
Analysis of Structural Changes in the Protein near the Phosphorylation Site

 ,
,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Study Design
- -
- The amino acid sequence of the protein must include at least 15 residues (excluding small peptides);
- -
- The protein pair must have a common unique Uniprot ID (excluding comparisons with homologous proteins from different organisms in the intact/modified structure pair);
- -
- The intact form of the pair should not contain any non-canonical amino acid residues (unless the proteins containing non-canonical amino acid variants are matched to the intact protein structure);
- -
- The length of the proteins in a pair should not differ by more than five amino acid residues (excluding comparisons between proteins with significantly different lengths in the intact/modified structure pair);
- -
- The amino acid sequence of the proteins (FASTA) in a pair should match at least 90% (selecting identical sequences in the intact/modified structure pair);
- -
- If several structures are found in the PDB for the modified form, the three-dimensional structure with the lowest RMSD, regarding the intact protein, is selected (reducing the number of comparison variants);
- -
- The number of phosphate groups in the modified form should be ≤3;
- -
- Each protein chain in one crystal with a modifying group is considered a separate structure;
- -
- Structure is identified by a unique combination of UniProt ID and PTM locus (to avoid redundancy in the dataset).
2.2. Calculating Geometric Indicators
- -
- Before calculating the geometric parameters, all the atoms not included in the structure of polymer chains were removed from the structures;
- -
- Hydrogen atoms were removed and then added using PyMol (h_add command) to avoid the error caused by the possible presence of hydrogen atoms in one of the compared structures while being absent in the other one.
- root-mean square distance between Cα-atoms in the intact and modified protein forms (RMSD, Å);
- solvent-accessible surface area (SASA, Å2);
- radius of gyration (Rg, nm);
- displacement between Cα-atoms in individual amino acid residues (Cα displacement, Å).
2.3. Analysis of Geometric Indicators
3. Results
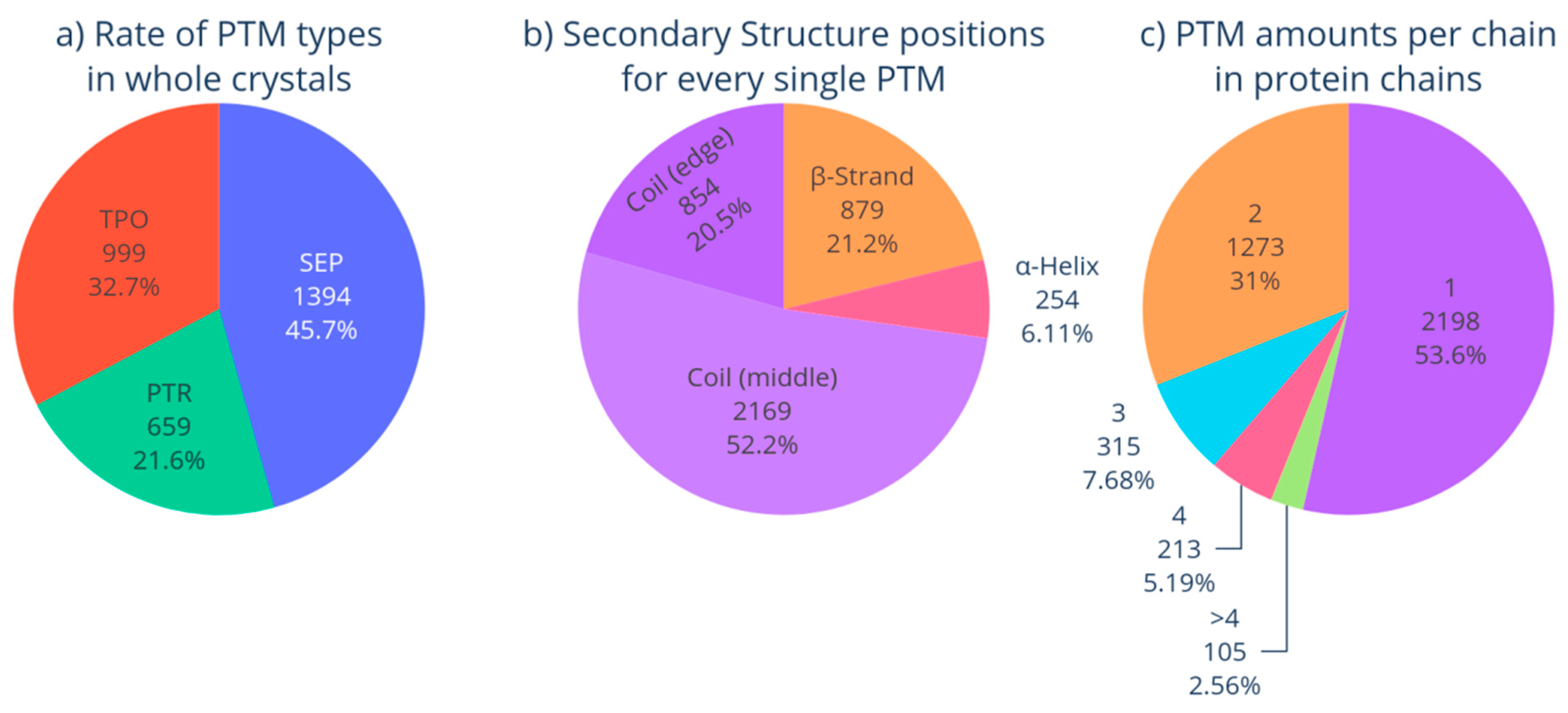
3.1. Phosphorylated Proteins in PDB
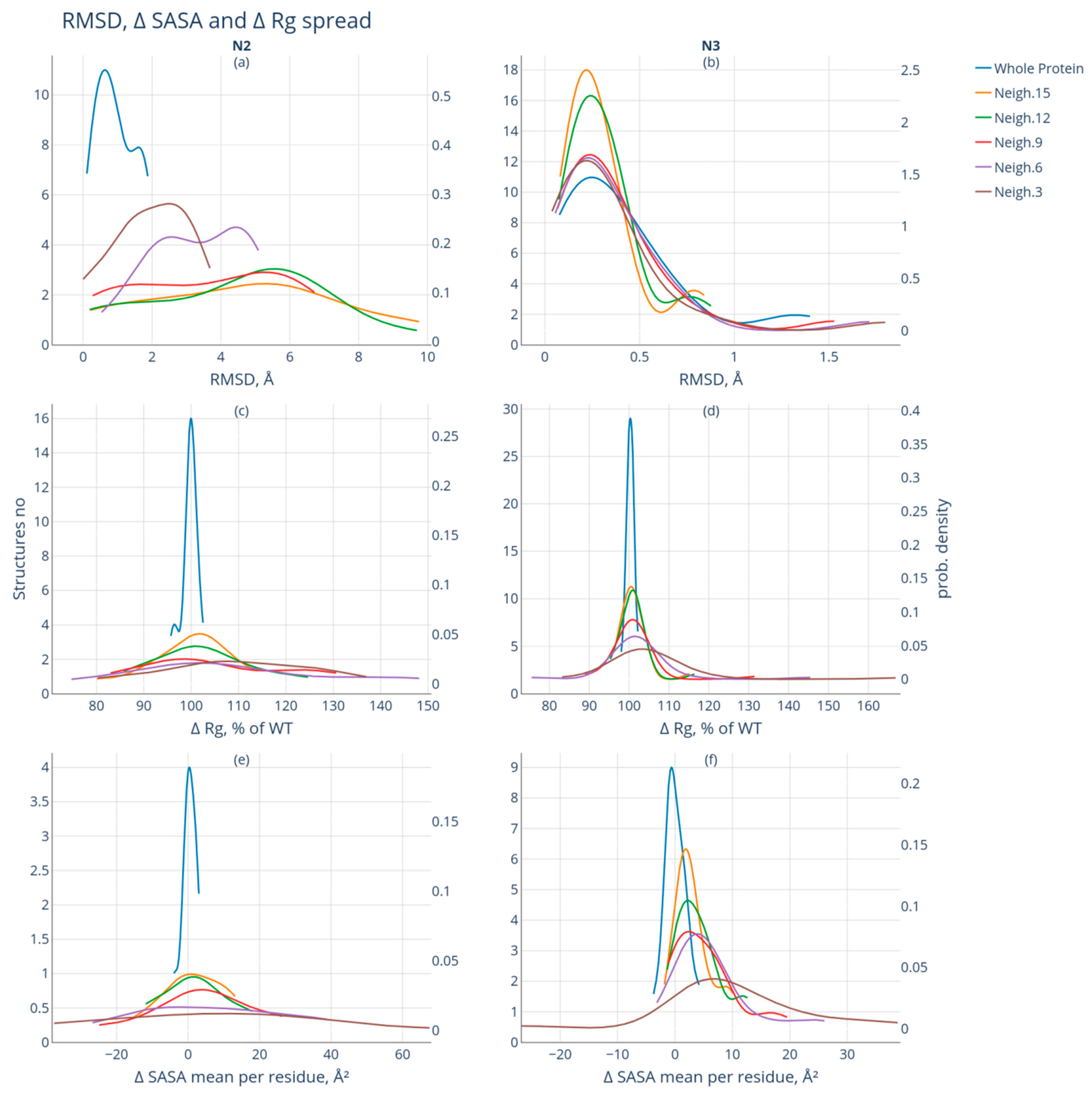
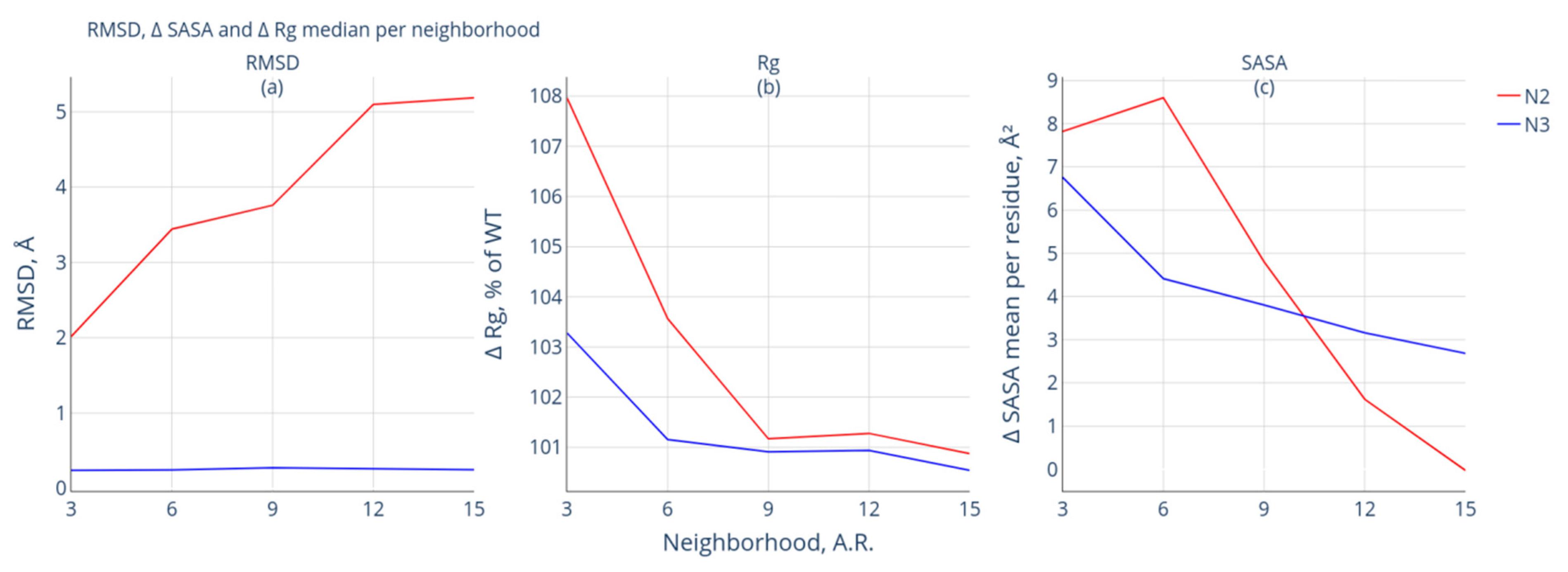
3.2. Comparative Analysis of the Geometry of Intact and Modified Protein Forms
- -
- Group N1 includes protein pairs that are characterized by RMSD > 2Å. Due to significant differences, it is impossible to compare these forms of proteins (see Supplementary Materials Table S2—proteins with RMSD > 2 Å). Fifteen such protein pairs were found. This group of protein pairs was excluded from further consideration;
- -
- Group N2 includes protein pairs of high similarity for which the RMSD value is <2 Å. The protein pairs of this group are characterized by significant spatial differences in the neighborhood of the modification site: the RMSD value of at least one of the studied neighborhoods of the modification site exceeds 2 Å. Nineteen such protein pairs were selected;
- -
- Group N3 also includes protein pairs of high similarity with RMSD <2 Å. The neighborhoods of the modification site are also characterized by high similarity: the calculated RMSD values do not exceed 2 Å for the intact and modified forms. This group included 29 protein pairs.
4. Discussion
5. Limitations
- -
- Filtering without considering differences in structure length with alignment based on the smaller structure;
- -
- Selecting modified and intact protein forms based on sequence homology without considering UniProt ID;
- -
- Disregarding the presence of amino acid substitutions and non-canonical amino acids outside the binding site;
- -
- Considering ligands present in the structures;
- -
- Identifying gaps in the amino acid sequence;
- -
- Selecting protein pairs where both structures were obtained using the same experimental method (X-Ray, NMR, or EM);
- -
- Averaging atomic positions across all three-dimensional structures of a given protein;
- -
- Analyzing local and global structural changes in a protein while considering b-factor values instead of RMSD.
6. Conclusions
- -
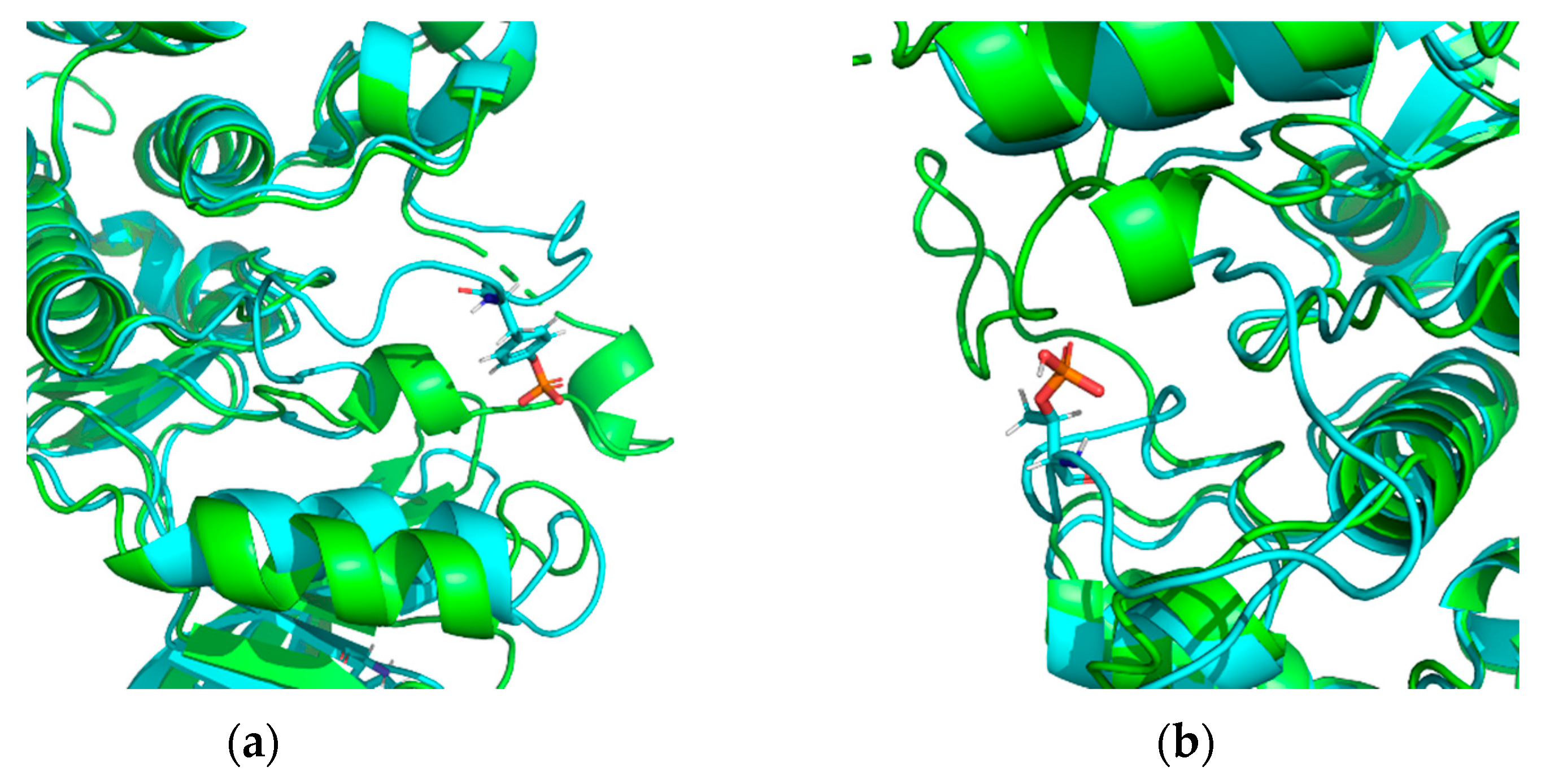
- unweaving or stretching of the α-helix turn, changes in β-strand length, and, therefore, changes in length and conformation of the unstructured region;
- -
- increased Rg values in the studied neighborhood;
- -
- significant surge in Cα displacement values for amino acid residues located near the modification site;
- -
- the calculated RMSD values for the neighborhood of the modification site exceed those for the whole protein.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quezada, H.; Guzmán-Ortiz, A.L.; Díaz-Sánchez, H.; Valle-Rios, R.; Aguirre-Hernández, J. Omics-Based Biomarkers: Current Status and Potential Use in the Clinic. Bol. Méd. Hosp. Infant. México Engl. Ed. 2017, 74, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.M.; Albrecht, V.; Mann, M. MS-Based Proteomics of Body Fluids: The End of the Beginning. Mol. Cell. Proteomics 2023, 22, 100577. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Sahu, M.; Srivastava, D.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Post-Translational Modifications: Regulators of Neurodegenerative Proteinopathies. Ageing Res. Rev. 2021, 68, 101336. [Google Scholar] [CrossRef]
- Shafi, S.; Singh, A.; Gupta, P.; Chawla, P.A.; Fayaz, F.; Sharma, A.; Pottoo, F.H. Deciphering the Role of Aberrant Protein Post-Translational Modification in the Pathology of Neurodegeneration. CNS Neurol. Disord. Drug Targets 2021, 20, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Q.; Kang, Y.; Xu, S.; Pang, D. Unconventional Protein Post-Translational Modifications: The Helmsmen in Breast Cancer. Cell Biosci. 2022, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Audagnotto, M.; Dal Peraro, M. Protein Post-Translational Modifications: In Silico Prediction Tools and Molecular Modeling. Comput. Struct. Biotechnol. J. 2017, 15, 307–319. [Google Scholar] [CrossRef]
- Zhong, Q.; Xiao, X.; Qiu, Y.; Xu, Z.; Chen, C.; Chong, B.; Zhao, X.; Hai, S.; Li, S.; An, Z.; et al. Protein Posttranslational Modifications in Health and Diseases: Functions, Regulatory Mechanisms, and Therapeutic Implications. MedComm 2023, 4, e261. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-Translational Modifications in Proteins: Resources, Tools and Prediction Methods. Database J. Biol. Databases Curation 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Doyle, H.A.; Mamula, M.J. Autoantigenesis: The Evolution of Protein Modifications in Autoimmune Disease. Curr. Opin. Immunol. 2012, 24, 112–118. [Google Scholar] [CrossRef]
- Mastrangelo, A.; Colasanti, T.; Barbati, C.; Pecani, A.; Sabatinelli, D.; Pendolino, M.; Truglia, S.; Massaro, L.; Mancini, R.; Miranda, F.; et al. The Role of Posttranslational Protein Modifications in Rheumatological Diseases: Focus on Rheumatoid Arthritis. J. Immunol. Res. 2015, 2015, 712490. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Guadagnin, G.; Cappello, P.; Novelli, F. Post-Translational Modifications in Tumor-Associated Antigens as a Platform for Novel Immuno-Oncology Therapies. Cancers 2022, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, L.-N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Grubisha, M.J.; Sweet, R.A.; MacDonald, M.L. Investigating Post-Translational Modifications in Neuropsychiatric Disease: The Next Frontier in Human Post-Mortem Brain Research. Front. Mol. Neurosci. 2021, 14, 689495. [Google Scholar] [CrossRef]
- Dai, X.; Zhang, J.; North, B.J.; Guo, J. Editorial: Post-Translational Modifications of Proteins in Cancer Immunity and Immunotherapy. Front. Immunol. 2022, 13, 1006145. [Google Scholar] [CrossRef]
- Pakhrin, S.C.; Pokharel, S.; Saigo, H.; Kc, D.B. Deep Learning-Based Advances In Protein Posttranslational Modification Site and Protein Cleavage Prediction. In Computational Methods for Predicting Post-Translational Modification Sites. Methods in Molecular Biology; Humana: New York, NY, USA, 2022; Volume 2499, pp. 285–322. [Google Scholar] [CrossRef]
- Sharma, B.S.; Prabhakaran, V.; Desai, A.P.; Bajpai, J.; Verma, R.J.; Swain, P.K. Post-Translational Modifications (PTMs), from a Cancer Perspective: An Overview. Oncogen 2019, 2, 12. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Lin, S.; Deng, W.; Peng, D.; Cui, Q.; Xue, Y. PTMD: A Database of Human Disease-Associated Post-Translational Modifications. Genomics Proteomics Bioinformatics 2018, 16, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 Years of PhosphoSitePlus®: Integrating Post-Translationally Modified Sites, Disease Variants and Isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef]
- Li, Z.; Li, S.; Luo, M.; Jhong, J.-H.; Li, W.; Yao, L.; Pang, Y.; Wang, Z.; Wang, R.; Ma, R.; et al. dbPTM in 2022: An Updated Database for Exploring Regulatory Networks and Functional Associations of Protein Post-Translational Modifications. Nucleic Acids Res. 2022, 50, D471–D479. [Google Scholar] [CrossRef]
- Müller, M.M. Post-Translational Modifications of Protein Backbones: Unique Functions, Mechanisms, and Challenges. Biochemistry 2018, 57, 177–185. [Google Scholar] [CrossRef]
- Lee, J.M.; Hammarén, H.M.; Savitski, M.M.; Baek, S.H. Control of Protein Stability by Post-Translational Modifications. Nat. Commun. 2023, 14, 201. [Google Scholar] [CrossRef]
- Strumillo, M.; Beltrao, P. Towards the Computational Design of Protein Post-Translational Regulation. Bioorg. Med. Chem. 2015, 23, 2877–2882. [Google Scholar] [CrossRef] [PubMed]
- Barber, K.W.; Rinehart, J. The ABCs of PTMs. Nat. Chem. Biol. 2018, 14, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Blasie, J.K.; Pascolini, D.; Asturias, F.; Herbette, L.G.; Pierce, D.; Scarpa, A. Large-Scale Structural Changes in the Sarcoplasmic Reticulum ATPase Appear Essential for Calcium Transport. Biophys. J. 1990, 58, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Menet, J.S.; Rosbash, M. A New Twist on Clock Protein Phosphorylation: A Conformational Change Leads to Protein Degradation. Mol. Cell 2011, 43, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Birck, C.; Mourey, L.; Gouet, P.; Fabry, B.; Schumacher, J.; Rousseau, P.; Kahn, D.; Samama, J.P. Conformational Changes Induced by Phosphorylation of the FixJ Receiver Domain. Struct. Lond. Engl. 1993 1999, 7, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, D.; Rahdar, M.; Tu-Sekine, B.; Sivakumaren, S.C.; Raben, D.; Amzel, L.M.; Devreotes, P.; Gabelli, S.B.; Cole, P. Phosphorylation-Mediated PTEN Conformational Closure and Deactivation Revealed with Protein Semisynthesis. eLife 2013, 2, e00691. [Google Scholar] [CrossRef]
- Colson, B.A.; Gruber, S.J.; Thomas, D.D. Structural Dynamics of Muscle Protein Phosphorylation. J. Muscle Res. Cell Motil. 2012, 33, 419–429. [Google Scholar] [CrossRef]
- Stock, J.; Da Re, S. Signal Transduction: Response Regulators on and Off. Curr. Biol. CB 2000, 10, R420–R424. [Google Scholar] [CrossRef]
- Xin, F.; Radivojac, P. Post-Translational Modifications Induce Significant yet Not Extreme Changes to Protein Structure. Bioinformatics 2012, 28, 2905–2913. [Google Scholar] [CrossRef]
- Pacheco-García, J.L.; Loginov, D.S.; Naganathan, A.N.; Vankova, P.; Cano-Muñoz, M.; Man, P.; Pey, A.L. Loss of Stability and Unfolding Cooperativity in hPGK1 upon Gradual Structural Perturbation of Its N-Terminal Domain Hydrophobic Core. Sci. Rep. 2022, 12, 17200. [Google Scholar] [CrossRef]
- Lopez, E.D.; Burastero, O.; Arcon, J.P.; Defelipe, L.A.; Ahn, N.G.; Marti, M.A.; Turjanski, A.G. Kinase Activation by Small Conformational Changes. J. Chem. Inf. Model. 2020, 60, 821–832. [Google Scholar] [CrossRef]
- Chaptal, V.; Larivière, L.; Gueguen-Chaignon, V.; Galinier, A.; Nessler, S.; Moréra, S. X-ray Structure of a Domain-Swapped Dimer of Ser46-Phosphorylated Crh from Bacillus Subtilis. Proteins 2006, 63, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Groban, E.S.; Narayanan, A.; Jacobson, M.P. Conformational Changes in Protein Loops and Helices Induced by Post-Translational Phosphorylation. PLoS Comput. Biol. 2006, 2, e32. [Google Scholar] [CrossRef] [PubMed]
- Shrake, A.; Rupley, J.A. Environment and Exposure to Solvent of Protein Atoms. Lysozyme and Insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Bio.PDB.SASA Module—Biopython 1.82.dev0 Documentation. Available online: https://biopython.org/docs/dev/api/Bio.PDB.SASA.html (accessed on 11 August 2023).
- Command: Align [PyMOL Documentation]. Available online: https://pymol.org/dokuwiki/doku.php?id=command:align (accessed on 22 September 2023).
- Radius of gyration—PyMOLWiki. Available online: https://pymolwiki.org/index.php/Radius_of_gyration (accessed on 11 August 2023).
- Scipy.stats.gaussian_kde—SciPy v1.11.1 Manual. Available online: https://docs.scipy.org/doc/scipy/reference/generated/scipy.stats.gaussian_kde.html (accessed on 11 August 2023).
- Zhang, Y.; Sagui, C. Secondary Structure Assignment for Conformationally Irregular Peptides: Comparison between DSSP, STRIDE and KAKSI. J. Mol. Graph. Model. 2015, 55, 72–84. [Google Scholar] [CrossRef]
- Rudnev, V.R.; Nikolsky, K.S.; Petrovsky, D.V.; Kulikova, L.I.; Kargatov, A.M.; Malsagova, K.A.; Stepanov, A.A.; Kopylov, A.T.; Kaysheva, A.L.; Efimov, A.V. 3β-Corner Stability by Comparative Molecular Dynamics Simulations. Int. J. Mol. Sci. 2022, 23, 11674. [Google Scholar] [CrossRef]
- Krissinel, E. On the relationship between sequence and structure similarities in proteomics. Bioinformatics 2007, 23, 717–723. [Google Scholar] [CrossRef]
- Lazar, T.; Guharoy, M.; Vranken, W.; Rauscher, S.; Wodak, S.J.; Tompa, P. Distance-Based Metrics for Comparing Conformational Ensembles of Intrinsically Disordered Proteins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nikolsky, K. Cα-displacement plots and visualizations for N2 and N3 subsets of PTM alignments research. Figshare 2023. [Google Scholar] [CrossRef]
- Yang, A.; Cho, K.; Park, H.-S. Chemical Biology Approaches for Studying Posttranslational Modifications. RNA Biol. 2017, 15, 427–440. [Google Scholar] [CrossRef]
- Craveur, P.; Narwani, T.J.; Rebehmed, J.; de Brevern, A.G. Investigation of the Impact of PTMs on the Protein Backbone Conformation. Amino Acids 2019, 51, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gram, H.; Zhao, M.; New, L.; Gu, J.; Feng, L.; Di Padova, F.; Ulevitch, R.J.; Han, J. Characterization of the Structure and Function of the Fourth Member of P38 Group Mitogen-Activated Protein Kinases, P38delta. J. Biol. Chem. 1997, 272, 30122–30128. [Google Scholar] [CrossRef] [PubMed]
- Dodson, C.A.; Bayliss, R. Activation of Aurora-A Kinase by Protein Partner Binding and Phosphorylation Are Independent and Synergistic. J. Biol. Chem. 2012, 287, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Menegay, H.J.; Myers, M.P.; Moeslein, F.M.; Landreth, G.E. Biochemical Characterization and Localization of the Dual Specificity Kinase CLK1. J. Cell Sci. 2000, 113 Pt 18, 3241–3253. [Google Scholar] [CrossRef] [PubMed]
- Krishnankutty, A.; Kimura, T.; Saito, T.; Aoyagi, K.; Asada, A.; Takahashi, S.-I.; Ando, K.; Ohara-Imaizumi, M.; Ishiguro, K.; Hisanaga, S. In Vivo Regulation of Glycogen Synthase Kinase 3β Activity in Neurons and Brains. Sci. Rep. 2017, 7, 8602. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PTM | Number of Protein Structures (PDB) | Mean Resolution, Å | Method | Origin |
|---|---|---|---|---|
| SEP | 1394 | 2.22 ± 0.8 | X-ray: 92% NMR: 3% EM: 5% | Homo sapiens: 53.5% Other eukaryote: 33.2% Bacteria: 6.8% Virus: 0.3% Archaea: 0.3% Other: 1.3% No data: 4.6% |
| TPO | 999 | 2.21 ± 0.6 | X-ray: 93% NMR: 3% EM: 4% | Homo sapiens: 49.1% Other eukaryote: 38.9% Bacteria: 3.9% Virus: 0.5% Archaea: 0.5% Other: 1.7% No data: 5.4% |
| PTR | 659 | 2.25 ± 0.5 | X-ray: 92% NMR: <8% EM: <0.5% | Homo sapiens: 72.9% Other eukaryote: 6.5% Bacteria: 2.4% Virus: 1.7% Other: 0.9% No data: 15.6% |
| Neighborhood | Min, (Å) | Max, (Å) | Mean, (Å) | Median, (Å) | Std, (Å) |
|---|---|---|---|---|---|
| 3 | 5.14 | 7.61 | 6.55 | 6.74 | 0.57 |
| 6 | 8.77 | 17.87 | 13.64 | 13.86 | 2.21 |
| 9 | 10.47 | 26.17 | 17.8 | 17.8 | 3.43 |
| 12 | 11.23 | 32.86 | 21 | 20.14 | 4.4 |
| 15 | 13.11 | 37.79 | 23 | 20.77 | 5.78 |
| Position | Group N2 | Group N3 |
|---|---|---|
| α-helix | 1 | 9 |
| Coil (middle) | 13 | 7 |
| β-strand | 3 | 2 |
| Coil (edge) | 2 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolsky, K.S.; Kulikova, L.I.; Petrovskiy, D.V.; Rudnev, V.R.; Malsagova, K.A.; Kaysheva, A.L. Analysis of Structural Changes in the Protein near the Phosphorylation Site. Biomolecules 2023, 13, 1564. https://doi.org/10.3390/biom13111564
Nikolsky KS, Kulikova LI, Petrovskiy DV, Rudnev VR, Malsagova KA, Kaysheva AL. Analysis of Structural Changes in the Protein near the Phosphorylation Site. Biomolecules. 2023; 13(11):1564. https://doi.org/10.3390/biom13111564
Chicago/Turabian StyleNikolsky, Kirill S., Liudmila I. Kulikova, Denis V. Petrovskiy, Vladimir R. Rudnev, Kristina A. Malsagova, and Anna L. Kaysheva. 2023. "Analysis of Structural Changes in the Protein near the Phosphorylation Site" Biomolecules 13, no. 11: 1564. https://doi.org/10.3390/biom13111564