
The Insulin-Degrading Enzyme from Structure to Allosteric Modulation: New Perspectives for Drug Design

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Discovery of the IDE and Its Role in Insulin Catabolism
2. Pills on IDE Multiple Roles in Health and Disease
2.1. The IDE and Type 2 Diabetes Mellitus (T2DM)
2.2. The IDE and Alzheimer’s Disease
Involvement of the Insulin-Degrading Enzyme in Retina Pathology
2.3. The IDE as a Playmaker in the Regulation of Proteostasis
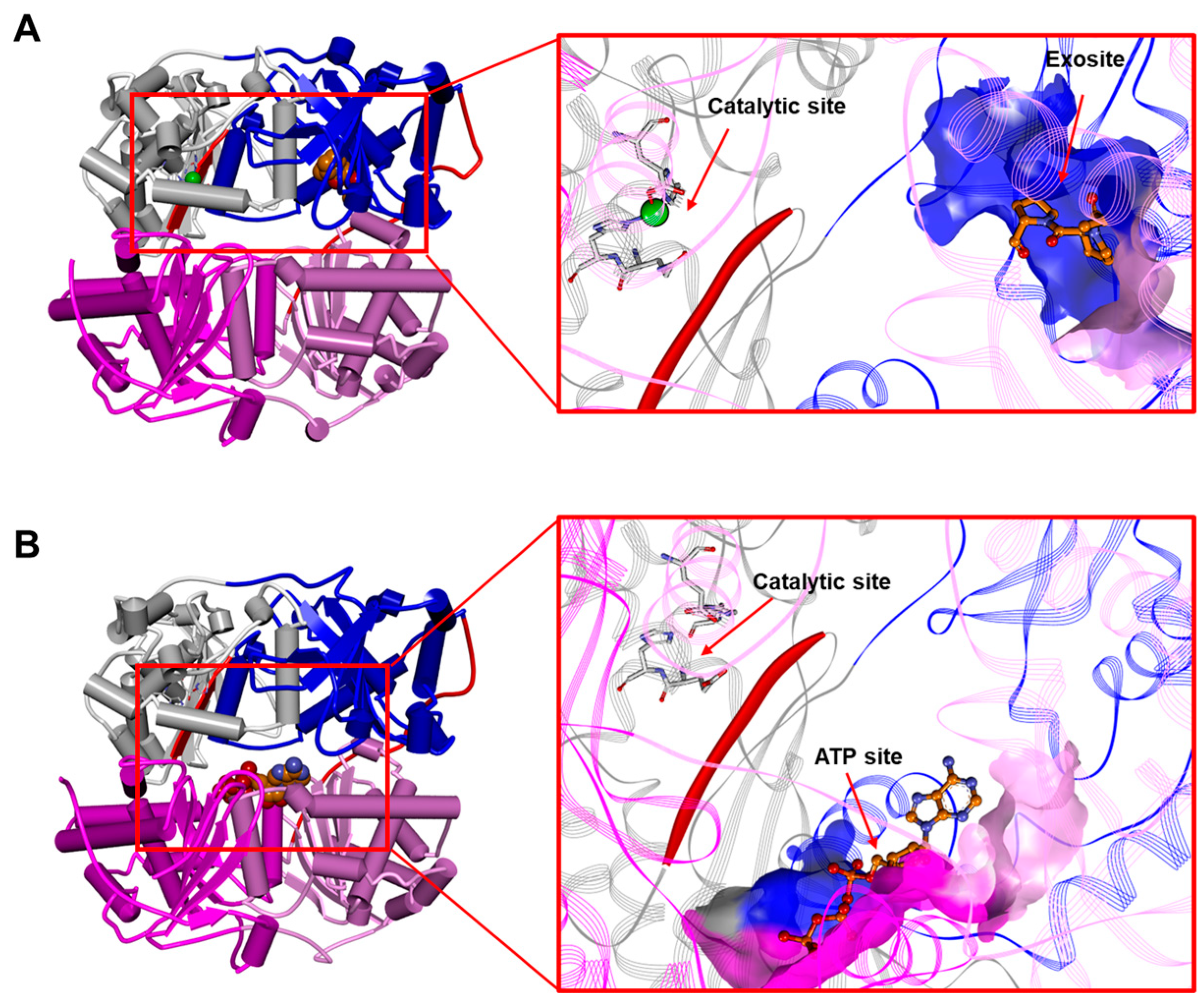
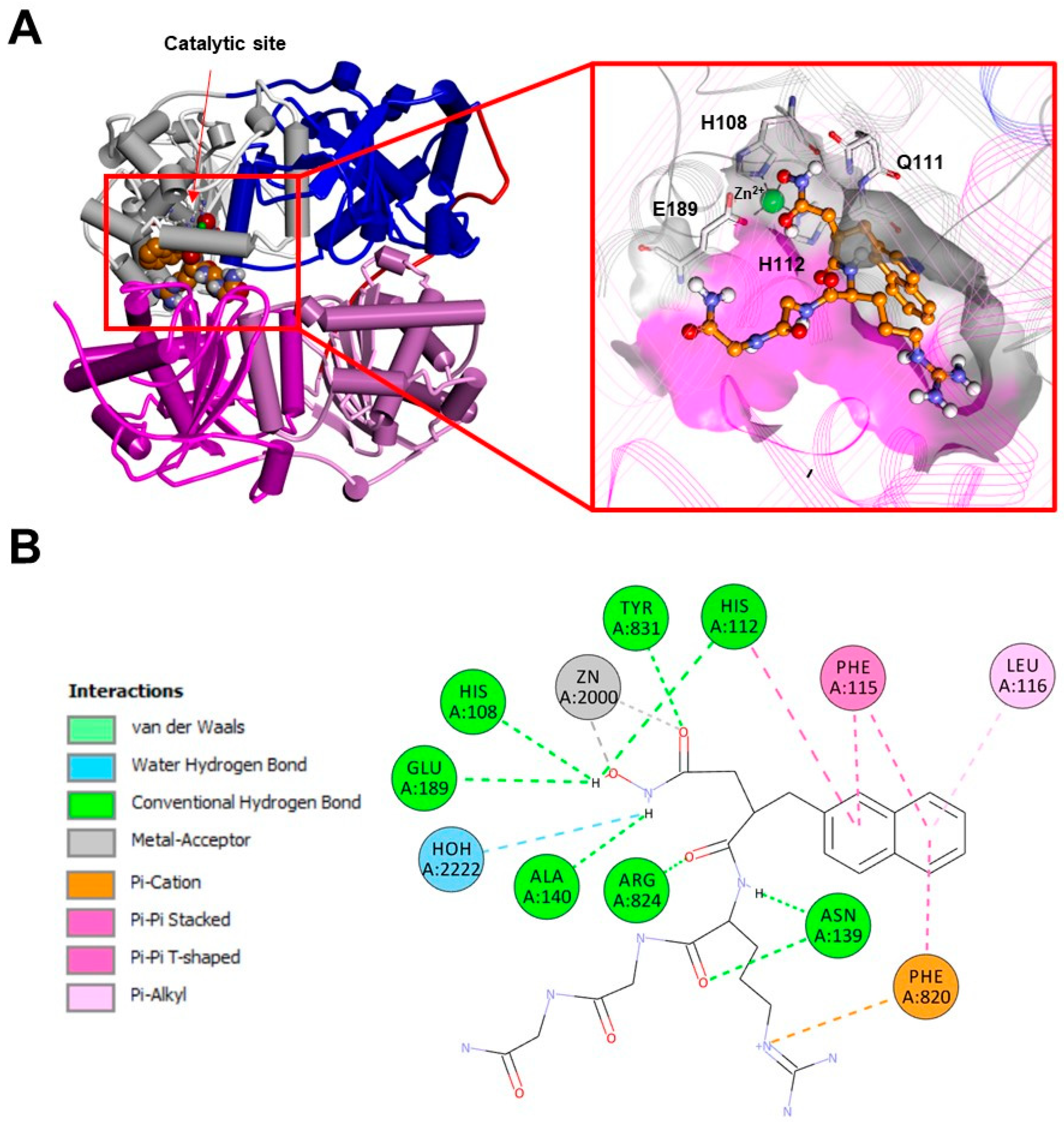
3. The “Atypical Structure” of the IDE
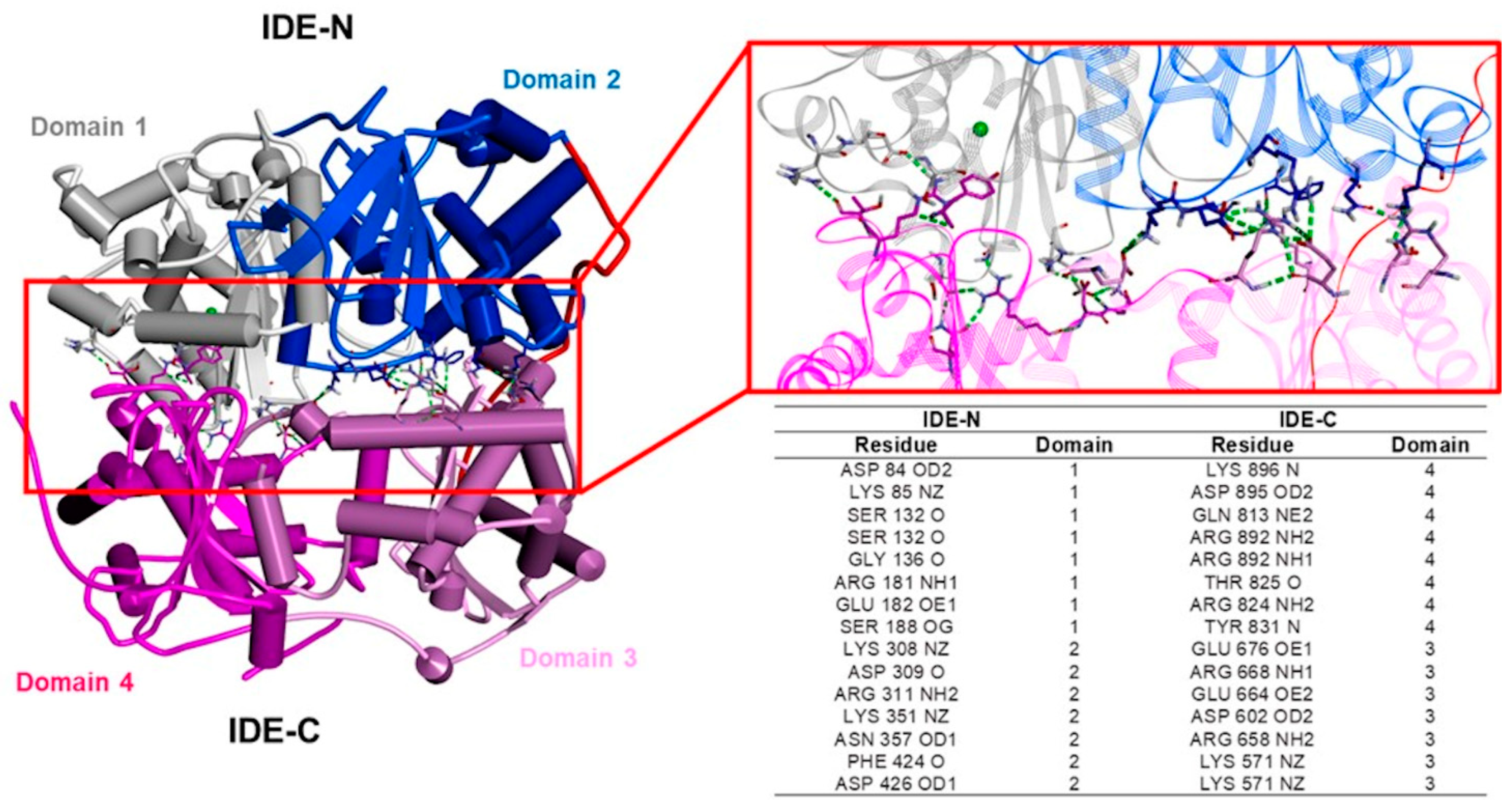
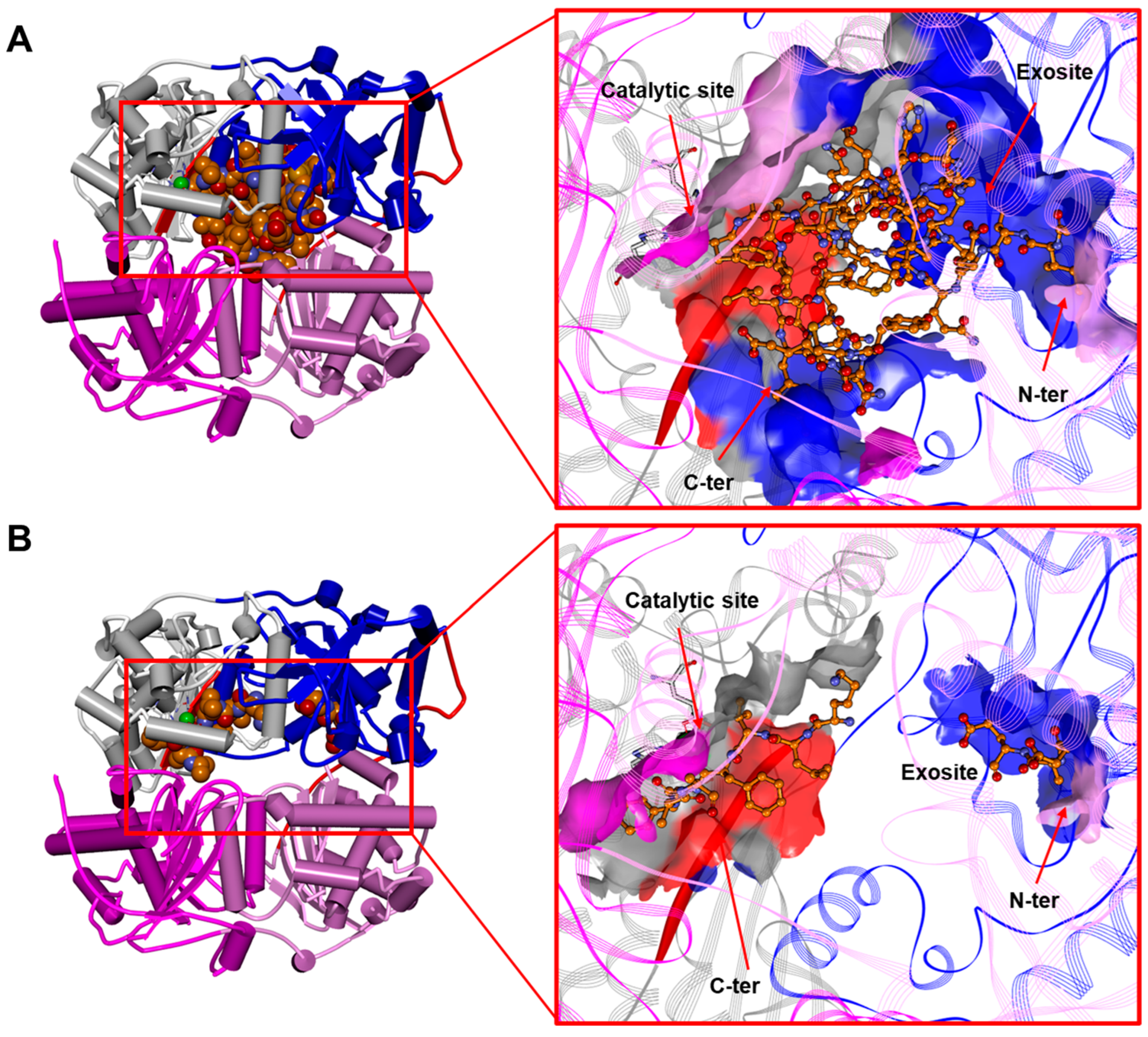
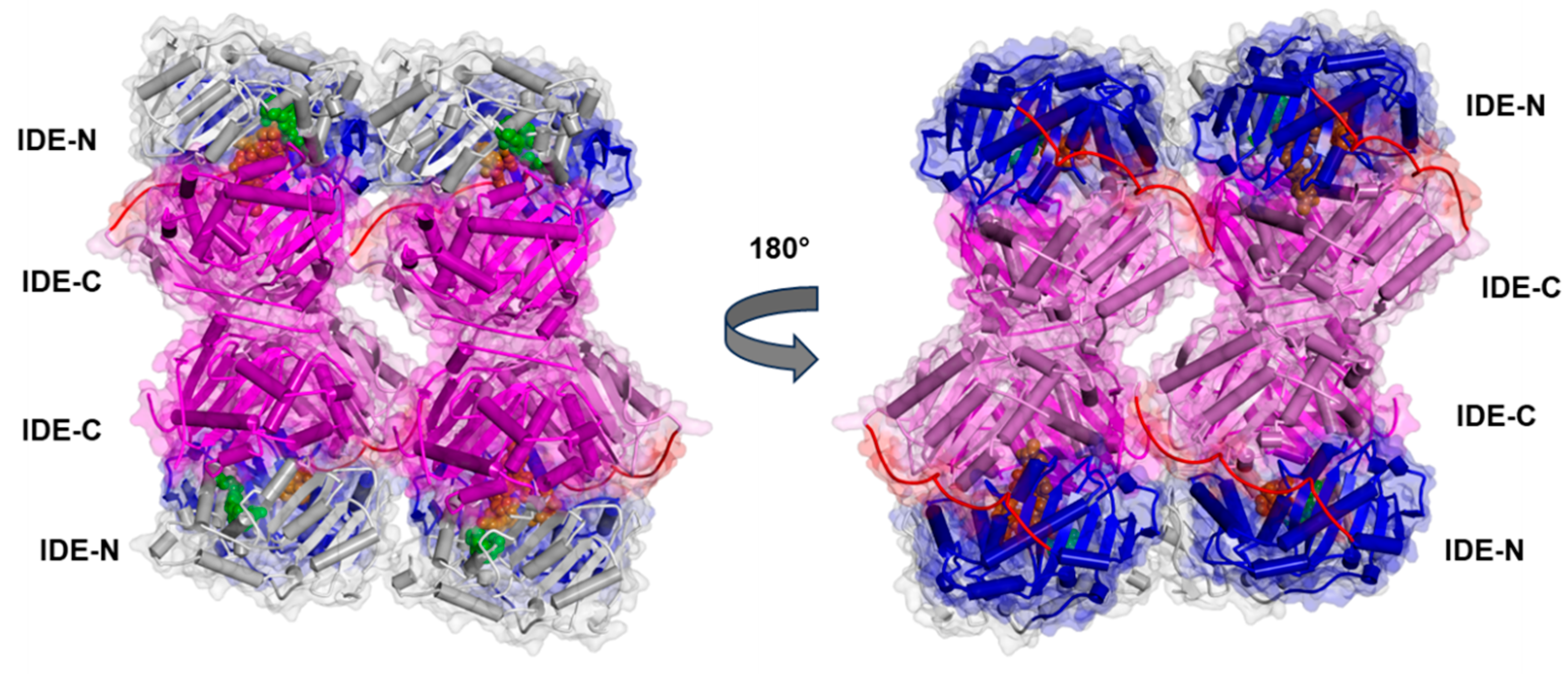
3.1. The IDE Structure and Biochemical Properties Relationship
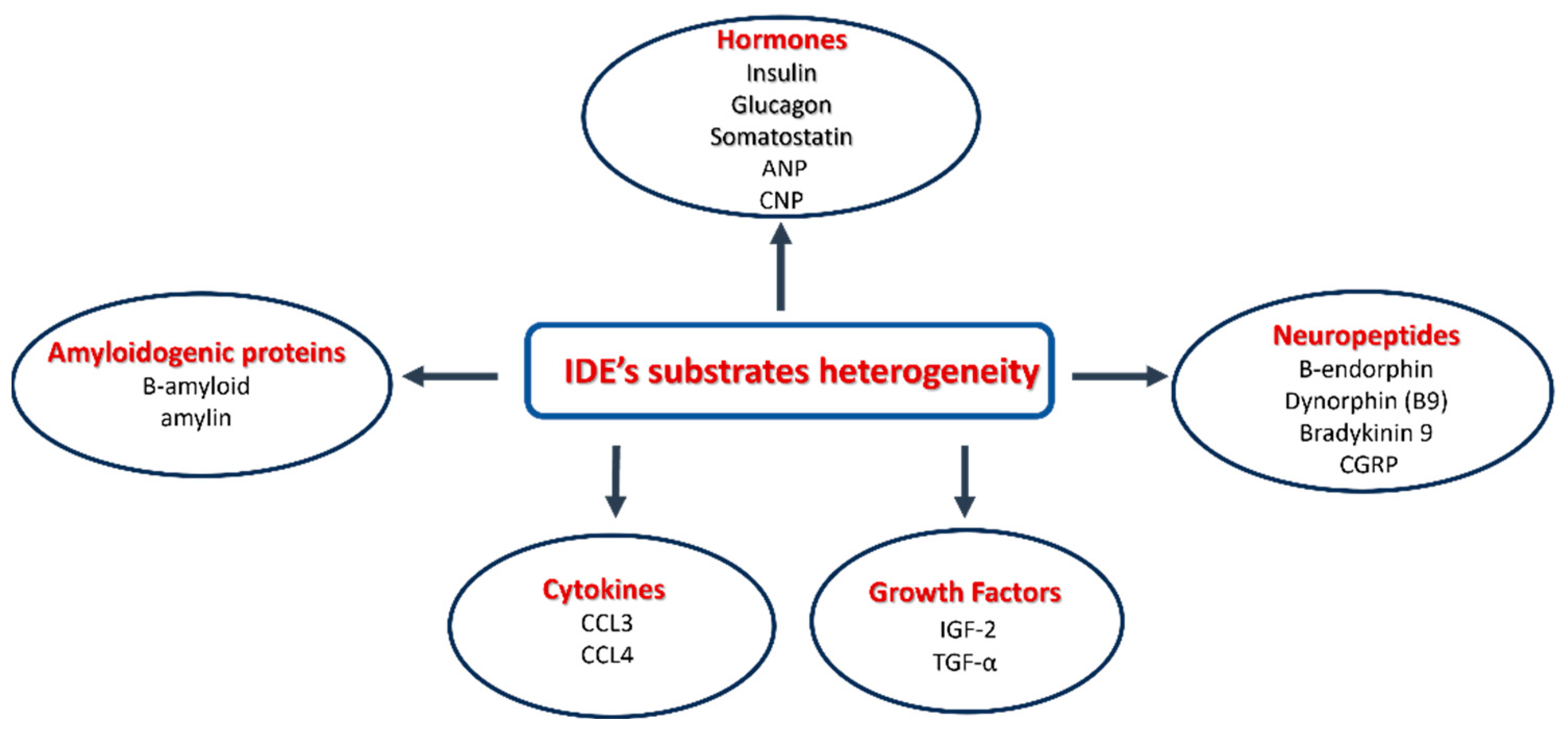
3.2. The Heterogeneity of IDE Substrates
3.3. The IDE as an Allosteric Enzyme
Metal Ions Affect IDE Activity
4. Pharmacological Modulation of the IDE: A Therapeutic Perspective
4.1. The IDE Inhibitors
4.2. The IDE Activators
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sbardella, D.; Fasciglione, G.F.; Gioia, M.; Ciaccio, C.; Tundo, G.R.; Marini, S.; Coletta, M. Human Matrix Metalloproteinases: An Ubiquitarian Class of Enzymes Involved in Several Pathological Processes. Mol. Asp. Med. 2012, 33, 119–208. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Grasso, G.; Gioia, M.; Coletta, A.; Polticelli, F.; Di Pierro, D.; Milardi, D.; Van Endert, P.; et al. Multiple Functions of Insulin-Degrading Enzyme: A Metabolic Crosslight? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 554–582. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, W.C.; Heinemann, M.A.; Kitabchi, A.E. Purification of Insulin-Specific Protease by Affinity Chromatography. Proc. Natl. Acad. Sci. USA 1972, 69, 3698–3702. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin Degradation: Progress and Potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar] [CrossRef]
- Affholter, J.A.; Fried, V.A.; Roth, R.A. Human Insulin-Degrading Enzyme Shares Structural and Functional Homologies with E. coli Protease III. Science 1988, 242, 1415–1418. [Google Scholar] [CrossRef]
- Fernández-Gamba, A.; Leal, M.C.; Morelli, L.; Castaño, E.M. Insulin-Degrading Enzyme: Structure-Function Relationship and Its Possible Roles in Health and Disease. Curr. Pharm. Des. 2009, 15, 3644–3655. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Belbin, O.; Zou, F.; Allen, M.; Ertekin-Taner, N.; Ansari, M.; Wilcox, S.L.; Kashino, M.R.; Ma, L.; Younkin, L.H.; et al. Concordant Association of Insulin Degrading Enzyme Gene (IDE) Variants with IDE mRNA, Abeta, and Alzheimer’s Disease. PLoS ONE 2010, 5, e8764. [Google Scholar] [CrossRef]
- Affholter, J.A.; Hsieh, C.L.; Francke, U.; Roth, R.A. Insulin-Degrading Enzyme: Stable Expression of the Human Complementary DNA, Characterization of Its Protein Product, and Chromosomal Mapping of the Human and Mouse Genes. Mol. Endocrinol. 1990, 4, 1125–1135. [Google Scholar] [CrossRef]
- Hersh, L.B. The Insulysin (Insulin Degrading Enzyme) Enigma. Cell. Mol. Life Sci. 2006, 63, 2432–2434. [Google Scholar] [CrossRef]
- Leissring, M.A.; Farris, W.; Wu, X.; Christodoulou, D.C.; Haigis, M.C.; Guarente, L.; Selkoe, D.J. Alternative Translation Initiation Generates a Novel Isoform of Insulin-Degrading Enzyme Targeted to Mitochondria. Biochem. J. 2004, 383, 439–446. [Google Scholar] [CrossRef]
- Morita, M.; Kurochkin, I.V.; Motojima, K.; Goto, S.; Takano, T.; Okamura, S.; Sato, R.; Yokota, S.; Imanaka, T. Insulin-Degrading Enzyme Exists inside of Rat Liver Peroxisomes and Degrades Oxidized Proteins. Cell Struct. Funct. 2000, 25, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-Degrading Enzyme Regulates Extracellular Levels of Amyloid Beta-Protein by Degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Folstein, M.F. Insulin, Insulin-Degrading Enzyme and Amyloid-Beta Peptide in Alzheimer’s Disease: Review and Hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Cha, M.-Y.; Choi, H.; Kang, S.; Choi, H.; Lee, M.-S.; Park, S.A.; Mook-Jung, I. Insulin-Degrading Enzyme Secretion from Astrocytes Is Mediated by an Autophagy-Based Unconventional Secretory Pathway in Alzheimer Disease. Autophagy 2016, 12, 784–800. [Google Scholar] [CrossRef] [PubMed]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. Insulin-Degrading Enzyme Is Not Secreted from Cultured Cells. Sci. Rep. 2018, 8, 2335. [Google Scholar] [CrossRef]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons Regulate Extracellular Levels of Amyloid Beta-Protein via Proteolysis by Insulin-Degrading Enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [CrossRef]
- Zhao, J.; Li, L.; Leissring, M.A. Insulin-Degrading Enzyme Is Exported via an Unconventional Protein Secretion Pathway. Mol. Neurodegener. 2009, 4, 4. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-Degrading Enzyme Regulates the Levels of Insulin, Amyloid Beta-Protein, and the Beta-Amyloid Precursor Protein Intracellular Domain in Vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin-Degrading Enzyme in the Fight against Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef]
- Bennett, R.G.; Hamel, F.G.; Duckworth, W.C. Insulin Inhibits the Ubiquitin-Dependent Degrading Activity of the 26S Proteasome. Endocrinology 2000, 141, 2508–2517. [Google Scholar] [CrossRef]
- Ciaccio, C.; Tundo, G.R.; Grasso, G.; Spoto, G.; Marasco, D.; Ruvo, M.; Gioia, M.; Rizzarelli, E.; Coletta, M. Somatostatin: A Novel Substrate and a Modulator of Insulin-Degrading Enzyme Activity. J. Mol. Biol. 2009, 385, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Lesire, L.; Leroux, F.; Deprez-Poulain, R.; Deprez, B. Insulin-Degrading Enzyme, an Under-Estimated Potential Target to Treat Cancer? Cells 2022, 11, 1228. [Google Scholar] [CrossRef] [PubMed]
- Tundo, G.; Ciaccio, C.; Sbardella, D.; Boraso, M.; Viviani, B.; Coletta, M.; Marini, S. Somatostatin Modulates Insulin-Degrading-Enzyme Metabolism: Implications for the Regulation of Microglia Activity in AD. PLoS ONE 2012, 7, e34376. [Google Scholar] [CrossRef]
- Grasso, G.; Lanza, V.; Malgieri, G.; Fattorusso, R.; Pietropaolo, A.; Rizzarelli, E.; Milardi, D. The Insulin Degrading Enzyme Activates Ubiquitin and Promotes the Formation of K48 and K63 Diubiquitin. Chem. Commun. 2015, 51, 15724–15727. [Google Scholar] [CrossRef]
- Sbardella, D.; Tundo, G.R.; Coletta, A.; Marcoux, J.; Koufogeorgou, E.I.; Ciaccio, C.; Santoro, A.M.; Milardi, D.; Grasso, G.; Cozza, P.; et al. The Insulin-Degrading Enzyme Is an Allosteric Modulator of the 20S Proteasome and a Potential Competitor of the 19S. Cell. Mol. Life Sci. 2018, 75, 3441–3456. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Li, Q.; Fischer, E.R.; Cohen, J.I. The Insulin Degrading Enzyme Binding Domain of Varicella-Zoster Virus (VZV) Glycoprotein E Is Important for Cell-to-Cell Spread and VZV Infectivity, While a Glycoprotein I Binding Domain Is Essential for Infection. Virology 2009, 386, 270–279. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Jackson, W.; de Souza, G.A.; Haarr, L.; Grose, C. Insulin-Degrading Enzyme Binds to the Nonglycosylated Precursor of Varicella-Zoster Virus gE Protein Found in the Endoplasmic Reticulum. J. Virol. 2010, 84, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ali, M.A.; Cohen, J.I. Insulin Degrading Enzyme Is a Cellular Receptor Mediating Varicella-Zoster Virus Infection and Cell-to-Cell Spread. Cell 2006, 127, 305–316. [Google Scholar] [CrossRef]
- Li, Q.; Ali, M.A.; Wang, K.; Sayre, D.; Hamel, F.G.; Fischer, E.R.; Bennett, R.G.; Cohen, J.I. Insulin Degrading Enzyme Induces a Conformational Change in Varicella-Zoster Virus gE, and Enhances Virus Infectivity and Stability. PLoS ONE 2010, 5, e11327. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The Proteasome as a Druggable Target with Multiple Therapeutic Potentialities: Cutting and Non-Cutting Edges. Pharmacol. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef]
- Mirsky, I.A.; Broh-Kahn, R.H. The Inactivation of Insulin by Tissue Extracts; the Distribution and Properties of Insulin Inactivating Extracts. Arch. Biochem. 1949, 20, 1–9. [Google Scholar]
- Brush, J.S. Purification and Characterization of a Protease with Specificity for Insulin from Rat Muscle. Diabetes 1971, 20, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, W.C.; Garcia, J.V.; Liepnieks, J.J.; Hamel, F.G.; Hermodson, M.A.; Frank, B.H.; Rosner, M.R. Drosophila Insulin Degrading Enzyme and Rat Skeletal Muscle Insulin Protease Cleave Insulin at Similar Sites. Biochemistry 1989, 28, 2471–2477. [Google Scholar] [CrossRef] [PubMed]
- Hamel, F.G.; Peavy, D.E.; Ryan, M.P.; Duckworth, W.C. High Performance Liquid Chromatographic Analysis of Insulin Degradation by Rat Skeletal Muscle Insulin Protease. Endocrinology 1986, 118, 328–333. [Google Scholar] [CrossRef]
- Shii, K.; Yokono, K.; Baba, S.; Roth, R.A. Purification and Characterization of Insulin-Degrading Enzyme from Human Erythrocytes. Diabetes 1986, 35, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Stentz, F.B.; Kitabchi, A.E.; Schilling, J.W.; Schronk, L.R.; Seyer, J.M. Identification of Insulin Intermediates and Sites of Cleavage of Native Insulin by Insulin Protease from Human Fibroblasts. J. Biol. Chem. 1989, 264, 20275–20282. [Google Scholar] [CrossRef]
- Burghen, G.A.; Kitabchi, A.E.; Brush, J.S. Characterization of a Rat Liver Protease with Specificity for Insulin. Endocrinology 1972, 91, 633–642. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Livingston, J.N. Insulin Degradation by Adipose Tissue. Studies at Several Levels of Cellular Organization. Biochem. J. 1980, 186, 351–360. [Google Scholar] [CrossRef]
- Stentz, F.B.; Harris, H.L.; Kitabchi, A.E. Characterization of Insulin-Degrading Activity of Intact and Subcellular Components of Human Fibroblasts. Endocrinology 1985, 116, 926–934. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Stentz, F.B.; Heinemann, M.; Kitabchi, A.E. Initial Site of Insulin Cleavage by Insulin Protease. Proc. Natl. Acad. Sci. USA 1979, 76, 635–639. [Google Scholar] [CrossRef]
- Leissring, M.A.; González-Casimiro, C.M.; Merino, B.; Suire, C.N.; Perdomo, G. Targeting Insulin-Degrading Enzyme in Insulin Clearance. Int. J. Mol. Sci. 2021, 22, 2235. [Google Scholar] [CrossRef]
- Blackard, W.G.; Ludeman, C.; Stillman, J. Role of Hepatocyte Plasma Membrane in Insulin Degradation. Am. J. Physiol. 1985, 248, E194–E202. [Google Scholar] [CrossRef]
- Grasso, G.; Rizzarelli, E.; Spoto, G. The Proteolytic Activity of Insulin-Degrading Enzyme: A Mass Spectrometry Study. J. Mass. Spectrom. 2009, 44, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Shii, K.; Roth, R.A. Inhibition of Insulin Degradation by Hepatoma Cells after Microinjection of Monoclonal Antibodies to a Specific Cytosolic Protease. Proc. Natl. Acad. Sci. USA 1986, 83, 4147–4151. [Google Scholar] [CrossRef] [PubMed]
- Peavy, D.E.; Hamel, F.G.; Kincke, V.L.; Duckworth, W.C. Evidence That Bacitracin Alters Intracellular Insulin Metabolism in Isolated Rat Hepatocytes. Diabetes 1985, 34, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, K.; Yokono, K.; Shii, K.; Hari, J.; Yaso, S.; Amano, K.; Sakamoto, T.; Kawase, Y.; Akiyama, H.; Nagata, M. Insulin-Degrading Enzyme Is Capable of Degrading Receptor-Bound Insulin. Biochem. Biophys. Res. Commun. 1988, 150, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Kagawa, S.; Tamura, T.; Shimbara, N.; Takashina, M.; Kristensen, P.; Hendil, K.B.; Tanaka, K.; Ichihara, A. Replacement of Proteasome Subunits X and Y by LMP7 and LMP2 Induced by Interferon-Gamma for Acquirement of the Functional Diversity Responsible for Antigen Processing. FEBS Lett. 1994, 343, 85–88. [Google Scholar] [CrossRef]
- Hamel, F.G.; Mahoney, M.J.; Duckworth, W.C. Degradation of Intraendosomal Insulin by Insulin-Degrading Enzyme without Acidification. Diabetes 1991, 40, 436–443. [Google Scholar] [CrossRef]
- Yokono, K.; Roth, R.A.; Baba, S. Identification of Insulin-Degrading Enzyme on the Surface of Cultured Human Lymphocytes, Rat Hepatoma Cells, and Primary Cultures of Rat Hepatocytes. Endocrinology 1982, 111, 1102–1108. [Google Scholar] [CrossRef]
- Bellia, F.; Pietropaolo, A.; Grasso, G. Formation of Insulin Fragments by Insulin-Degrading Enzyme: The Role of Zinc(II) and Cystine Bridges. J. Mass. Spectrom. 2013, 48, 135–140. [Google Scholar] [CrossRef]
- Grasso, G.; Satriano, C.; Milardi, D. A Neglected Modulator of Insulin-Degrading Enzyme Activity and Conformation: The pH. Biophys. Chem. 2015, 203–204, 33–40. [Google Scholar] [CrossRef]
- Authier, F.; Posner, B.I.; Bergeron, J.J. Insulin-Degrading Enzyme. Clin. Investig. Med. 1996, 19, 149–160. [Google Scholar]
- Authier, F.; Posner, B.I.; Bergeron, J.J. Endosomal Proteolysis of Internalized Proteins. FEBS Lett. 1996, 389, 55–60. [Google Scholar] [CrossRef]
- Clot, J.P.; Janicot, M.; Fouque, F.; Desbuquois, B.; Haumont, P.Y.; Lederer, F. Characterization of Insulin Degradation Products Generated in Liver Endosomes: In Vivo and in Vitro Studies. Mol. Cell. Endocrinol. 1990, 72, 175–185. [Google Scholar] [CrossRef]
- Hamel, F.G.; Posner, B.I.; Bergeron, J.J.; Frank, B.H.; Duckworth, W.C. Isolation of Insulin Degradation Products from Endosomes Derived from Intact Rat Liver. J. Biol. Chem. 1988, 263, 6703–6708. [Google Scholar] [CrossRef] [PubMed]
- Seabright, P.J.; Smith, G.D. The Characterization of Endosomal Insulin Degradation Intermediates and Their Sequence of Production. Biochem. J. 1996, 320 Pt 3, 947–956. [Google Scholar] [CrossRef]
- González-Casimiro, C.M.; Merino, B.; Casanueva-Álvarez, E.; Postigo-Casado, T.; Cámara-Torres, P.; Fernández-Díaz, C.M.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines 2021, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hay, S.O.; Kang, D.; McBride, M.; Li, L.; Zhao, J.; Leissring, M.A. Deletion of Insulin-Degrading Enzyme Elicits Antipodal, Age-Dependent Effects on Glucose and Insulin Tolerance. PLoS ONE 2011, 6, e20818. [Google Scholar] [CrossRef] [PubMed]
- Merino, B.; Fernández-Díaz, C.M.; Parrado-Fernández, C.; González-Casimiro, C.M.; Postigo-Casado, T.; Lobatón, C.D.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Hepatic Insulin-Degrading Enzyme Regulates Glucose and Insulin Homeostasis in Diet-Induced Obese Mice. Metab. Clin. Exp. 2020, 113, 154352. [Google Scholar] [CrossRef]
- Miller, B.C.; Eckman, E.A.; Sambamurti, K.; Dobbs, N.; Chow, K.M.; Eckman, C.B.; Hersh, L.B.; Thiele, D.L. Amyloid-Beta Peptide Levels in Brain are Inversely Correlated with Insulysin Activity Levels in Vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 6221–6226. [Google Scholar] [CrossRef]
- Steneberg, P.; Bernardo, L.; Edfalk, S.; Lundberg, L.; Backlund, F.; Ostenson, C.-G.; Edlund, H. The Type 2 Diabetes-Associated Gene Ide Is Required for Insulin Secretion and Suppression of α-Synuclein Levels in β-Cells. Diabetes 2013, 62, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Villa-Pérez, P.; Merino, B.; Fernández-Díaz, C.M.; Cidad, P.; Lobatón, C.D.; Moreno, A.; Muturi, H.T.; Ghadieh, H.E.; Najjar, S.M.; Leissring, M.A.; et al. Liver-Specific Ablation of Insulin-Degrading Enzyme Causes Hepatic Insulin Resistance and Glucose Intolerance, without Affecting Insulin Clearance in Mice. Metab. Clin. Exp. 2018, 88, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.; Guarda, M.; Meneses, M.J.; Macedo, M.P.; Vicente Miranda, H. Insulin-Degrading Enzyme: An Ally against Metabolic and Neurodegenerative Diseases. J. Pathol. 2021, 255, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, S.H.; Schwartz, S.S. Diabetic Retinopathy—An Underdiagnosed and Undertreated Inflammatory, Neuro-Vascular Complication of Diabetes. Front. Endocrinol. 2019, 10, 843. [Google Scholar] [CrossRef]
- Broh-Kahn, R.H.; Simkin, B.; Mirsky, I.A. The Inactivation of Insulin by Tissue Extracts; Changes in Insulin Sensitivity in Rabbits Induced by Previous Injections of Rat Liver Extracts. Arch. Biochem. 1950, 25, 157–167. [Google Scholar] [PubMed]
- Pivovarova, O.; Höhn, A.; Grune, T.; Pfeiffer, A.F.H.; Rudovich, N. Insulin-Degrading Enzyme: New Therapeutic Target for Diabetes and Alzheimer’s Disease? Ann. Med. 2016, 48, 614–624. [Google Scholar] [CrossRef]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-Degrading Enzyme as a Downstream Target of Insulin Receptor Signaling Cascade: Implications for Alzheimer’s Disease Intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef]
- Rezende, L.F.; Camargo, R.L.; Branco, R.C.S.; Cappelli, A.P.G.; Boschero, A.C.; Carneiro, E.M. Reduced Insulin Clearance and Lower Insulin-Degrading Enzyme Expression in the Liver Might Contribute to the Thrifty Phenotype of Protein-Restricted Mice. Br. J. Nutr. 2014, 112, 900–907. [Google Scholar] [CrossRef]
- Costes, S.; Butler, P.C. Insulin Degrading Enzyme Inhibition, a Novel Therapy for Type 2 Diabetes? Cell Metab. 2014, 20, 201–203. [Google Scholar] [CrossRef]
- Kullenberg, H.; Rossen, J.; Johansson, U.-B.; Hagströmer, M.; Nyström, T.; Kumlin, M.; Svedberg, M.M. Increased Levels of Insulin-Degrading Enzyme in Patients with Type 2 Diabetes Mellitus. Endocrine 2022, 77, 561–565. [Google Scholar] [CrossRef]
- Maianti, J.P.; Tan, G.A.; Vetere, A.; Welsh, A.J.; Wagner, B.K.; Seeliger, M.A.; Liu, D.R. Substrate-Selective Inhibitors that Reprogram the Activity of Insulin-Degrading Enzyme. Nat. Chem. Biol. 2019, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- González-Casimiro, C.M.; Cámara-Torres, P.; Merino, B.; Diez-Hermano, S.; Postigo-Casado, T.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Effects of Fasting and Feeding on Transcriptional and Posttranscriptional Regulation of Insulin-Degrading Enzyme in Mice. Cells 2021, 10, 2446. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kodamullil, A.T.; Hofmann-Apitius, M. Comorbidity Analysis between Alzheimer’s Disease and Type 2 Diabetes Mellitus (T2DM) Based on Shared Pathways and the Role of T2DM Drugs. J. Alzheimer’s Dis. 2017, 60, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.-J. Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2016, 27, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.A. Unraveling Alzheimer’s: Making Sense of the Relationship between Diabetes and Alzheimer’s Disease1. J. Alzheimer’s Dis. 2016, 51, 961–977. [Google Scholar] [CrossRef]
- Kuo, W.L.; Montag, A.G.; Rosner, M.R. Insulin-Degrading Enzyme Is Differentially Expressed and Developmentally Regulated in Various Rat Tissues. Endocrinology 1993, 132, 604–611. [Google Scholar] [CrossRef]
- Sánchez-Cruz, A.; Hernández-Fuentes, M.D.; Murillo-Gómez, C.; de la Rosa, E.J.; Hernández-Sánchez, C. Possible Role of Insulin-Degrading Enzyme in the Physiopathology of Retinitis Pigmentosa. Cells 2022, 11, 1621. [Google Scholar] [CrossRef]
- Kochkina, E.G.; Plesneva, S.A.; Vasilev, D.S.; Zhuravin, I.A.; Turner, A.J.; Nalivaeva, N.N. Effects of Ageing and Experimental Diabetes on Insulin-Degrading Enzyme Expression in Male Rat Tissues. Biogerontology 2015, 16, 473–484. [Google Scholar] [CrossRef]
- Press, M.; Jung, T.; König, J.; Grune, T.; Höhn, A. Protein Aggregates and Proteostasis in Aging: Amylin and β-Cell Function. Mech. Ageing Dev. 2019, 177, 46–54. [Google Scholar] [CrossRef]
- Crous-Bou, M.; Minguillón, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s Disease Prevention: From Risk Factors to Early Intervention. Alzheimers Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Koya, J.; Reznik, S.E. Insulin Resistance Exacerbates Alzheimer Disease via Multiple Mechanisms. Front. Neurosci. 2021, 15, 687157. [Google Scholar] [CrossRef]
- Walsh, D.M.; Tseng, B.P.; Rydel, R.E.; Podlisny, M.B.; Selkoe, D.J. The Oligomerization of Amyloid Beta-Protein Begins Intracellularly in Cells Derived from Human Brain. Biochemistry 2000, 39, 10831–10839. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G. The Use of Mass Spectrometry to Study Amyloid-β Peptides. Mass. Spectrom. Rev. 2011, 30, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Malito, E.; Hulse, R.E.; Tang, W.-J. Amyloid Beta-Degrading Cryptidases: Insulin Degrading Enzyme, Presequence Peptidase, and Neprilysin. Cell. Mol. Life Sci. 2008, 65, 2574–2585. [Google Scholar] [CrossRef]
- Portelius, E.; Brinkmalm, G.; Pannee, J.; Zetterberg, H.; Blennow, K.; Dahlén, R.; Brinkmalm, A.; Gobom, J. Proteomic Studies of Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease: An Update. Expert Rev. Proteom. 2017, 14, 1007–1020. [Google Scholar] [CrossRef]
- Stargardt, A.; Gillis, J.; Kamphuis, W.; Wiemhoefer, A.; Kooijman, L.; Raspe, M.; Benckhuijsen, W.; Drijfhout, J.W.; Hol, E.M.; Reits, E. Reduced Amyloid-β Degradation in Early Alzheimer’s Disease but Not in the APPswePS1dE9 and 3xTg-AD Mouse Models. Aging Cell 2013, 12, 499–507. [Google Scholar] [CrossRef]
- Miners, J.S.; Baig, S.; Palmer, J.; Palmer, L.E.; Kehoe, P.G.; Love, S. Abeta-Degrading Enzymes in Alzheimer’s Disease. Brain Pathol. 2008, 18, 240–252. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Ansorge, S.; Riederer, P.; Reiser, M.; Frölich, L.; Bogerts, B. Insulin-Degrading Enzyme in the Alzheimer’s Disease Brain: Prominent Localization in Neurons and Senile Plaques. Neurosci. Lett. 1999, 263, 161–164. [Google Scholar] [CrossRef]
- Leal, M.C.; Dorfman, V.B.; Gamba, A.F.; Frangione, B.; Wisniewski, T.; Castaño, E.M.; Sigurdsson, E.M.; Morelli, L. Plaque-Associated Overexpression of Insulin-Degrading Enzyme in the Cerebral Cortex of Aged Transgenic Tg2576 Mice with Alzheimer Pathology. J. Neuropathol. Exp. Neurol. 2006, 65, 976–987. [Google Scholar] [CrossRef]
- Shinall, H.; Song, E.S.; Hersh, L.B. Susceptibility of Amyloid Beta Peptide Degrading Enzymes to Oxidative Damage: A Potential Alzheimer’s Disease Spiral. Biochemistry 2005, 44, 15345–15350. [Google Scholar] [CrossRef] [PubMed]
- Vepsäläinen, S.; Hiltunen, M.; Helisalmi, S.; Wang, J.; van Groen, T.; Tanila, H.; Soininen, H. Increased Expression of Abeta Degrading Enzyme IDE in the Cortex of Transgenic Mice with Alzheimer’s Disease-like Neuropathology. Neurosci. Lett. 2008, 438, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Oddo, S.; Sugarman, M.C.; Akbari, Y.; LaFerla, F.M. Age- and Region-Dependent Alterations in Abeta-Degrading Enzymes: Implications for Abeta-Induced Disorders. Neurobiol. Aging 2005, 26, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.W.; Sanz-Blasco, S.; Dolatabadi, N.; Parker, J.; Chon, K.; Lee, M.S.; Soussou, W.; McKercher, S.R.; Ambasudhan, R.; Nakamura, T.; et al. Elevated Glucose and Oligomeric β-Amyloid Disrupt Synapses via a Common Pathway of Aberrant Protein S-Nitrosylation. Nat. Commun. 2016, 7, 10242. [Google Scholar] [CrossRef] [PubMed]
- Cordes, C.M.; Bennett, R.G.; Siford, G.L.; Hamel, F.G. Nitric Oxide Inhibits Insulin-Degrading Enzyme Activity and Function through S-Nitrosylation. Biochem. Pharmacol. 2009, 77, 1064–1073. [Google Scholar] [CrossRef]
- Kummer, M.P.; Hülsmann, C.; Hermes, M.; Axt, D.; Heneka, M.T. Nitric Oxide Decreases the Enzymatic Activity of Insulin Degrading Enzyme in APP/PS1 Mice. J. Neuroimmune Pharmacol. 2012, 7, 165–172. [Google Scholar] [CrossRef]
- Ralat, L.A.; Ren, M.; Schilling, A.B.; Tang, W.-J. Protective Role of Cys-178 against the Inactivation and Oligomerization of Human Insulin-Degrading Enzyme by Oxidation and Nitrosylation. J. Biol. Chem. 2009, 284, 34005–34018. [Google Scholar] [CrossRef]
- Björk, B.F.; Katzov, H.; Kehoe, P.; Fratiglioni, L.; Winblad, B.; Prince, J.A.; Graff, C. Positive Association between Risk for Late-Onset Alzheimer Disease and Genetic Variation in IDE. Neurobiol. Aging 2007, 28, 1374–1380. [Google Scholar] [CrossRef]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.-W.; Kovacina, K.S.; Craft, S. Reduced Hippocampal Insulin-Degrading Enzyme in Late-Onset Alzheimer’s Disease Is Associated with the Apolipoprotein E-Epsilon4 Allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- Wang, S.; He, F.; Wang, Y. Association between Polymorphisms of the Insulin-Degrading Enzyme Gene and Late-Onset Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2015, 28, 94–98. [Google Scholar] [CrossRef]
- Šerý, O.; Zeman, T.; Hálová, A.; Janout, V.; Janoutová, J.; Lochman, J.; Balcar, V.J. Polymorphism Rs2421943 of the Insulin-Degrading Enzyme Gene and the Risk of Late-Onset Alzheimer’s Disease. Curr. Alzheimer Res. 2022, 19, 236–245. [Google Scholar] [CrossRef]
- Diniz Pereira, J.; Gomes Fraga, V.; Morais Santos, A.L.; Carvalho, M.D.G.; Caramelli, P.; Braga Gomes, K. Alzheimer’s Disease and Type 2 Diabetes Mellitus: A Systematic Review of Proteomic Studies. J. Neurochem. 2021, 156, 753–776. [Google Scholar] [CrossRef] [PubMed]
- Kopf, D.; Frölich, L. Risk of Incident Alzheimer’s Disease in Diabetic Patients: A Systematic Review of Prospective Trials. J. Alzheimer’s Dis. 2009, 16, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, W. Molecular Links between Alzheimer’s Disease and Diabetes Mellitus. Neuroscience 2013, 250, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Isiegas, C.; Marinich-Madzarevich, J.A.; Marchena, M.; Ruiz, J.M.; Cano, M.J.; de la Villa, P.; Hernández-Sánchez, C.; de la Rosa, E.J.; de Pablo, F. Intravitreal Injection of Proinsulin-Loaded Microspheres Delays Photoreceptor Cell Death and Vision Loss in the Rd10 Mouse Model of Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3610–3618. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-Syndromic Retinitis Pigmentosa. Progress Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Barnea, E.R.; Almogi-Hazan, O.; Or, R.; Mueller, M.; Ria, F.; Weiss, L.; Paidas, M.J. Immune Regulatory and Neuroprotective Properties of Preimplantation Factor: From Newborn to Adult. Pharmacol. Ther. 2015, 156, 10–25. [Google Scholar] [CrossRef]
- Hayrabedyan, S.; Todorova, K.; Spinelli, M.; Barnea, E.R.; Mueller, M. The Core Sequence of PIF Competes for Insulin/Amyloid β in Insulin Degrading Enzyme: Potential Treatment for Alzheimer’s Disease. Oncotarget 2018, 9, 33884–33895. [Google Scholar] [CrossRef]
- Tullio, M.B.; Castelletto, V.; Hamley, I.W.; Martino Adami, P.V.; Morelli, L.; Castaño, E.M. Proteolytically Inactive Insulin-Degrading Enzyme Inhibits Amyloid Formation Yielding Non-Neurotoxic Aβ Peptide Aggregates. PLoS ONE 2013, 8, e59113. [Google Scholar] [CrossRef]
- Sharma, S.K.; Chorell, E.; Steneberg, P.; Vernersson-Lindahl, E.; Edlund, H.; Wittung-Stafshede, P. Insulin-Degrading Enzyme Prevents α-Synuclein Fibril Formation in a Nonproteolytical Manner. Sci. Rep. 2015, 5, 12531. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chorell, E.; Wittung-Stafshede, P. Insulin-Degrading Enzyme Is Activated by the C-Terminus of α-Synuclein. Biochem. Biophys. Res. Commun. 2015, 466, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Tullio, M.B.; Morelli, L.; Castaño, E.M. The Irreversible Binding of Amyloid Peptide Substrates to Insulin-Degrading Enzyme. Prion 2008, 2, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Kurochkin, I.V. Insulin-Degrading Enzyme: Embarking on Amyloid Destruction. Trends Biochem. Sci. 2001, 26, 421–425. [Google Scholar] [CrossRef]
- Ramaraju, B.; Nelson, S.L.; Zheng, W.; Ghirlando, R.; Deshmukh, L. Quantitative NMR Study of Insulin-Degrading Enzyme Using Amyloid-β and HIV-1 P6 Elucidates Its Chaperone Activity. Biochemistry 2021, 60, 2519–2523. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, D.; Tundo, G.R.; Sciandra, F.; Bozzi, M.; Gioia, M.; Ciaccio, C.; Tarantino, U.; Brancaccio, A.; Coletta, M.; Marini, S. Proteasome Activity Is Affected by Fluctuations in Insulin-Degrading Enzyme Distribution. PLoS ONE 2015, 10, e0132455. [Google Scholar] [CrossRef] [PubMed]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Bianculli, A.; Orlandi, A.; Desimio, M.G.; Arcuri, G.; Coletta, M.; Marini, S. Insulin-Degrading Enzyme (IDE): A Novel Heat Shock-like Protein. J. Biol. Chem. 2013, 288, 2281–2289. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Oddone, F.; Kudriaeva, A.A.; Lacal, P.M.; Belogurov, A.A.; Graziani, G.; Marini, S. At the Cutting Edge against Cancer: A Perspective on Immunoproteasome and Immune Checkpoints Modulation as a Potential Therapeutic Intervention. Cancers 2021, 13, 4852. [Google Scholar] [CrossRef]
- Tundo, G.R.; Cascio, P.; Milardi, D.; Santoro, A.M.; Graziani, G.; Lacal, P.M.; Bocedi, A.; Oddone, F.; Parravano, M.; Coletta, A.; et al. Targeting Immunoproteasome in Neurodegeneration: A Glance to the Future. Pharmacol. Ther. 2023, 241, 108329. [Google Scholar] [CrossRef]
- Leestemaker, Y.; Ovaa, H. Tools to Investigate the Ubiquitin Proteasome System. Drug Discov. Today Technol. 2017, 26, 25–31. [Google Scholar] [CrossRef]
- Shen, Y.; Joachimiak, A.; Rosner, M.R.; Tang, W.-J. Structures of Human Insulin-Degrading Enzyme Reveal a New Substrate Recognition Mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Selkoe, D.J. Structural Biology: Enzyme Target to Latch on To. Nature 2006, 443, 761–762. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kuo, W.-L.; Yousef, M.; Rosner, M.R.; Tang, W.-J. The C-Terminal Domain of Human Insulin Degrading Enzyme Is Required for Dimerization and Substrate Recognition. Biochem. Biophys. Res. Commun. 2006, 343, 1032–1037. [Google Scholar] [CrossRef]
- Perlman, R.K.; Gehm, B.D.; Kuo, W.L.; Rosner, M.R. Functional Analysis of Conserved Residues in the Active Site of Insulin-Degrading Enzyme. J. Biol. Chem. 1993, 268, 21538–21544. [Google Scholar] [CrossRef]
- Abramov-Harpaz, K.; Miller, Y. Insights into Non-Proteolytic Inhibitory Mechanisms of Polymorphic Early-Stage Amyloid β Oligomers by Insulin Degrading Enzyme. Biomolecules 2022, 12, 1886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liang, W.G.; Bailey, L.J.; Tan, Y.Z.; Wei, H.; Wang, A.; Farcasanu, M.; Woods, V.A.; McCord, L.A.; Lee, D.; et al. Ensemble cryoEM Elucidates the Mechanism of Insulin Capture and Degradation by Human Insulin Degrading Enzyme. eLife 2018, 7, e33572. [Google Scholar] [CrossRef]
- Noinaj, N.; Bhasin, S.K.; Song, E.S.; Scoggin, K.E.; Juliano, M.A.; Juliano, L.; Hersh, L.B.; Rodgers, D.W. Identification of the Allosteric Regulatory Site of Insulysin. PLoS ONE 2011, 6, e20864. [Google Scholar] [CrossRef]
- McCord, L.A.; Liang, W.G.; Dowdell, E.; Kalas, V.; Hoey, R.J.; Koide, A.; Koide, S.; Tang, W.-J. Conformational States and Recognition of Amyloidogenic Peptides of Human Insulin-Degrading Enzyme. Proc. Natl. Acad. Sci. USA 2013, 110, 13827–13832. [Google Scholar] [CrossRef]
- Noinaj, N.; Song, E.S.; Bhasin, S.; Alper, B.J.; Schmidt, W.K.; Hersh, L.B.; Rodgers, D.W. Anion Activation Site of Insulin-Degrading Enzyme. J. Biol. Chem. 2012, 287, 48–57. [Google Scholar] [CrossRef]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. A Monomeric Variant of Insulin Degrading Enzyme (IDE) Loses Its Regulatory Properties. PLoS ONE 2010, 5, e9719. [Google Scholar] [CrossRef]
- Bennett, R.G.; Duckworth, W.C.; Hamel, F.G. Degradation of Amylin by Insulin-Degrading Enzyme. J. Biol. Chem. 2000, 275, 36621–36625. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Mielczarek, P.; Niedziolka, M.; Silberring, J. Metabolism of Cryptic Peptides Derived from Neuropeptide FF Precursors: The Involvement of Insulin-Degrading Enzyme. Int. J. Mol. Sci. 2014, 15, 16787–16799. [Google Scholar] [CrossRef]
- Stefanidis, L.; Fusco, N.D.; Cooper, S.E.; Smith-Carpenter, J.E.; Alper, B.J. Molecular Determinants of Substrate Specificity in Human Insulin-Degrading Enzyme. Biochemistry 2018, 57, 4903–4914. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Schulze, C.; Baumeister, H.; Buck, F.; Richter, D. Rat Insulin-Degrading Enzyme: Cleavage Pattern of the Natriuretic Peptide Hormones ANP, BNP, and CNP Revealed by HPLC and Mass Spectrometry. Biochemistry 1992, 31, 11138–11143. [Google Scholar] [CrossRef]
- Im, H.; Manolopoulou, M.; Malito, E.; Shen, Y.; Zhao, J.; Neant-Fery, M.; Sun, C.-Y.; Meredith, S.C.; Sisodia, S.S.; Leissring, M.A.; et al. Structure of Substrate-Free Human Insulin-Degrading Enzyme (IDE) and Biophysical Analysis of ATP-Induced Conformational Switch of IDE. J. Biol. Chem. 2007, 282, 25453–25463. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Chang, T.; Saghatelian, A. Substrate-Selective Enzyme Inhibitors. Trends Pharmacol. Sci. 2019, 40, 716–718. [Google Scholar] [CrossRef]
- Durham, T.B.; Toth, J.L.; Klimkowski, V.J.; Cao, J.X.C.; Siesky, A.M.; Alexander-Chacko, J.; Wu, G.Y.; Dixon, J.T.; McGee, J.E.; Wang, Y.; et al. Dual Exosite-Binding Inhibitors of Insulin-Degrading Enzyme Challenge Its Role as the Primary Mediator of Insulin Clearance in Vivo. J. Biol. Chem. 2015, 290, 20044–20059. [Google Scholar] [CrossRef]
- Guo, Q.; Manolopoulou, M.; Bian, Y.; Schilling, A.B.; Tang, W.-J. Molecular Basis for the Recognition and Cleavages of IGF-II, TGF-Alpha, and Amylin by Human Insulin-Degrading Enzyme. J. Mol. Biol. 2010, 395, 430–443. [Google Scholar] [CrossRef]
- Ren, M.; Guo, Q.; Guo, L.; Lenz, M.; Qian, F.; Koenen, R.R.; Xu, H.; Schilling, A.B.; Weber, C.; Ye, R.D.; et al. Polymerization of MIP-1 Chemokine (CCL3 and CCL4) and Clearance of MIP-1 by Insulin-Degrading Enzyme. EMBO J. 2010, 29, 3952–3966. [Google Scholar] [CrossRef]
- Ralat, L.A.; Kalas, V.; Zheng, Z.; Goldman, R.D.; Sosnick, T.R.; Tang, W.-J. Ubiquitin Is a Novel Substrate for Human Insulin-Degrading Enzyme. J. Mol. Biol. 2011, 406, 454–466. [Google Scholar] [CrossRef]
- Grasso, G.; Rizzarelli, E.; Spoto, G. How the Binding and Degrading Capabilities of Insulin Degrading Enzyme Are Affected by Ubiquitin. Biochim. Biophys. Acta 2008, 1784, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Loison, L. Monod before Monod: Enzymatic Adaptation, Lwoff, and the Legacy of General Biology. Hist. Philos. Life Sci. 2013, 35, 167–192. [Google Scholar] [PubMed]
- Monod, J.; Wyman, J.; Changeux, J.P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef] [PubMed]
- Song, E.-S.; Juliano, M.A.; Juliano, L.; Hersh, L.B. Substrate Activation of Insulin-Degrading Enzyme (Insulysin). A Potential Target for Drug Development. J. Biol. Chem. 2003, 278, 49789–49794. [Google Scholar] [CrossRef]
- Tundo, G.R.; Di Muzio, E.; Ciaccio, C.; Sbardella, D.; Di Pierro, D.; Polticelli, F.; Coletta, M.; Marini, S. Multiple Allosteric Sites Are Involved in the Modulation of Insulin-Degrading-Enzyme Activity by Somatostatin. FEBS J. 2016, 283, 3755–3770. [Google Scholar] [CrossRef]
- Camberos, M.C.; Pérez, A.A.; Udrisar, D.P.; Wanderley, M.I.; Cresto, J.C. ATP Inhibits Insulin-Degrading Enzyme Activity. Exp. Biol. Med. 2001, 226, 334–341. [Google Scholar] [CrossRef]
- Del Carmen Camberos, M.; Cresto, J.C. Insulin-Degrading Enzyme Hydrolyzes ATP. Exp. Biol. Med. 2007, 232, 281–292. [Google Scholar]
- Song, E.S.; Juliano, M.A.; Juliano, L.; Fried, M.G.; Wagner, S.L.; Hersh, L.B. ATP Effects on Insulin-Degrading Enzyme Are Mediated Primarily through Its Triphosphate Moiety. J. Biol. Chem. 2004, 279, 54216–54220. [Google Scholar] [CrossRef]
- Cabrol, C.; Huzarska, M.A.; Dinolfo, C.; Rodriguez, M.C.; Reinstatler, L.; Ni, J.; Yeh, L.-A.; Cuny, G.D.; Stein, R.L.; Selkoe, D.J.; et al. Small-Molecule Activators of Insulin-Degrading Enzyme Discovered through High-Throughput Compound Screening. PLoS ONE 2009, 4, e5274. [Google Scholar] [CrossRef]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. Mixed Dimers of Insulin-Degrading Enzyme Reveal a Cis Activation Mechanism. J. Biol. Chem. 2011, 286, 13852–13858. [Google Scholar] [CrossRef]
- Cruz, C.H.B.; Seabra, G. Molecular Dynamics Simulations Reveal a Novel Mechanism for ATP Inhibition of Insulin Degrading Enzyme. J. Chem. Inf. Model. 2014, 54, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Song, E.S.; Ozbil, M.; Zhang, T.; Sheetz, M.; Lee, D.; Tran, D.; Li, S.; Prabhakar, R.; Hersh, L.B.; Rodgers, D.W. An Extended Polyanion Activation Surface in Insulin Degrading Enzyme. PLoS ONE 2015, 10, e0133114. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Giuffrida, M.L.; Rizzarelli, E. Metallostasis and Amyloid β-Degrading Enzymes. Metallomics 2012, 4, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Pietropaolo, A.; Spoto, G.; Pappalardo, G.; Tundo, G.R.; Ciaccio, C.; Coletta, M.; Rizzarelli, E. Copper(I) and Copper(II) Inhibit Aβ Peptides Proteolysis by Insulin-Degrading Enzyme Differently: Implications for Metallostasis Alteration in Alzheimer’s Disease. Chemistry 2011, 17, 2752–2762. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Salomone, F.; Tundo, G.R.; Pappalardo, G.; Ciaccio, C.; Spoto, G.; Pietropaolo, A.; Coletta, M.; Rizzarelli, E. Metal Ions Affect Insulin-Degrading Enzyme Activity. J. Inorg. Biochem. 2012, 117, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hay, S.O.; Bannister, T.D.; Wang, H.; Cameron, M.D.; Caulfield, T.R.; Masson, A.; Bertrand, J.; Howard, E.A.; McGuire, M.P.; Crisafulli, U.; et al. Selective Targeting of Extracellular Insulin-Degrading Enzyme by Quasi-Irreversible Thiol-Modifying Inhibitors. ACS Chem. Biol. 2015, 10, 2716–2724. [Google Scholar] [CrossRef]
- Leissring, M.A.; Malito, E.; Hedouin, S.; Reinstatler, L.; Sahara, T.; Abdul-Hay, S.O.; Choudhry, S.; Maharvi, G.M.; Fauq, A.H.; Huzarska, M.; et al. Designed Inhibitors of Insulin-Degrading Enzyme Regulate the Catabolism and Activity of Insulin. PLoS ONE 2010, 5, e10504. [Google Scholar] [CrossRef]
- Abdul-Hay, S.O.; Lane, A.L.; Caulfield, T.R.; Claussin, C.; Bertrand, J.; Masson, A.; Choudhry, S.; Fauq, A.H.; Maharvi, G.M.; Leissring, M.A. Optimization of Peptide Hydroxamate Inhibitors of Insulin-Degrading Enzyme Reveals Marked Substrate-Selectivity. J. Med. Chem. 2013, 56, 2246–2255. [Google Scholar] [CrossRef]
- Distefano, A.; Caruso, G.; Oliveri, V.; Bellia, F.; Sbardella, D.; Zingale, G.A.; Caraci, F.; Grasso, G. Neuroprotective Effect of Carnosine Is Mediated by Insulin-Degrading Enzyme. ACS Chem. Neurosci. 2022, 13, 1588–1593. [Google Scholar] [CrossRef]
- Adamek, R.N.; Suire, C.N.; Stokes, R.W.; Brizuela, M.K.; Cohen, S.M.; Leissring, M.A. Hydroxypyridinethione Inhibitors of Human Insulin-Degrading Enzyme. ChemMedChem 2021, 16, 1775–1787. [Google Scholar] [CrossRef]
- Maianti, J.P.; McFedries, A.; Foda, Z.H.; Kleiner, R.E.; Du, X.Q.; Leissring, M.A.; Tang, W.-J.; Charron, M.J.; Seeliger, M.A.; Saghatelian, A.; et al. Anti-Diabetic Activity of Insulin-Degrading Enzyme Inhibitors Mediated by Multiple Hormones. Nature 2014, 511, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Deprez-Poulain, R.; Hennuyer, N.; Bosc, D.; Liang, W.G.; Enée, E.; Marechal, X.; Charton, J.; Totobenazara, J.; Berte, G.; Jahklal, J.; et al. Catalytic Site Inhibition of Insulin-Degrading Enzyme by a Small Molecule Induces Glucose Intolerance in Mice. Nat. Commun. 2015, 6, 8250. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.S.; Wahiduzzaman, M.; Reyad-Ul-Ferdous, M.; Islam, M.N.; Roy, M. Inhibition of Insulin Degrading Enzyme to Control Diabetes Mellitus and Its Applications on Some Other Chronic Disease: A Critical Review. Pharm. Res. 2022, 39, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Kukday, S.S.; Manandhar, S.P.; Ludley, M.C.; Burriss, M.E.; Alper, B.J.; Schmidt, W.K. Cell-Permeable, Small-Molecule Activators of the Insulin-Degrading Enzyme. J. Biomol. Screen. 2012, 17, 1348–1361. [Google Scholar] [CrossRef]
- Krasinski, C.A.; Ivancic, V.A.; Zheng, Q.; Spratt, D.E.; Lazo, N.D. Resveratrol Sustains Insulin-Degrading Enzyme Activity toward Aβ42. ACS Omega 2018, 3, 13275–13282. [Google Scholar] [CrossRef]
- Grasso, G.; Bush, A.I.; D’Agata, R.; Rizzarelli, E.; Spoto, G. Enzyme Solid-State Support Assays: A Surface Plasmon Resonance and Mass Spectrometry Coupled Study of Immobilized Insulin Degrading Enzyme. Eur. Biophys. J. 2009, 38, 407–414. [Google Scholar] [CrossRef]
- Adessi, C.; Enderle, T.; Grueninger, F.; Roth, D. Activator for Insulin Degrading Enzyme. US Patent WO/2006/066847, 29 June 2006. [Google Scholar]
- Charton, J.; Gauriot, M.; Guo, Q.; Hennuyer, N.; Marechal, X.; Dumont, J.; Hamdane, M.; Pottiez, V.; Landry, V.; Sperandio, O.; et al. Imidazole-Derived 2-[N-Carbamoylmethyl-Alkylamino]Acetic Acids, Substrate-Dependent Modulators of Insulin-Degrading Enzyme in Amyloid-β Hydrolysis. Eur. J. Med. Chem. 2014, 79, 184–193. [Google Scholar] [CrossRef]
- Kraupner, N.; Dinh, C.P.; Wen, X.; Landry, V.; Herledan, A.; Leroux, F.; Bosc, D.; Charton, J.; Maillard, C.; Warenghem, S.; et al. Identification of Indole-Based Activators of Insulin Degrading Enzyme. Eur. J. Med. Chem. 2022, 228, 113982. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tundo, G.R.; Grasso, G.; Persico, M.; Tkachuk, O.; Bellia, F.; Bocedi, A.; Marini, S.; Parravano, M.; Graziani, G.; Fattorusso, C.; et al. The Insulin-Degrading Enzyme from Structure to Allosteric Modulation: New Perspectives for Drug Design. Biomolecules 2023, 13, 1492. https://doi.org/10.3390/biom13101492
Tundo GR, Grasso G, Persico M, Tkachuk O, Bellia F, Bocedi A, Marini S, Parravano M, Graziani G, Fattorusso C, et al. The Insulin-Degrading Enzyme from Structure to Allosteric Modulation: New Perspectives for Drug Design. Biomolecules. 2023; 13(10):1492. https://doi.org/10.3390/biom13101492
Chicago/Turabian StyleTundo, Grazia Raffaella, Giuseppe Grasso, Marco Persico, Oleh Tkachuk, Francesco Bellia, Alessio Bocedi, Stefano Marini, Mariacristina Parravano, Grazia Graziani, Caterina Fattorusso, and et al. 2023. "The Insulin-Degrading Enzyme from Structure to Allosteric Modulation: New Perspectives for Drug Design" Biomolecules 13, no. 10: 1492. https://doi.org/10.3390/biom13101492
APA StyleTundo, G. R., Grasso, G., Persico, M., Tkachuk, O., Bellia, F., Bocedi, A., Marini, S., Parravano, M., Graziani, G., Fattorusso, C., & Sbardella, D. (2023). The Insulin-Degrading Enzyme from Structure to Allosteric Modulation: New Perspectives for Drug Design. Biomolecules, 13(10), 1492. https://doi.org/10.3390/biom13101492