Encoding a Ouabain-Sensitive H,K-ATPase. Genomics 1996, 32, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Zajac, M.; Dreano, E.; Edwards, A.; Planelles, G.; Sermet-Gaudelus, I. Airway Surface Liquid PH Regulation in Airway Epithelium Current Understandings and Gaps in Knowledge. Int. J. Mol. Sci. 2021, 22, 3384. [Google Scholar] [CrossRef] [PubMed]
- Jaisser, F.; Beggah, A.T. The Nongastric H+-K+-ATPases: Molecular and Functional Properties. Am. J. Physiol. 1999, 276, F812–F824. [Google Scholar] [CrossRef]
- Gumz, M.L.; Lynch, I.J.; Greenlee, M.M.; Cain, B.D.; Wingo, C.S. The Renal H+-K+-ATPases: Physiology, Regulation, and Structure. Am. J. Physiol. Ren. Physiol. 2010, 298, F12–F21. [Google Scholar] [CrossRef] [PubMed]
- Pestov, N.B.; Korneenko, T.V.; Adams, G.; Tillekeratne, M.; Shakhparonov, M.I.; Modyanov, N.N. Nongastric H-K-ATPase in Rodent Prostate: Lobe-Specific Expression and Apical Localization. Am. J. Physiol. Cell Physiol. 2002, 282, C907–C916. [Google Scholar] [CrossRef] [PubMed]
- Pestov, N.B.; Korneenko, T.V.; Shakhparonov, M.I.; Shull, G.E.; Modyanov, N.N. Loss of Acidification of Anterior Prostate Fluids in ATP12A-Null Mutant Mice Indicates That Nongastric H-K-ATPase Functions as Proton Pump in Vivo. Am. J. Physiol. Cell Physiol. 2006, 291, C366–C374. [Google Scholar] [CrossRef]
- Favia, M.; Gerbino, A.; Notario, E.; Tragni, V.; Sgobba, M.N.; Dell’Aquila, M.E.; Pierri, C.L.; Guerra, L.; Ciani, E. The Non-Gastric H+/K+ ATPase (ATP12A) Is Expressed in Mammalian Spermatozoa. Int. J. Mol. Sci. 2022, 23, 1048. [Google Scholar] [CrossRef]
- Gorrieri, G.; Scudieri, P.; Caci, E.; Schiavon, M.; Tomati, V.; Sirci, F.; Napolitano, F.; Carrella, D.; Gianotti, A.; Musante, I.; et al. Goblet Cell Hyperplasia Requires High Bicarbonate Transport to Support Mucin Release. Sci. Rep. 2016, 6, 36016. [Google Scholar] [CrossRef]
- Scudieri, P.; Musante, I.; Caci, E.; Venturini, A.; Morelli, P.; Walter, C.; Tosi, D.; Palleschi, A.; Martin-Vasallo, P.; Sermet-Gaudelus, I.; et al. Increased Expression of ATP12A Proton Pump in Cystic Fibrosis Airways. JCI Insight 2018, 3, e123616. [Google Scholar] [CrossRef]
- Coakley, R.D.; Grubb, B.R.; Paradiso, A.M.; Gatzy, J.T.; Johnson, L.G.; Kreda, S.M.; O’Neal, W.K.; Boucher, R.C. Abnormal Surface Liquid pH Regulation by Cultured Cystic Fibrosis Bronchial Epithelium. Proc. Natl. Acad. Sci. USA 2003, 100, 16083–16088. [Google Scholar] [CrossRef]
- Abou Alaiwa, M.H.; Reznikov, L.R.; Gansemer, N.D.; Sheets, K.A.; Horswill, A.R.; Stoltz, D.A.; Zabner, J.; Welsh, M.J. PH Modulates the Activity and Synergism of the Airway Surface Liquid Antimicrobials β-Defensin-3 and LL-37. Proc. Natl. Acad. Sci. USA 2014, 111, 18703–18708. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Villacreses, R.; Thornell, I.M.; Noriega, J.; Mather, S.; Brommel, C.M.; Lu, L.; Zabner, A.; Ehler, A.; Meyerholz, D.K.; et al. V-Type ATPase Mediates Airway Surface Liquid Acidification in Pig Small Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2021, 65, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Young, V.C.; Nakanishi, H.; Meyer, D.J.; Nishizawa, T.; Oshima, A.; Artigas, P.; Abe, K. Structure and Function of H+/K+ Pump Mutants Reveal Na+/K+ Pump Mechanisms. Nat. Commun. 2022, 13, 5270. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.V.; Caplan, M.J. ATP1AL1, a Member of the Non-Gastric H,K-ATPase Family, Functions as a Sodium Pump. J. Biol. Chem. 1998, 273, 27772–27778. [Google Scholar] [CrossRef]
- Shah, V.S.; Meyerholz, D.K.; Tang, X.X.; Reznikov, L.; Abou Alaiwa, M.; Ernst, S.E.; Karp, P.H.; Wohlford-Lenane, C.L.; Heilmann, K.P.; Leidinger, M.R.; et al. Airway Acidification Initiates Host Defense Abnormalities in Cystic Fibrosis Mice. Science 2016, 351, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic Fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Grubb, B.R.; Boucher, R.C. Pathophysiology of Gene-Targeted Mouse Models for Cystic Fibrosis. Physiol. Rev. 1999, 79, S193–S214. [Google Scholar] [CrossRef] [PubMed]
- Guilbault, C.; Saeed, Z.; Downey, G.P.; Radzioch, D. Cystic Fibrosis Mouse Models. Am. J. Respir. Cell Mol. Biol. 2007, 36, 1–7. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced Airway Surface pH Impairs Bacterial Killing in the Porcine Cystic Fibrosis Lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired Mucus Detachment Disrupts Mucociliary Transport in a Piglet Model of Cystic Fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef]
- Birket, S.E.; Chu, K.K.; Liu, L.; Houser, G.H.; Diephuis, B.J.; Wilsterman, E.J.; Dierksen, G.; Mazur, M.; Shastry, S.; Li, Y.; et al. A Functional Anatomic Defect of the Cystic Fibrosis Airway. Am. J. Respir. Crit. Care Med. 2014, 190, 421–432. [Google Scholar] [CrossRef]
- Randell, S.H.; Boucher, R.C. Effective Mucus Clearance Is Essential for Respiratory Health. Am. J. Respir. Cell Mol. Biol. 2006, 35, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.T.; Coburn, S.L.; Leech, J.A.; Heidrich, E.M.; Kleyman, T.R.; Wenzel, S.E.; Pilewski, J.M.; Corcoran, T.E.; Myerburg, M.M. ATP12A Promotes Mucus Dysfunction during Type 2 Airway Inflammation. Sci. Rep. 2018, 8, 2109. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Ocampo, C.J.; Stevens, W.W.; Price, C.P.E.; Thompson, C.F.; Homma, T.; Huang, J.H.; Norton, J.E.; Suh, L.A.; Pothoven, K.L.; et al. Proton Pump Inhibitors Decrease Eotaxin-3/CCL26 Expression in Patients with Chronic Rhinosinusitis with Nasal Polyps: Possible Role of the Nongastric H,K-ATPase. J. Allergy Clin. Immunol. 2017, 139, 130–141.e11. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Puvvadi, R.; Borisov, S.M.; Shaw, N.C.; Klimant, I.; Berry, L.J.; Montgomery, S.T.; Nguyen, T.; Kreda, S.M.; Kicic, A.; et al. Airway Surface Liquid pH Is Not Acidic in Children with Cystic Fibrosis. Nat. Commun. 2017, 8, 1409. [Google Scholar] [CrossRef]
- Massey, M.K.; Reiterman, M.J.; Mourad, J.; Luckie, D.B. Is CFTR an Exchanger?: Regulation of HCO3− Transport and Extracellular pH by CFTR. Biochem. Biophys. Rep. 2020, 25, 100863. [Google Scholar] [CrossRef]
- Garland, A.L.; Walton, W.G.; Coakley, R.D.; Tan, C.D.; Gilmore, R.C.; Hobbs, C.A.; Tripathy, A.; Clunes, L.A.; Bencharit, S.; Stutts, M.J.; et al. Molecular Basis for pH-Dependent Mucosal Dehydration in Cystic Fibrosis Airways. Proc. Natl. Acad. Sci. USA 2013, 110, 15973–15978. [Google Scholar] [CrossRef]
- Shah, V.S.; Ernst, S.; Tang, X.X.; Karp, P.H.; Parker, C.P.; Ostedgaard, L.S.; Welsh, M.J. Relationships among CFTR Expression, HCO3− Secretion, and Host Defense May Inform Gene- and Cell-Based Cystic Fibrosis Therapies. Proc. Natl. Acad. Sci. USA 2016, 113, 5382–5387. [Google Scholar] [CrossRef]
- Guidone, D.; Buccirossi, M.; Scudieri, P.; Genovese, M.; Sarnataro, S.; De Cegli, R.; Cresta, F.; Terlizzi, V.; Planelles, G.; Crambert, G.; et al. Airway Surface Hyperviscosity and Defective Mucociliary Transport by IL-17/TNF-α Are Corrected by Beta-Adrenergic Stimulus. JCI Insight 2022, 7, e164944. [Google Scholar] [CrossRef]
- Delpiano, L.; Thomas, J.J.; Yates, A.R.; Rice, S.J.; Gray, M.A.; Saint-Criq, V. Esomeprazole Increases Airway Surface Liquid pH in Primary Cystic Fibrosis Epithelial Cells. Front. Pharmacol. 2018, 9, 1462. [Google Scholar] [CrossRef]
- Abdelgied, M.; Uhl, K.; Chen, O.G.; Schultz, C.; Tripp, K.; Peraino, A.M.; Paithankar, S.; Chen, B.; Kakazu, M.T.; Bahena, A.C.; et al. Targeting ATP12A, a Non-Gastric Proton Pump Alpha Subunit, for Idiopathic Pulmonary Fibrosis Treatment. Am. J. Respir. Cell Mol. Biol. 2023, 68, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Zhang, Q.; Zhao, W.; Ding, W.; Liu, J.; Zhao, Y. A 12-month Follow-up Study on the Preventive Effect of Oral Lansoprazole on Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Int. J. Exp. Pathol. 2016, 97, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in Epithelial Physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef]
- Weiler, C.A.; Drumm, M.L. Genetic Influences on Cystic Fibrosis Lung Disease Severity. Front. Pharmacol. 2013, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Collaco, J.M.; Cutting, G.R. Update on Gene Modifiers in Cystic Fibrosis. Curr. Opin. Pulm. Med. 2008, 14, 559–566. [Google Scholar] [CrossRef]
- Marson, F.A.L. Disease-Modifying Genetic Factors in Cystic Fibrosis. Curr. Opin. Pulm. Med. 2018, 24, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wang, F.; Xiao, B.; Panjwani, N.; Lin, F.; Keenan, K.; Avolio, J.; Esmaeili, M.; Zhang, L.; He, G.; et al. Genetic Association and Transcriptome Integration Identify Contributing Genes and Tissues at Cystic Fibrosis Modifier Loci. PLoS Genet. 2019, 15, e1008007. [Google Scholar] [CrossRef]
- Sathe, M.; Houwen, R. Meconium Ileus in Cystic Fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2017, 16, S32–S39. [Google Scholar] [CrossRef] [PubMed]
- Quinton, P.M. Cystic Fibrosis: Impaired Bicarbonate Secretion and Mucoviscidosis. Lancet Lond. Engl. 2008, 372, 415–417. [Google Scholar] [CrossRef]
- Scheele, G.A.; Fukuoka, S.I.; Kern, H.F.; Freedman, S.D. Pancreatic Dysfunction in Cystic Fibrosis Occurs as a Result of Impairments in Luminal pH, Apical Trafficking of Zymogen Granule Membranes, and Solubilization of Secretory Enzymes. Pancreas 1996, 12, 1–9. [Google Scholar] [CrossRef]
- Singh, V.K.; Schwarzenberg, S.J. Pancreatic Insufficiency in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16, S70–S78. [Google Scholar] [CrossRef]
- Wang, J.; Barbuskaite, D.; Tozzi, M.; Giannuzzo, A.; Sørensen, C.E.; Novak, I. Proton Pump Inhibitors Inhibit Pancreatic Secretion: Role of Gastric and Non-Gastric H+/K+-ATPases. PLoS ONE 2015, 10, e0126432. [Google Scholar] [CrossRef] [PubMed]
- Simonin, J.; Bille, E.; Crambert, G.; Noel, S.; Dreano, E.; Edwards, A.; Hatton, A.; Pranke, I.; Villeret, B.; Cottart, C.-H.; et al. Airway Surface Liquid Acidification Initiates Host Defense Abnormalities in Cystic Fibrosis. Sci. Rep. 2019, 9, 6516. [Google Scholar] [CrossRef]
- Grishin, A.V.; Bevensee, M.O.; Modyanov, N.N.; Rajendran, V.; Boron, W.F.; Caplan, M.J. Functional expression of the cDNA encoded by the human ATP1AL1 gene. Am. J. Physiol. 1996, 271, F539–F551. [Google Scholar] [CrossRef]
- Oshima, T.; Miwa, H. Potent Potassium-Competitive Acid Blockers: A New Era for the Treatment of Acid-Related Diseases. J. Neurogastroenterol. Motil. 2018, 24, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Robinson, J.M.; Wood, R.E.; Pue, C.A.; Mentz, W.M.; Wager, G.C.; Gatzy, J.T.; Boucher, R.C. Ion composition of airway surface liquid of patients with cystic fibrosis as compared with normal and disease-control subjects. J. Clin. Investig. 1997, 100, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Namkung, W.; Song, Y.; Mills, A.D.; Padmawar, P.; Finkbeiner, W.E.; Verkman, A.S. In situ measurement of airway surface liquid [K+] using a ratioable K+-sensitive fluorescent dye. J. Biol. Chem. 2009, 284, 15916–15926. [Google Scholar] [CrossRef]
- Sui, H.; Xu, X.; Su, Y.; Gong, Z.; Yao, M.; Liu, X.; Zhang, T.; Jiang, Z.; Bai, T.; Wang, J.; et al. Gene Therapy for Cystic Fibrosis: Challenges and Prospects. Front. Pharmacol. 2022, 13, 1015926. [Google Scholar] [CrossRef]
- Perricone, M.A.; Morris, J.E.; Pavelka, K.; Plog, M.S.; O’Sullivan, B.P.; Joseph, P.M.; Dorkin, H.; Lapey, A.; Balfour, R.; Meeker, D.P.; et al. Aerosol and Lobar Administration of a Recombinant Adenovirus to Individuals with Cystic Fibrosis. II. Transfection Efficiency in Airway Epithelium. Hum. Gene Ther. 2001, 12, 1383–1394. [Google Scholar] [CrossRef]
- Joseph, P.M.; O’Sullivan, B.P.; Lapey, A.; Dorkin, H.; Oren, J.; Balfour, R.; Perricone, M.A.; Rosenberg, M.; Wadsworth, S.C.; Smith, A.E.; et al. Aerosol and Lobar Administration of a Recombinant Adenovirus to Individuals with Cystic Fibrosis. I. Methods, Safety, and Clinical Implications. Hum. Gene Ther. 2001, 12, 1369–1382. [Google Scholar] [CrossRef]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.M.; Johnstone, E.C. The Clinical Landscape for AAV Gene Therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Kochergin-Nikitsky, K.; Belova, L.; Lavrov, A.; Smirnikhina, S. Tissue and Cell-Type-Specific Transduction Using RAAV Vectors in Lung Diseases. J. Mol. Med. 2021, 99, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- Loring, H.S.; ElMallah, M.K.; Flotte, T.R. Development of RAAV2-CFTR: History of the First RAAV Vector Product to Be Used in Humans. Hum. Gene Ther. Methods. 2016, 27, 49–58. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.L.; de Oliveira, G.P.; Kim, N.; Cruz, F.F.; Kitoko, J.Z.; Blanco, N.G.; Martini, S.V.; Hanes, J.; Rocco, P.R.M.; Suk, J.S.; et al. Nanoparticle-Based Thymulin Gene Therapy Therapeutically Reverses Key Pathology of Experimental Allergic Asthma. Sci. Adv. 2020, 6, eaay7973. [Google Scholar] [CrossRef]
- Cecchin, R.; Troyer, Z.; Witwer, K.; Morris, K.V. Extracellular Vesicles: The next Generation in Gene Therapy Delivery. Mol. Ther. 2023, 31, 1225–1230. [Google Scholar] [CrossRef]
- Bardoliwala, D.; Patel, V.; Javia, A.; Ghosh, S.; Patel, A.; Misra, A. Nanocarriers in Effective Pulmonary Delivery of SiRNA: Current Approaches and Challenges. Ther. Deliv. 2019, 10, 311–332. [Google Scholar] [CrossRef]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In Silico Methods and Tools for Drug Discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dębczyński, M.; Gorrieri, G.; Mojsak, D.; Guida, F.; Zara, F.; Scudieri, P. ATP12A Proton Pump as an Emerging Therapeutic Target in Cystic Fibrosis and Other Respiratory Diseases. Biomolecules 2023, 13, 1455. https://doi.org/10.3390/biom13101455
Dębczyński M, Gorrieri G, Mojsak D, Guida F, Zara F, Scudieri P. ATP12A Proton Pump as an Emerging Therapeutic Target in Cystic Fibrosis and Other Respiratory Diseases. Biomolecules. 2023; 13(10):1455. https://doi.org/10.3390/biom13101455
Chicago/Turabian StyleDębczyński, Michał, Giulia Gorrieri, Damian Mojsak, Floriana Guida, Federico Zara, and Paolo Scudieri. 2023. "ATP12A Proton Pump as an Emerging Therapeutic Target in Cystic Fibrosis and Other Respiratory Diseases" Biomolecules 13, no. 10: 1455. https://doi.org/10.3390/biom13101455