

Antioxidant, LC-MS Analysis, and Cholinesterase Inhibitory Potentials of Phoenix dactylifera Cultivar Khudari: An In Vitro Enzyme Kinetics and In Silico Study
 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection and Preparation of Dates Extract
2.2. Total Phenolic Content (TPC) and Phytochemical Analysis
2.3. Antioxidant Property Assay
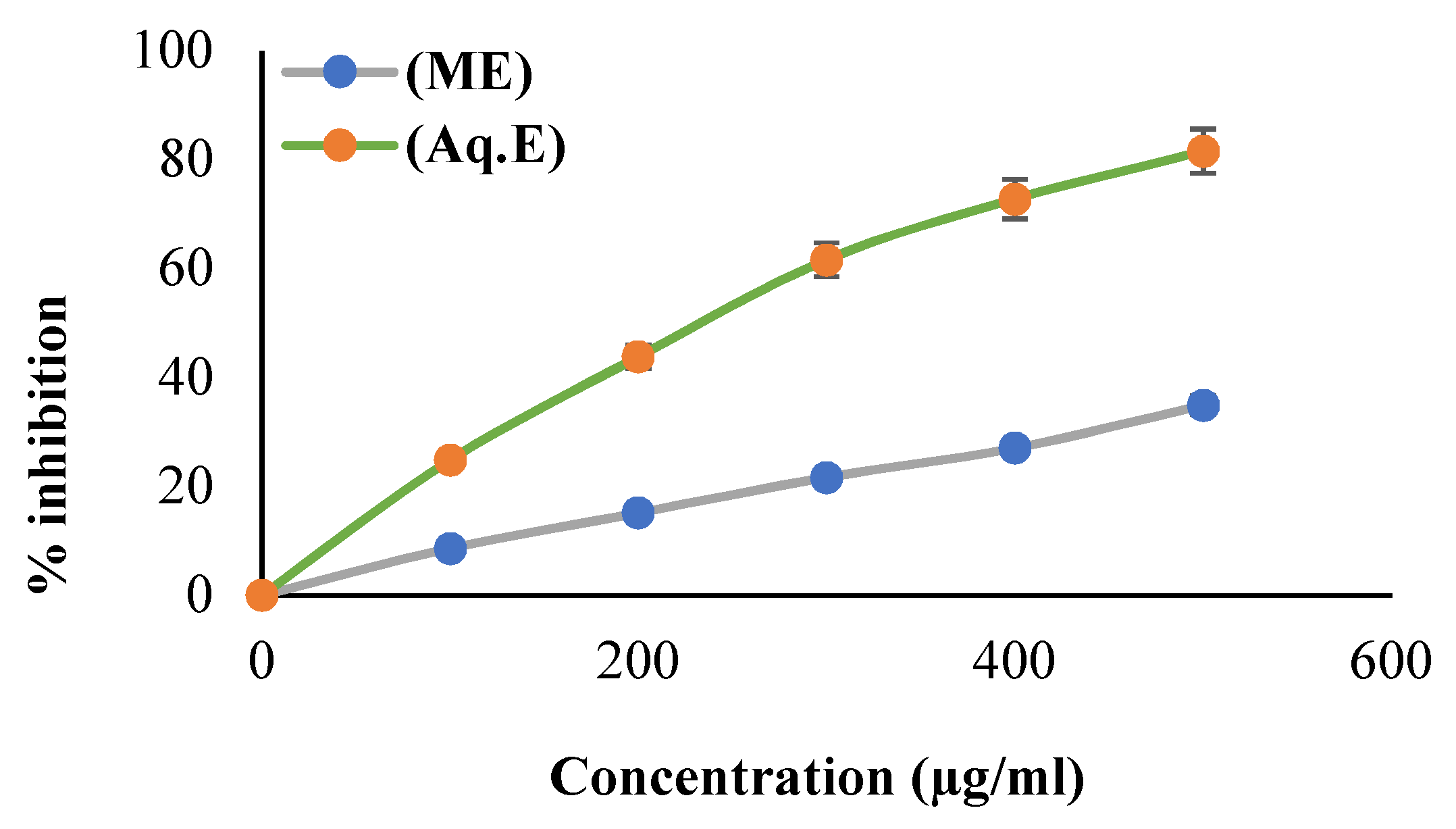
2.3.1. Scavenging Activity of DPPH Radical
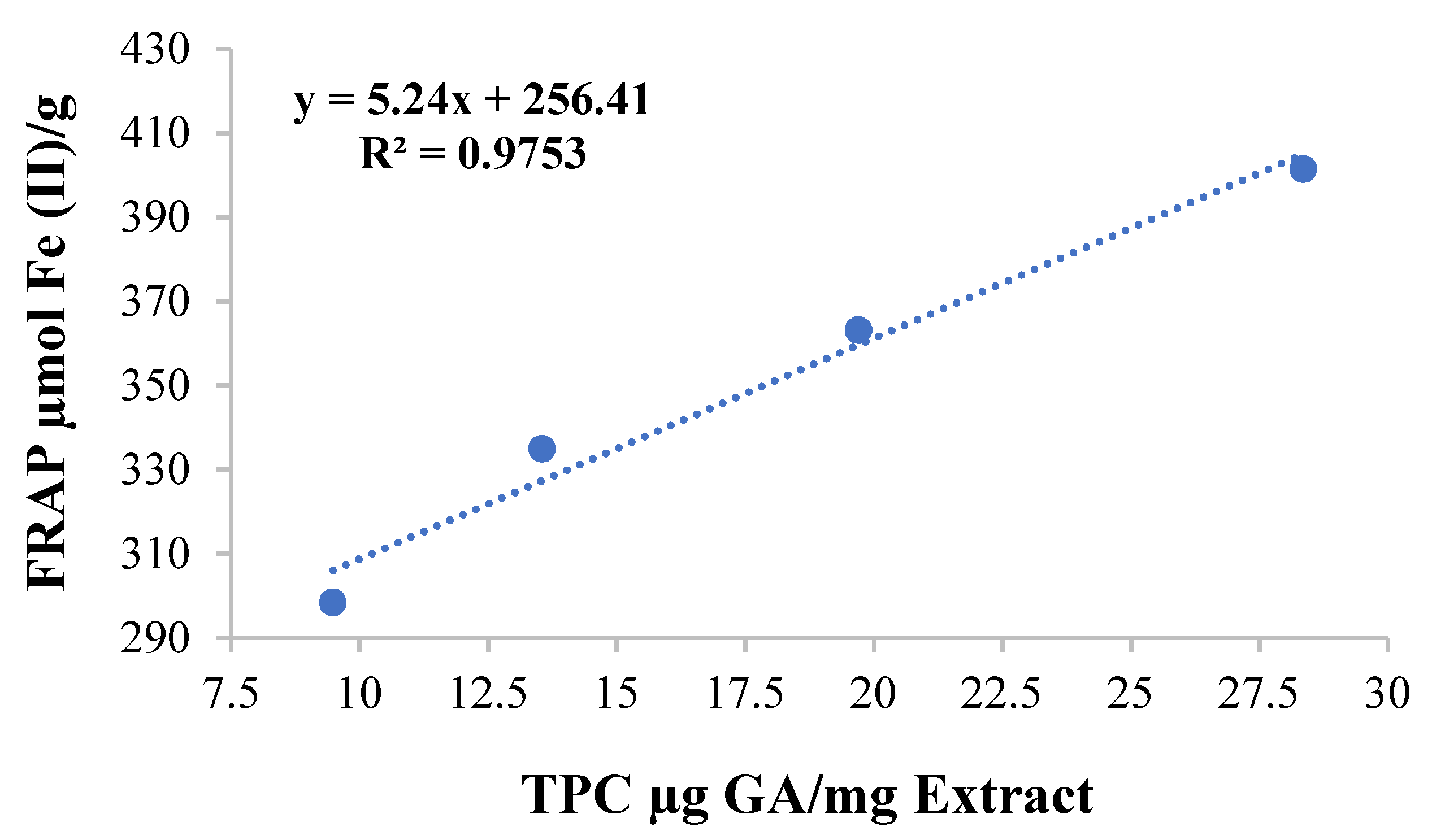
2.3.2. Analysis of Ferric-Reducing Antioxidant Power
2.4. Cell-Free Enzyme Inhibition Assay
2.4.1. Acetylcholinesterase Inhibition Assay
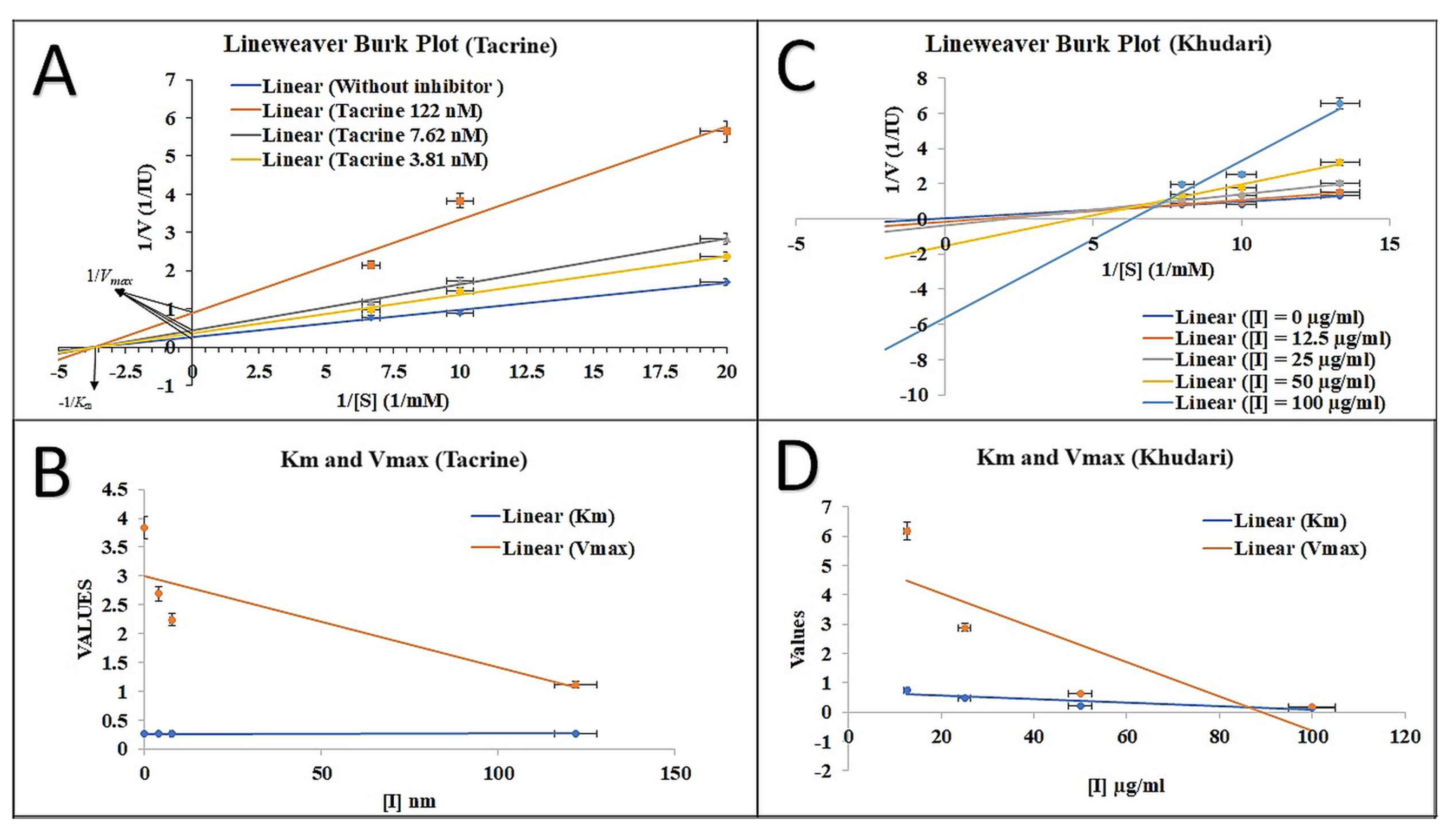
2.4.2. Spectrometric Study of the Enzyme Inhibition Kinetics
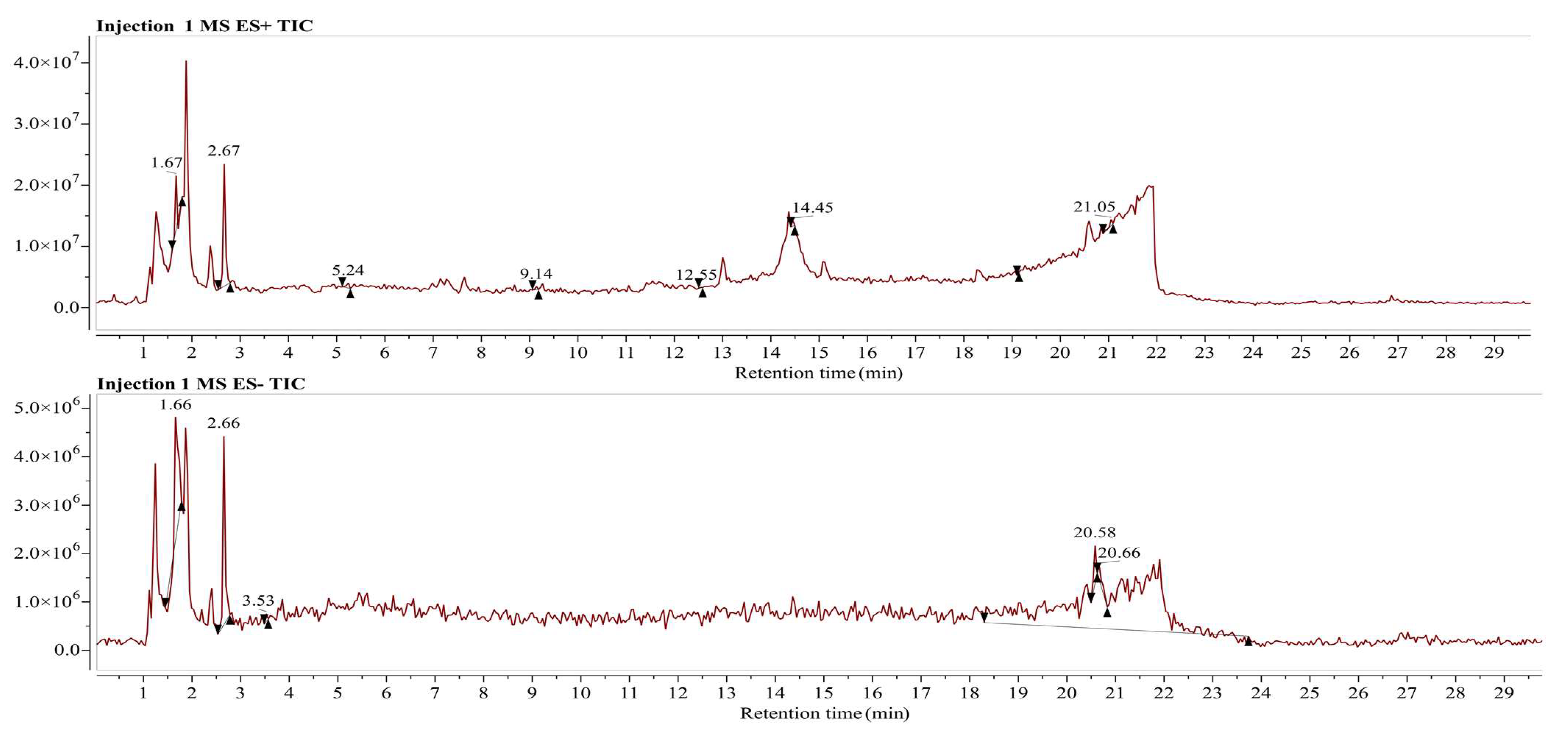
2.4.3. LC-MS (Liquid Chromatography–Mass Spectrometry) Analysis
2.5. Computational Study
2.5.1. Platform of In Silico Computational Study
2.5.2. Retrieval and Preparation of 3D Structure of Ligands
2.5.3. Retrieval and Preparation of Target Protein
2.5.4. Molecular Docking Studies of Selected Ligands against AChE
2.5.5. Pharmacokinetics and Drug-Likeness Profiling of Ligands
2.5.6. Predicted Toxicity Assessment of the Selected Ligands
2.5.7. Molecular Dynamics Simulation Analysis
2.5.8. Calculations of Free Energy (Prime-MM/GBSA)
3. Results and Discussion
3.1. Phytochemical Estimation of Khudari Fruit Pulp
3.2. Scavenging Property of DPPH Radical
3.3. Ferric-Ions-Reducing Potential and Total Phenolic Content
3.4. AChE Inhibition Property and Mode of Inhibition
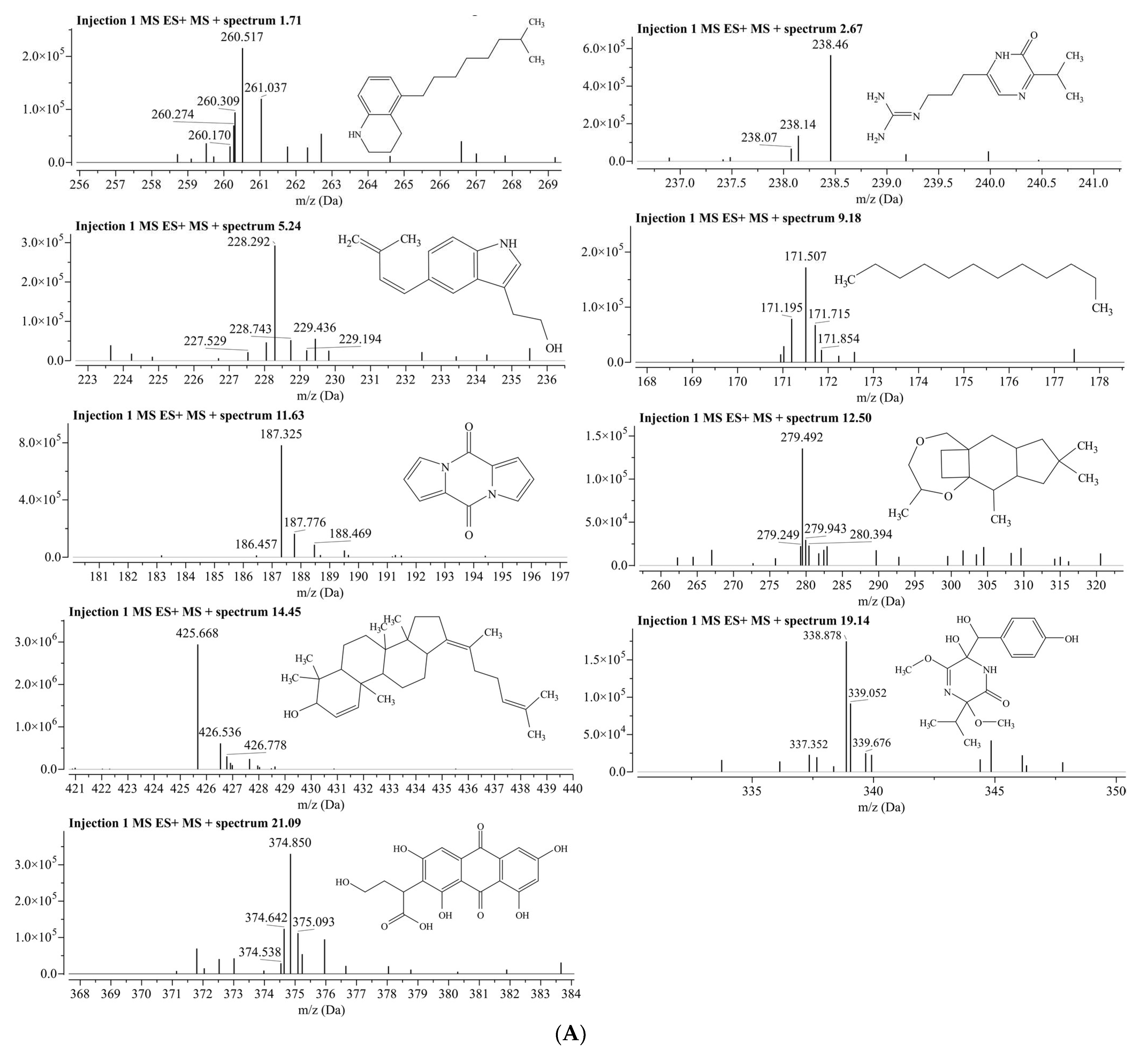
3.5. LC/MS Analysis for Identification of Phytochemical Compounds in Aqueous Fraction of Phoenix dactylifera (Khudari Cultivar)
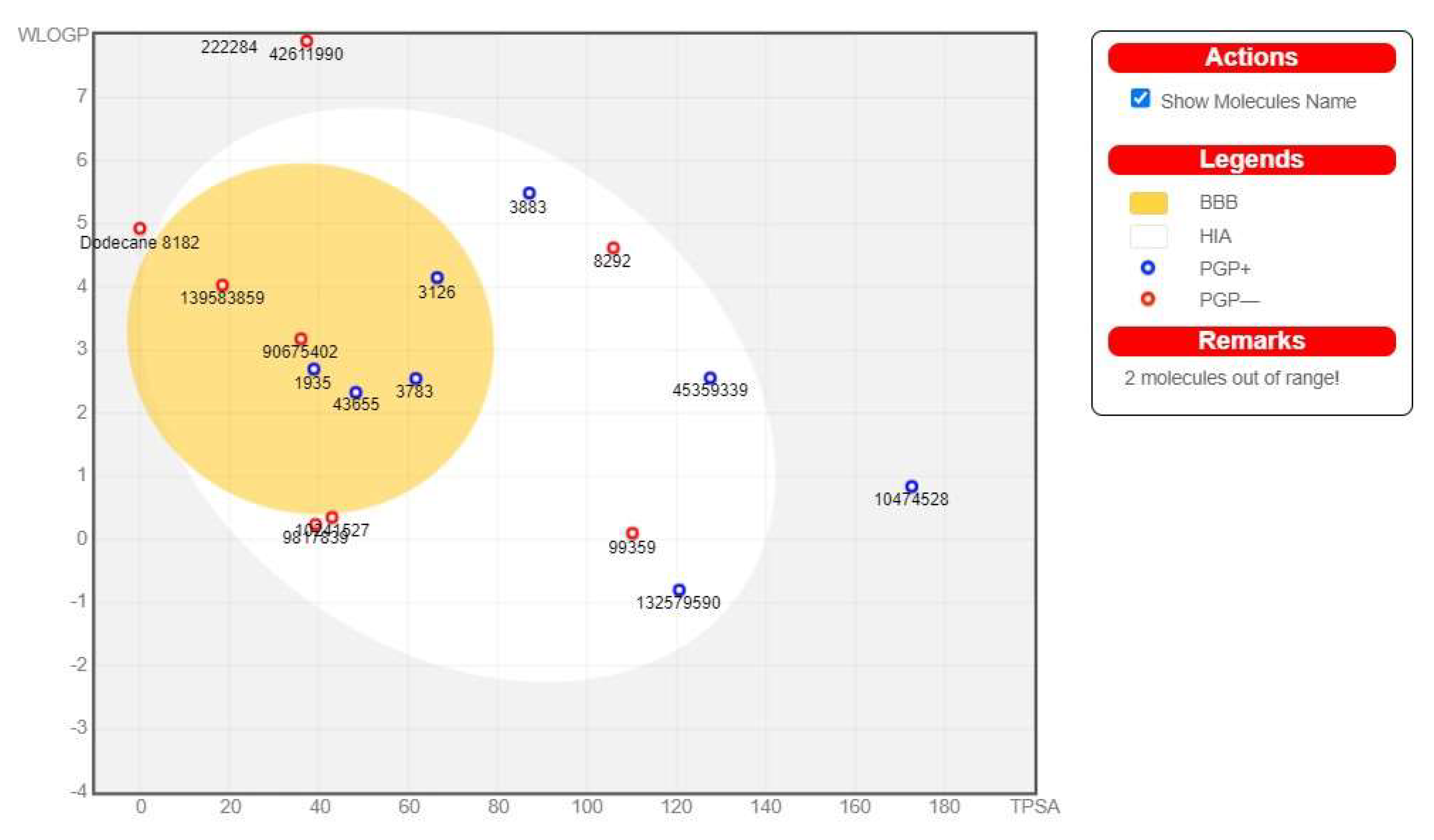
3.6. ADME Pharmacokinetic Profiling of the Detected Compounds
3.7. Drug-Likeness Properties of Predicted Compounds
3.8. Assessment of Toxicity of Selected Compounds
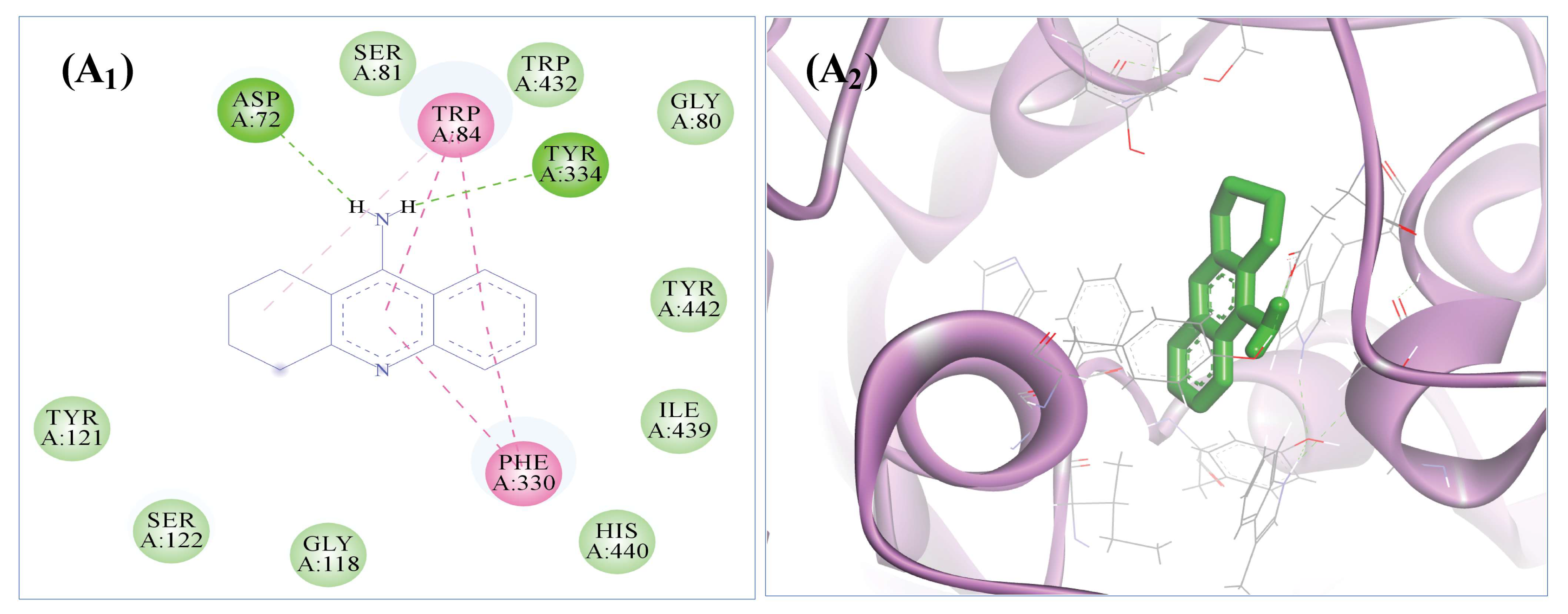
3.9. The Selected Five Compounds Strongly Bind to the Active Pocket of AChE
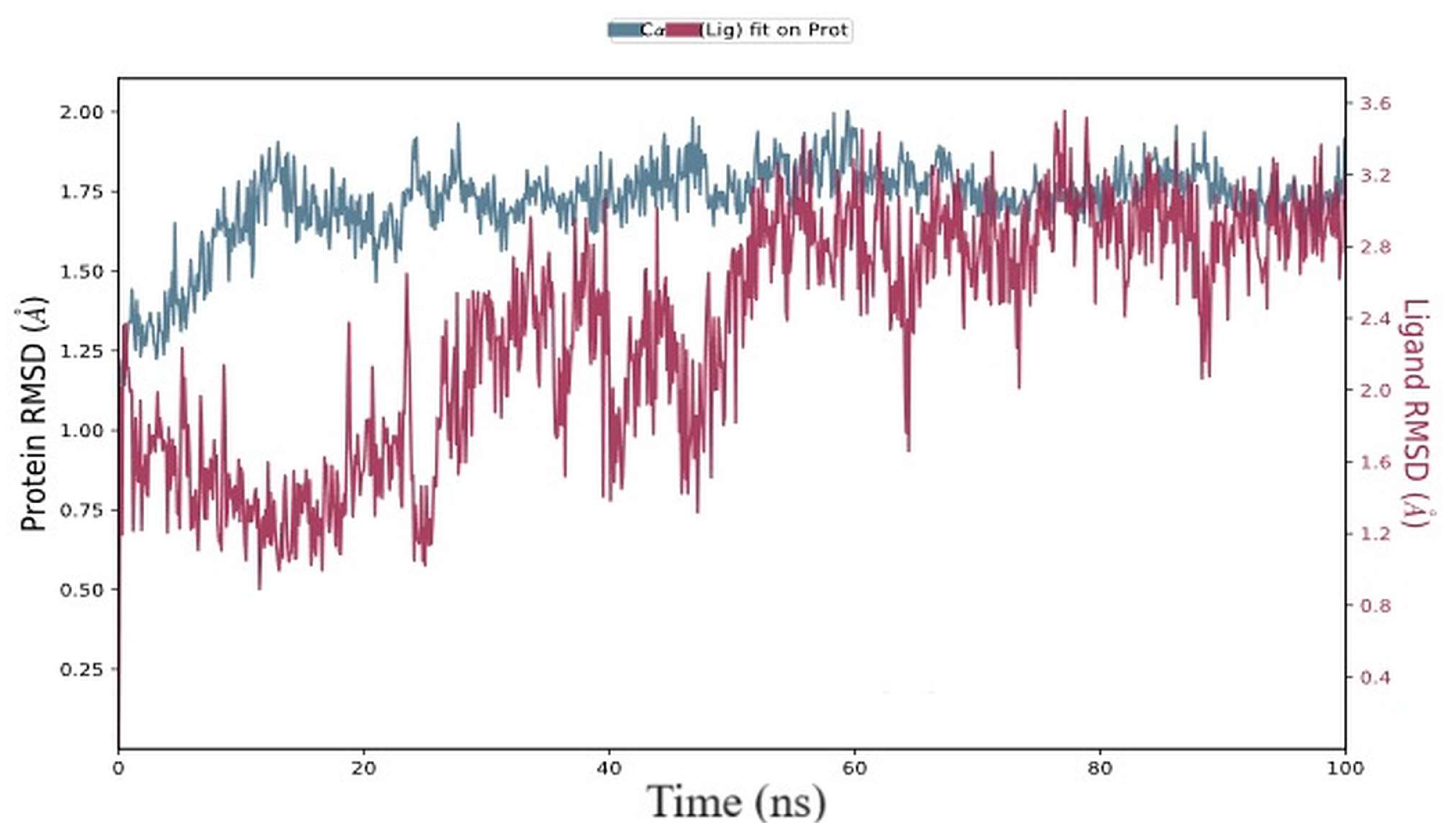
3.10. Molecular Dynamics Simulation (MDS) Analysis
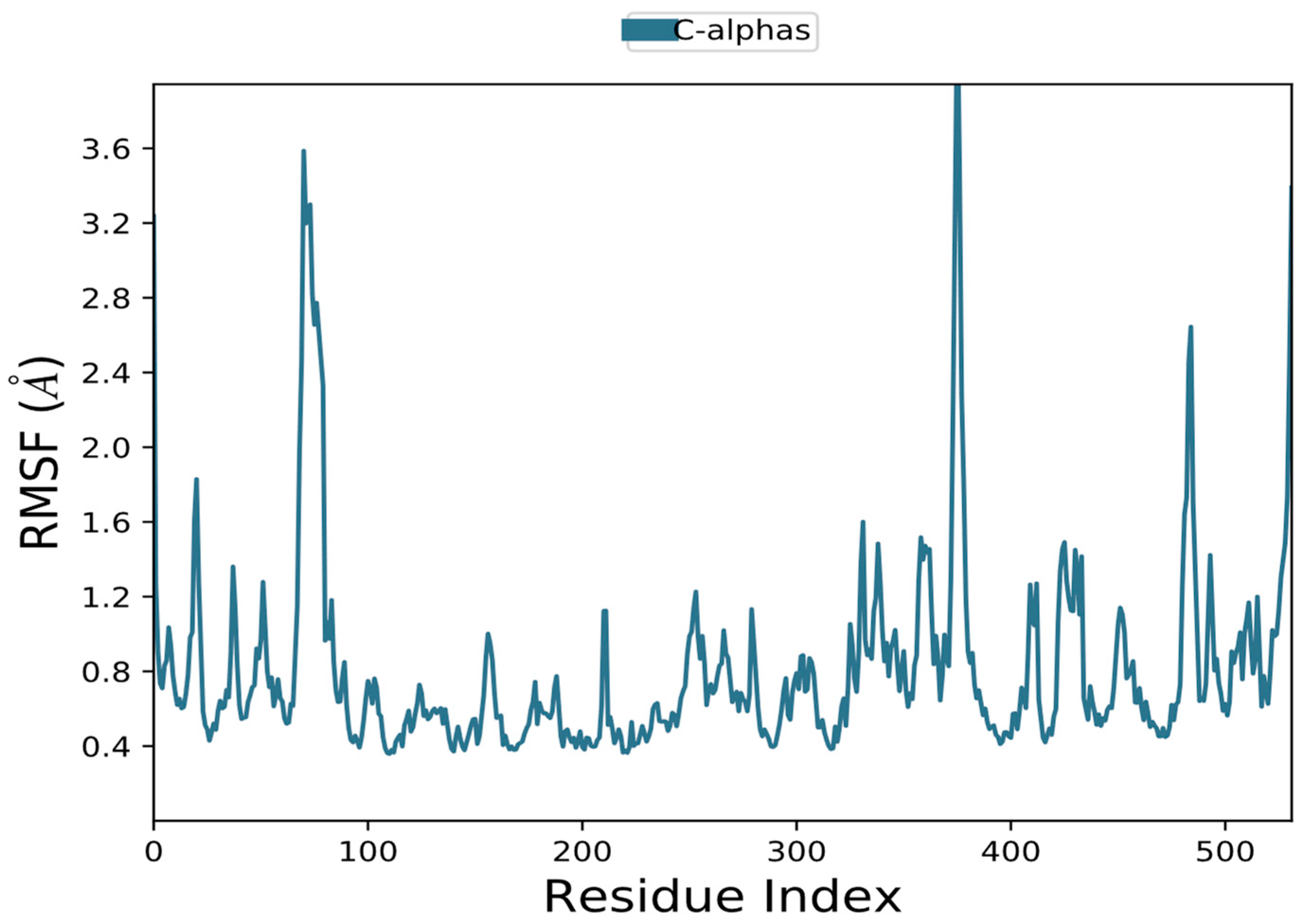
3.10.1. RMSD and RMSF Data Analysis
3.10.2. The Solvent-Accessible Surface Area (SASA) and the Gyro-Radius (Rg)
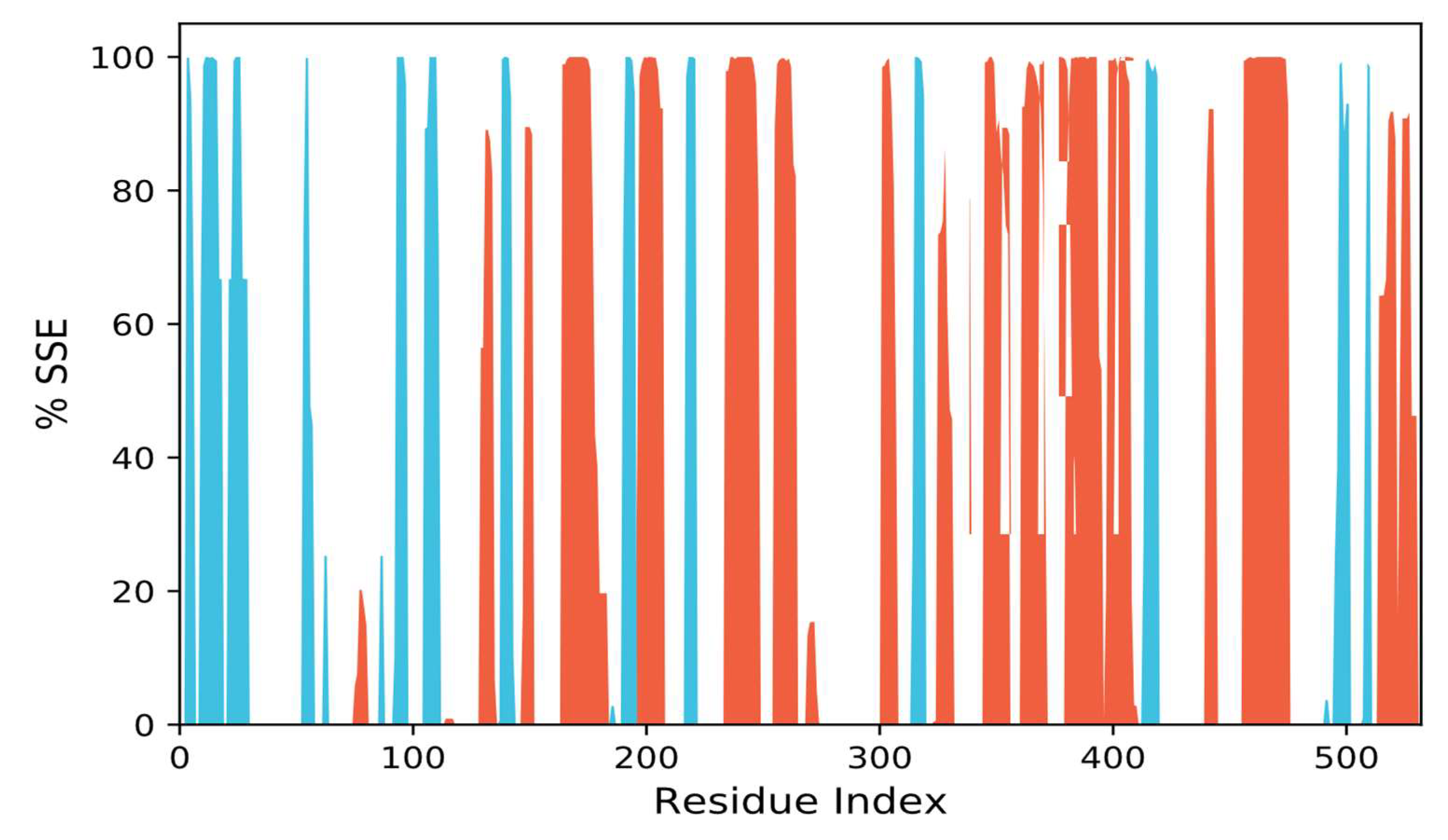
3.10.3. SSEs, i.e., Secondary Structural Elements Study
3.10.4. Interactions Analysis
3.11. Prime/MM-GBSA Free Energy Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Geda, Y.E.; Roberts, R.O.; Mielke, M.M.; Knopman, D.S.; Christianson, T.J.H.; Pankratz, V.S.; Boeve, B.F.; Sochor, O.; Tangalos, E.G.; Petersen, R.C.; et al. Baseline Neuropsychiatric Symptoms and the Risk of Incident Mild Cognitive Impairment: A Population-Based Study. Am. J. Psychiatry 2014, 171, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Bezoari, M.D.; Boothe, G. Determination of Potential Multi-Target Inhibitors of Alzheimer’s Disease In Silico. J. Undergrad. Chem. Res. 2019, 18, 21. [Google Scholar]
- Leon, C.; Hill, J.S.; Wasan, K.M. Potential Role of Acyl-Coenzyme A:Cholesterol Transferase (ACAT) Inhibitors as Hypolipidemic and Antiatherosclerosis Drugs. Pharm. Res. 2005, 22, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Sarter, M.; Parikh, V. Choline Transporters, Cholinergic Transmission and Cognition. Nat. Rev. Neurosci. 2005, 6, 48–56. [Google Scholar] [CrossRef]
- Rosenberg, P.B.; Nowrangi, M.A.; Lyketsos, C.G. Neuropsychiatric Symptoms in Alzheimer’s Disease: What Might Be Associated Brain Circuits? Mol. Asp. Med. 2015, 43–44, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Dubey, R. Target Enzyme in Alzheimer’s Disease: Acetylcholinesterase Inhibitors. Curr. Top. Med. Chem. 2019, 19, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Dou, K.-X.; Tan, M.-S.; Tan, C.-C.; Cao, X.-P.; Hou, X.-H.; Guo, Q.-H.; Tan, L.; Mok, V.; Yu, J.-T. Comparative Safety and Effectiveness of Cholinesterase Inhibitors and Memantine for Alzheimer’s Disease: A Network Meta-Analysis of 41 Randomized Controlled Trials. Alzheimer’s Res. Ther. 2018, 10, 126. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Ahmad, J. Evaluation of Antioxidant and Antimicrobial Activity of Ficus Carica Leaves: An In Vitro Approach. J. Plant Pathol. Microb. 2012, 4, 1–4. [Google Scholar] [CrossRef]
- Akhter, F.; Alvi, S.S.; Ahmad, P.; Iqbal, D.; Alshehri, B.M.; Khan, M.S. Therapeutic Efficacy of Boerhaavia diffusa (Linn.) Root Methanolic Extract in Attenuating Streptozotocin-Induced Diabetes, Diabetes-Linked Hyperlipidemia and Oxidative-Stress in Rats. Biomed. Res. Ther. 2019, 6, 3293–3306. [Google Scholar] [CrossRef]
- Alvi, S.; Ahmad, P.; Ishrat, M.; Iqbal, D.; Khan, S. Secondary Metabolites from Rosemary (Rosmarinus officinalis L.): Structure, Biochemistry and Therapeutic Implications Against Neurodegenerative Diseases. In Natural Bio-Active Compounds: Volume 2: Chemistry, Pharmacology and Health Care Practices; Springer Nature: Singapore, 2019; pp. 1–24. ISBN 9789811372049. [Google Scholar]
- Iqbal, D.; Khan, M.S.; Khan, M.S.; Ahmad, S.; Srivastava, A.K. An in Vitro and Molecular Informatics Study to Evaluate the Antioxidative and β-Hydroxy-β-Methylglutaryl-CoA Reductase Inhibitory Property of Ficus Virens Ait. Phytother. Res. 2014, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, D.; Khan, M.S.; Khan, A.; Ahmad, S. Extenuating the Role of Ficus Virens Ait and Its Novel Bioactive Compound on Antioxidant Defense System and Oxidative Damage in Cigarette Smoke Exposed Rats. Biomed. Res. Ther. 2016, 3, 723–732. [Google Scholar] [CrossRef]
- Iqbal, D.; Khan, A.; A Ansari, I.; Khan, M.S. Investigating The Role of Novel Bioactive Compound from Ficus Virens Ait on Cigarette Smoke Induced Oxidative Stress and Hyperlipidemia in Rats. Iran J. Pharm. Res. 2017, 16, 1089–1103. [Google Scholar]
- Jahan, S.; Ansari, U.A.; Siddiqui, A.J.; Iqbal, D.; Khan, J.; Banawas, S.; Alshehri, B.; Alshahrani, M.M.; Alsagaby, S.A.; Redhu, N.S.; et al. Nobiletin Ameliorates Cellular Damage and Stress Response and Restores Neuronal Identity Altered by Sodium Arsenate Exposure in Human iPSCs-Derived hNPCs. Pharmaceuticals 2022, 15, 593. [Google Scholar] [CrossRef]
- Jana, A.; Bhattacharjee, A.; Das, S.S.; Srivastava, A.; Choudhury, A.; Bhattacharjee, R.; De, S.; Perveen, A.; Iqbal, D.; Gupta, P.K.; et al. Molecular Insights into Therapeutic Potentials of Hybrid Compounds Targeting Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 3512–3528. [Google Scholar] [CrossRef]
- Khatoon, A.; Khan, F.; Ahmad, N.; Shaikh, S.; Rizvi, S.M.D.; Shakil, S.; Al-Qahtani, M.H.; Abuzenadah, A.M.; Tabrez, S.; Ahmed, A.B.F.; et al. Silver Nanoparticles from Leaf Extract of Mentha Piperita: Eco-Friendly Synthesis and Effect on Acetylcholinesterase Activity. Life Sci. 2018, 209, 430–434. [Google Scholar] [CrossRef]
- Khushtar, M.; Siddiqui, H.H.; Dixit, R.K.; Khan, M.S.; Iqbal, D.; Rahman, M.A. Amelioration of Gastric Ulcers Using a Hydro-Alcoholic Extract of Triphala in Indomethacin-Induced Wistar Rats. Eur. J. Integr. Med. 2016, 8, 546–551. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Das, S.S.; Biswal, S.S.; Nath, A.; Das, D.; Basu, A.; Malik, S.; Kumar, L.; Kar, S.; Singh, S.K.; et al. Mechanistic Role of HPV-Associated Early Proteins in Cervical Cancer: Molecular Pathways and Targeted Therapeutic Strategies. Crit. Rev. Oncol./Hematol. 2022, 174, 103675. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, P.; Alvi, S.S.; Iqbal, D.; Khan, M.S. Insights into Pharmacological Mechanisms of Polydatin in Targeting Risk Factors-Mediated Atherosclerosis. Life Sci. 2020, 254, 117756. [Google Scholar] [CrossRef] [PubMed]
- El-Seedi, H.R.; Khalifa, S.A.M.; Yosri, N.; Khatib, A.; Chen, L.; Saeed, A.; Efferth, T.; Verpoorte, R. Plants Mentioned in the Islamic Scriptures (Holy Qur’ân and Ahadith): Traditional Uses and Medicinal Importance in Contemporary Times. J. Ethnopharmacol. 2019, 243, 112007. [Google Scholar] [CrossRef] [PubMed]
- Echegaray, N.; Pateiro, M.; Gullón, B.; Amarowicz, R.; Misihairabgwi, J.M.; Lorenzo, J.M. Phoenix dactylifera Products in Human Health—A Review. Trends Food Sci. Technol. 2020, 105, 238–250. [Google Scholar] [CrossRef]
- Mandal, K.; Joshi, B.C.; Dobhal, Y. Phytopharmacological Review on Date Palm (Phoenix dactylifera). Indian J. Pharm. Sci. 2022, 84, 261–267. [Google Scholar] [CrossRef]
- El-Far, A.H.; Ragab, R.F.; Mousa, S.A. Date Palm Bioactive Compounds: Nutraceuticals, Functional Nutrients, and Pharmaceuticals. In The Date Palm Genome, Volume 2: Omics and Molecular Breeding; Al-Khayri, J.M., Jain, S.M., Johnson, D.V., Eds.; Compendium of Plant Genomes; Springer International Publishing: Cham, Switzerland, 2021; pp. 27–50. ISBN 978-3-030-73750-4. [Google Scholar]
- Assirey, E.A. The Chemical Composition, Total Phenolic and Antioxidant Content of Four Date Palm Saudi Cultivars. J. Taibah Univ. Sci. 2021, 15, 282–287. [Google Scholar] [CrossRef]
- Amin, E.; Mohamed, E.I.A.; Alenezi, A.S.; Aldwesh, M.A.; Sebak, M.; Naguib, I.A.; Bukhari, S.I.; Bukhari, K.; Zaki, M.A.; Afifi, N. Pattern Recognition of Phytoconstituents and Bioactivities of Date Pit Extracts from Different Cultivars Grown in the Qassim Area. Separations 2023, 10, 102. [Google Scholar] [CrossRef]
- Assirey, E.A.R. Nutritional Composition of Fruit of 10 Date Palm (Phoenix dactylifera L.) Cultivars Grown in Saudi Arabia. J. Taibah Univ. Sci. 2015, 9, 75–79. [Google Scholar] [CrossRef]
- Paulikienė, S.; Raila, A.; Žvirdauskienė, R.; Zvicevičius, E. Application of an Environmentally Friendly Preventive Measure for the Preservation of Fresh Vegetables. J. Food Sci. Technol. 2019, 56, 2147–2157. [Google Scholar] [CrossRef]
- Abdel-Magied, N.; Ahmed, A.G.; Abo Zid, N. Possible Ameliorative Effect of Aqueous Extract of Date (Phoenix dactylifera) Pits in Rats Exposed to Gamma Radiation. Int. J. Radiat. Biol. 2018, 94, 815–824. [Google Scholar] [CrossRef]
- Ipatova, O.M.; Prozorovskaia, N.N.; Rusina, I.F.; Prozorovskiĭ, V.N. Antioxidant properties of a leaf extract from Aronia (Aronia melanocarba) containing proanthocyanidins. Biomed. Khim. 2003, 49, 165–176. [Google Scholar]
- Srinivasan, M.; Rukkumani, R.; Ram Sudheer, A.; Menon, V.P. Ferulic Acid, a Natural Protector against Carbon Tetrachloride-Induced Toxicity. Fundam. Clin. Pharmacol. 2005, 19, 491–496. [Google Scholar] [CrossRef]
- Subash, S.; Essa, M.M.; Braidy, N.; Awlad-Thani, K.; Vaishnav, R.; Al-Adawi, S.; Al-Asmi, A.; Guillemin, G.J. Diet Rich in Date Palm Fruits Improves Memory, Learning and Reduces Beta Amyloid in Transgenic Mouse Model of Alzheimer’s Disease. J. Ayurveda Integr. Med. 2015, 6, 111–120. [Google Scholar] [CrossRef]
- Pujari, R.; Vyawahare, N.; Thakurdesai, P. Neuroprotective and Antioxidant Role of Phoenix dactylifera in Permanent Bilateral Common Carotid Occlusion in Rats. J. Acute Dis. 2014, 3, 104–114. [Google Scholar] [CrossRef]
- Uddin, I.; Vandana, A.V.A.; Kavya, G.K.G.; Syed, Y.H. Systematic Study on Protective Role of Date Palm (Phoenix dactylifera L.) on Central Nervous System Disorders. Ann. Phytomed. 2020, 9, 58–65. [Google Scholar] [CrossRef]
- Alsagaby, S.A.; Iqbal, D.; Ahmad, I.; Patel, H.; Mir, S.A.; Madkhali, Y.A.; Oyouni, A.A.A.; Hawsawi, Y.M.; Alhumaydhi, F.A.; Alshehri, B.; et al. In silico investigations identified Butyl Xanalterate to competently target CK2α (CSNK2A1) for therapy of chronic lymphocytic leukemia. Sci Rep. 2022, 12, 17648. [Google Scholar] [CrossRef] [PubMed]
- Jabir, N.R.; Shakil, S.; Tabrez, S.; Khan, M.S.; Rehman, M.T.; Ahmed, B.A. In Silico Screening of Glycogen Synthase Kinase-3β Targeted Ligands against Acetylcholinesterase and Its Probable Relevance to Alzheimer’s Disease. J. Biomol. Struct. Dyn. 2021, 39, 5083–5092. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; AlAjmi, M.F.; Hussain, A.; Rather, G.M.; Khan, M.A. High-Throughput Virtual Screening, Molecular Dynamics Simulation, and Enzyme Kinetics Identified ZINC84525623 as a Potential Inhibitor of NDM-1. Int. J. Mol. Sci. 2019, 20, E819. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Khan, M.S.; Shahwan, M.; Husain, F.M.; Rehman, M.T.; Hassan, M.I.; Ahmad, F.; Islam, A. Unraveling Binding Mechanism of Alzheimer’s Drug Rivastigmine Tartrate with Human Transferrin: Molecular Docking and Multi-Spectroscopic Approach towards Neurodegenerative Diseases. Biomolecules 2019, 9, E495. [Google Scholar] [CrossRef]
- Iqbal, D.; Rehman, M.T.; Bin Dukhyil, A.; Rizvi, S.M.D.; Al Ajmi, M.F.; Alshehri, B.M.; Banawas, S.; Khan, M.S.; Alturaiki, W.; Alsaweed, M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals 2021, 14, 937. [Google Scholar] [CrossRef]
- Harborne, A. Phytochemical Methods a Guide to Modern Techniques of Plant Analysis; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1998; ISBN 0-412-57270-2. [Google Scholar]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. Analysis of Total Phenols and Other Oxidation Substrates and Antioxidants by Means of Folin-Ciocalteu Reagent. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1999; Volume 299, pp. 152–178. ISBN 0076-6879. [Google Scholar]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a Free Radical Method to Evaluate Antioxidant Activity. LWT—Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, D.; Khan, M.S.; Waiz, M.; Rehman, M.T.; Alaidarous, M.; Jamal, A.; Alothaim, A.S.; AlAjmi, M.F.; Alshehri, B.M.; Banawas, S.; et al. Exploring the Binding Pattern of Geraniol with Acetylcholinesterase through In Silico Docking, Molecular Dynamics Simulation, and In Vitro Enzyme Inhibition Kinetics Studies. Cells 2021, 10, 3533. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein–Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Alvi, S.S.; Ansari, I.A.; Khan, I.; Iqbal, J.; Khan, M.S. Potential Role of Lycopene in Targeting Proprotein Convertase Subtilisin/Kexin Type-9 to Combat Hypercholesterolemia. Free Radic. Biol. Med. 2017, 108, 394–403. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, D.; Rizvi, S.M.D.; Rehman, M.T.; Khan, M.S.; Bin Dukhyil, A.; AlAjmi, M.F.; Alshehri, B.M.; Banawas, S.; Zia, Q.; Alsaweed, M.; et al. Soyasapogenol-B as a Potential Multitarget Therapeutic Agent for Neurodegenerative Disorders: Molecular Docking and Dynamics Study. Entropy 2022, 24, 593. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Shakil, S. Molecular Interaction of Inhibitors with Human Brain Butyrylcholinesterase. EXCLI J. 2021, 20, 1597. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies Using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Koul, B.; Farooq, U.; Yadav, D.; Song, M. Phytochemicals: A Promising Alternative for the Prevention of Alzheimer’s Disease. Life 2023, 13, 999. [Google Scholar] [CrossRef]
- Sawikr, Y.; Yarla, N.S.; Peluso, I.; Kamal, M.A.; Aliev, G.; Bishayee, A. Neuroinflammation in Alzheimer’s Disease: The Preventive and Therapeutic Potential of Polyphenolic Nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017, 108, 33–57. [Google Scholar] [CrossRef]
- Shaw, F.H.; Bentley, G.A. The Pharmacology of Some New Anti-Cholinesterases. Aust. J. Exp. Biol. Med. Sci. 1953, 31, 573–576. [Google Scholar] [CrossRef]
- Marquis, J.K. Pharmacological Significance of Acetylcholinesterase Inhibition by Tetrahydroaminoacridine. Biochem. Pharmacol. 1990, 40, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, M.; Christian, L.; Inhester, T.; Petzold, S.; Gütschow, M. Kinetics of Inhibition of Acetylcholinesterase in the Presence of Acetonitrile. FEBS J. 2009, 276, 2292–2307. [Google Scholar] [CrossRef]
- Farag, M.A.; Gad, H.A.; Heiss, A.G.; Wessjohann, L.A. Metabolomics Driven Analysis of Six Nigella Species Seeds via UPLC-qTOF-MS and GC–MS Coupled to Chemometrics. Food Chem. 2014, 151, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking Drug Design in the Artificial Intelligence Era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Ma, X.L.; Chen, C.; Yang, J. Predictive Model of Blood-Brain Barrier Penetration of Organic Compounds. Acta Pharmacol. Sin. 2005, 26, 500–512. [Google Scholar] [CrossRef]
- Leão, R.P.; Cruz, J.V.; da Costa, G.V.; Cruz, J.N.; Ferreira, E.F.B.; Silva, R.C.; de Lima, L.R.; Borges, R.S.; Dos Santos, G.B.; Santos, C.B.R. Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach. Pharmaceuticals 2020, 13, 209. [Google Scholar] [CrossRef]
- Bittermann, K.; Goss, K.U. Predicting Apparent Passive Permeability of Caco-2 and MDCK Cell-Monolayers: A Mechanistic Model. PLoS ONE 2017, 12, e0190319. [Google Scholar] [CrossRef]
- Volpe, D.A. Variability in Caco-2 and MDCK Cell-Based Intestinal Permeability Assays. J. Pharm. Sci. 2008, 97, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Furubayashi, T.; Kataoka, M.; Sakane, T.; Sezaki, H.; Tokuda, H. Optimized Conditions for Prediction of Intestinal Drug Permeability Using Caco-2 Cells. Eur. J. Pharm. Sci. 2000, 10, 195–204. [Google Scholar] [CrossRef]
- Chen, C.-P.; Chen, C.-C.; Huang, C.-W.; Chang, Y.-C. Evaluating Molecular Properties Involved in Transport of Small Molecules in Stratum Corneum: A Quantitative Structure-Activity Relationship for Skin Permeability. Molecules 2018, 23, 911. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Pea, F.; Lipman, J. The Clinical Relevance of Plasma Protein Binding Changes. Clin. Pharmacokinet. 2013, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, K.G. Effect of Blood Protein Concentrations on Drug-Dosing Regimes: Practical Guidance. Theor. Biol. Med. Model. 2013, 10, 20. [Google Scholar] [CrossRef]
- Wang, B.; Yang, L.P.; Zhang, X.Z.; Huang, S.Q.; Bartlam, M.; Zhou, S.F. New Insights into the Structural Characteristics and Functional Relevance of the Human Cytochrome P450 2D6 Enzyme Structural Features of CYP2D6 B. Wang et Al. Drug Metab. Rev. 2009, 41, 573–643. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Boutina, D.; Beck, G.; Sherborne, B.; Cooper, I.; et al. Evaluation of Human Intestinal Absorption Data and Subsequent Derivation of a Quantitative Structure—Activity Relationship (QSAR) with the Abraham Descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef]
- Hessler, G.; Baringhaus, K.-H. Artificial Intelligence in Drug Design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; Association for Computing Machinery: New York, NY, USA, 2006; pp. 84–es. [Google Scholar]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K.S. Bringing Molecular Dynamics Simulation Data into View. Trends Biochem. Sci. 2019, 44, 902–913. [Google Scholar] [CrossRef]
- Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of Lead Compounds against Scm (Fms10) in Enterococcus Faecium Using Computer Aided Drug Designing. Life 2021, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Oyedele, A.-Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular Modeling in Drug Discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Karplus, M.; Petsko, G.A. Molecular Dynamics Simulations in Biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phytochemicals | Methanolic Extract of KH | Aqueous Extract of KH |
|---|---|---|
| Carbohydrates | ++ | +++ |
| Tannins | + | + |
| Saponins | - | - |
| Alkaloids | + | ++ |
| Flavonoids | ++ | +++ |
| Glycosides | + | ++ |
| Phenols | ++ | +++ |
| Terpenoids | + | - |
| Cardiac glycosides | - | ++ |
| Steroids | - | - |
| Compound ID | Compound Name | Chemical Formula | Match Score | RT | Adduct /Loss | Predicted m/z | Matched m/z | Fragmentations |
|---|---|---|---|---|---|---|---|---|
| 73053139 | 5-(7-Methyloctyl)-1,2,3,4-Tetrahydroquinoline | C18H29N | 0.905 | 1.71 | H+/− | 260.2373 | 260.5172 | 260.517, 260.170, 260.274, 260.309, 261.037 |
| 99359 | Argvalin | C11H19N5O | 0.944 | 2.67 | H+/− | 238.1662 | 238.4553 | 238.04, 238.14, 238.46 |
| 90675402 | CHEMBL3259981 | C15H17NO | 0.917 | 5.24 | H+/− | 228.1383 | 228.2916 | 228.292, 228.049, 227.529, 229.436, 229.818 |
| 8182 | N-Dodecane | C12H26 | 0.859 | 9.18 | H+/− | 171.2107 | 171.5068 | 171.507, 171.195, 171.715, 171.854 |
| 10241527 | Pyrocoll | C10H6N2O2 | 0.950 | 11.63 | H+/− | 187.0502 | 187.3247 | 187.325, 186.457, 187.776, 188.469 |
| 139583859 | Methyl hydroxysterpurate ethylidene cetal | C18H30O2 | 0.851 | 12.50 | H+/− | 279.2319 | 279.4917 | 279.492, 279.249, 279.943, 280.394 |
| 139586920 | 3β-Hydroxy-4β-methylfusida-17(20)(16,21-cis),24-diene | C30H48O | 0.943 | 14.45 | H+/− | 425.3778 | 425.6685 | 425.668, 426.536, 426.778 |
| 132579590 | Terezine E | C16H22N2O6 | 0.864 | 19.14 | H+/− | 339.1551 | 338.8782 | 338.878, 337.352, 339.052, 339.676, 339.919 |
| 10474528 | Paeciloquinone D | C18H14O9 | 0.855 | 21.09 | H+/− | 375.0711 | 374.8500 | 374.850, 374.642, 374.538, 375.093 |
| Compound ID | Compound Name | Chemical Formula | Match Score | RT | Adduct /Loss | Predicted m/z | Matched m/z | Fragmentations |
|---|---|---|---|---|---|---|---|---|
| 45359339 | MCULE-1146796980 | C27H37NO7 | 0.989 | 1.66 | -/H+ | 486.2486 | 486.477 | 486.477, 486.200, 486.986 |
| 8292 | Fensulfothion | C11H17O4PS2 | 0.874 | 1.66 | -/H+ | 307.0222 | 306.9764 | 306.976, 307.855 |
| 43655 | 8-Hydroxyloxapine | C18H18ClN3O2 | 0.872 | 1.70 | -/H+ | 342.1004 | 342.3123 | 342.312, 342.8912 |
| 42611990 | Gilvsin A | C30H48O3 | 0.859 | 2.66 | -/H+ | 453.3727 | 453.3151 | 453.315, 453.084 |
| 222284 | Sitosterol | C29H48O | 0.943 | 3.53 | -/H+ | 413.3778 | 413.3541 | 413.354 |
| 3883 | Lansoprazole | C16H14F3N3O2S | 0.971 | 20.58 | -/H+ | 368.0675 | 368.2130 | 368.213, 369.045 |
| 3783 | Isoxsuprine | C18H23NO3 | 0.967 | 20.58 | -/H+ | 300.1594 | 300.2700 | 300.270, 301.195 |
| 3126 | 2-Aminooctadecane | C18H39NO2 | 0.966 | 20.58 | -/H+ | 300.2897 | 300.2700 | 300.270, 301.195 |
| 9817839 | Dehydroevodiamine | C19H15N3O | 0.954 | 20.66 | -/H+ | 300.1131 | 299.8538 | 299.854, 300.131 |
| Compound Name | 2-Aminooctadecane | Isoxsuprine | 8-Hydroxyloxapine | 2-{5-[(1E)-3-methylbuta-1,3-dien-1-yl]-1H-indol-3-yl} Ethanol | Methyl_Hydroxysterpurate Ethylidene Acetal |
|---|---|---|---|---|---|
| ID | 3126 | 3783 | 43655 | 90675402 | 139583859 |
| BBB | 5.73 | 2.28 | 0.49 | 7.46 | 1.96 |
| Buffer Solubility mg/L | 6.46 | 541.43 | 54.65 | 8.46 | 66.19 |
| Pure Water Solubility mg/L | 2.32 | 629.45 | 21.31 | 25.39 | 22.11 |
| MDCK | 77.07 | 260.92 | 97.93 | 199.83 | 280.41 |
| Caco2 | 20.60 | 15.32 | 42.70 | 26.95 | 57.68 |
| Skin Permeability | −0.77 | −3.17 | −3.75 | −3.07 | −2.66 |
| Plasma Protein Binding | 100.00 | 84.22 | 76.91 | 83.53 | 100.00 |
| HIA | 87.18 | 90.24 | 96.12 | 91.77 | 100.00 |
| CYP_2D6_inhibition | Inhibitor | Inhibitor | Non | Non | Non |
| CYP_2D6_substrate | Substrate | Substrate | Substrate | Non | Non |
| Compound | miLogP | M. Wt. | TPSA | Hydrogen Bond Acceptor | Hydrogen Bond Donor | No. of Violations | No. of Rotatable Bond | Volume |
|---|---|---|---|---|---|---|---|---|
| 3126 | 5.04 | 301.51 | 66.48 | 3 | 4 | 1 | 16 | 342.20 |
| 3783 | 2.77 | 301.39 | 61.72 | 4 | 3 | 0 | 7 | 293.28 |
| 43655 | 3.75 | 343.81 | 52.74 | 5 | 1 | 0 | 1 | 296.90 |
| 90675402 | 2.98 | 213.28 | 36.02 | 2 | 2 | 0 | 4 | 209.79 |
| 139583859 | 4.22 | 278.44 | 18.47 | 2 | 0 | 0 | 0 | 287.92 |
| 1935 * | 3.05 | 198.27 | 38.91 | 2 | 2 | 0 | 0 | 191.53 |
| PubChem CID | Compound Name | Predicted LD50 mg/kg | Predicted Toxicity Class | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity |
|---|---|---|---|---|---|---|---|---|
| 3126 | 2-Aminooctadecane | 3500 | 5 | NT | NT | NT | NT | NT |
| 3783 | Isoxsuprine | 200 | 3 | NT | NT | NT | NT | NT |
| 43655 | 8-Hydroxyloxapine | 40 | 2 | NT | NT | T | NT | NT |
| 90675402 | 2-{5-[(1E)-3-methylbuta-1,3-dien-1-yl]-1H-indol-3-yl}ethanol | 1680 | 4 | NT | NT | T | NT | NT |
| 139583859 | Methyl Hydroxysterpurate Ethylidene Acetal | 7800 | 6 | NT | NT | NT | NT | NT |
| S. No. | Compound ID | Compound Name | Binding energy ∆G Kcal/mol | Interacting Amino Acids |
|---|---|---|---|---|
| 1 | 43655 | 8-Hydroxyloxapine | −9.49 | Asn85, Asp72, Glu199, Gly117, Gly118, His440, Ile439, Met436, Phe330, Ser122, Ser200, Ser81, Tyr70, Trp432, Trp84, Tyr121, Tyr334, Tyr442, Val71 |
| 2 | 3783 | Isoxsuprine | −9.13 | Arg28, Asp72, Asn85, Ile287, Phe288, Phe290, Phe330, Phe331, Tyr70, Trp84, Tyr121, Tyr334, Ser122, Trp279, Leu282, Ser286, Val71. |
| 3 | 90675402 | 2-{5-[(1E)-3-methylbuta-1,3-dien-1-yl]-1H-indol-3-yl} ethanol | −8.67 | Gly117, Gly118, Gly123, Gly441, Glu199, His440, Ile444, Ile439, Leu127, Phe330, Ser81, Ser200, Trp84, Tyr116, Ser124, Tyr130, Tyr334 Met436, Trp432, Tyr442, |
| 4 | 139583859 | Methyl hydroxysterpurate ethylidene acetal | −7.91 | Try121, Trp279, Ile287, Phe288, Arg289, Phe290, Phe330, Phe331, Tyr334, Gly335. |
| 5 | 3126 | 2-Aminooctadecane | −6.91 | Asp72, Asn85, Gly80, Gly117, Gly118, Glu199, Gly123, Gly441, His440, Ile439, Ile444, Phe330, Ser122, Ser81, Ser200, Tyr70, Trp84, Tyr121, Tyr130, Tyr334, Tyr442 |
| 6 | 1935* | Tacrine | −8.25 | Asp72, Gly80, Gly118, His440, Ile39, Phe330, Ser81, Trp84, Tyr121, Ser122, Tyr334, Trp432, Tyr442 |
| Title/Time | dG_Bind | dG_Bind_Coulomb | dG_Bind_Covalent | dG_Bind_vdW | dG_Bind_Lipo | dG_Bind_Hbond |
|---|---|---|---|---|---|---|
| 0 ns | −59.33 | −7.61 | 1.74 | −41.71 | −41.85 | −1.15 |
| 10 ns | −40.99 | −6.13 | 1.43 | −30.93 | −30.56 | −1.25 |
| 20 ns | −47.96 | −8.10 | 2.40 | −32.55 | −33.99 | −1.20 |
| 30 ns | −53.30 | 2.08 | 0.63 | −36.19 | −37.30 | −1.07 |
| 40 ns | −45.57 | −10.64 | 1.60 | −32.09 | −29.27 | −1.57 |
| 50 ns | −48.29 | −2.79 | 1.55 | −33.32 | −33.75 | −1.23 |
| 60 ns | −55.58 | −8.24 | 2.63 | −34.82 | −36.14 | −1.46 |
| 70 ns | −51.02 | −4.76 | 0.97 | −37.19 | −37.06 | −1.34 |
| 80 ns | −57.39 | −6.50 | 2.29 | −37.25 | −40.67 | −1.42 |
| 90 ns | −70.20 | −11.54 | 1.93 | −37.93 | −43.78 | −1.52 |
| 100 ns | −52.98 | −7.37 | 1.14 | −38.68 | −39.64 | −1.08 |
| AVG | −52.9643581 | −6.508649533 | 1.666593643 | −35.69480304 | −36.72741552 | −1.298206565 |
| SD | 7.463234925 | 3.575755222 | 0.591036946 | 3.119070994 | 4.377846677 | 0.166159877 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almalki, S.G.; Alqurashi, Y.E.; Alturaiki, W.; Almawash, S.; Khan, A.; Ahmad, P.; Iqbal, D. Antioxidant, LC-MS Analysis, and Cholinesterase Inhibitory Potentials of Phoenix dactylifera Cultivar Khudari: An In Vitro Enzyme Kinetics and In Silico Study. Biomolecules 2023, 13, 1474. https://doi.org/10.3390/biom13101474
Almalki SG, Alqurashi YE, Alturaiki W, Almawash S, Khan A, Ahmad P, Iqbal D. Antioxidant, LC-MS Analysis, and Cholinesterase Inhibitory Potentials of Phoenix dactylifera Cultivar Khudari: An In Vitro Enzyme Kinetics and In Silico Study. Biomolecules. 2023; 13(10):1474. https://doi.org/10.3390/biom13101474
Chicago/Turabian StyleAlmalki, Sami G., Yaser E. Alqurashi, Wael Alturaiki, Saud Almawash, Amir Khan, Parvej Ahmad, and Danish Iqbal. 2023. "Antioxidant, LC-MS Analysis, and Cholinesterase Inhibitory Potentials of Phoenix dactylifera Cultivar Khudari: An In Vitro Enzyme Kinetics and In Silico Study" Biomolecules 13, no. 10: 1474. https://doi.org/10.3390/biom13101474