
MicroRNA193a: An Emerging Mediator of Glomerular Diseases


Abstract
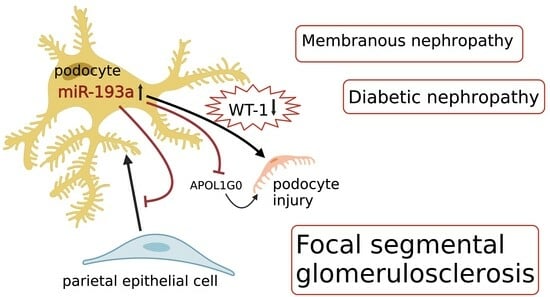
:
1. Introduction
2. What Are microRNAs?
3. miRNAs in Glomerular Diseases
4. Why Study microRNA?
- MicroRNAs are stable-in-body fluids such as plasma and urine in exosomes or vesicles. This makes them versatile as noninvasive biomarkers to diagnose diseases as they can be easily measured. miRNAs are secreted by cells in exosome-encapsulated form and can target other types of cells locally and distantly. Urinary miRNAs are either passively filtered by glomeruli or secreted by tubules.
- The role of a few miRNAs is well studied in the pathogenesis of human diseases. Specific miRNAs have been known to be involved in kidney disease for the last two decades. Therefore, miRNAs can be used as diagnostic, prognostic, surveillance, and predictive biomarkers in many human diseases.
- Chemical modification of miRNAs (oligonucleotide inhibitors), tandem repeats of miR-binding sites (decoy or sponge), and inhibition of miRNAs using nanoparticles/nucleic acids (anti-miRs) can be delivered to cells, and they can unmask the effects of miRNAs under different conditions. Therefore, miRNAs can be developed as therapeutic agents and could act as therapeutic targets as well.
5. microRNA-193a in Glomerular Diseases
5.1. A. miR-193a in FSGS
5.2. How Does miR-193a Cause Podocyte Injury?
5.3. Can miR-193a Differentiate Primary from Secondary FSGS?
5.4. APOL1 and miR-193a
5.5. miR-193a in Experimental Crescentic GN
5.6. miR-193a in Diabetic Nephropathy
5.7. miR-193a in Membranous Nephropathy
6. Clinical Application of miR-193a in Glomerular Disease
6.1. MiR-193a as a Biomarker for Diagnosis and Prognosis of Glomerular Diseases
6.2. MiR-193a as a Therapeutic Agent for Glomerular Disease
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Thurlow, J.S.; Joshi, M.; Yan, G.; Norris, K.C.; Agodoa, L.Y.; Yuan, C.M.; Nee, R. Global Epidemiology of End-Stage Kidney Disease and Disparities in Kidney Replacement Therapy. Am. J. Nephrol. 2021, 52, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Mahtal, N.; Lenoir, O.; Tinel, C.; Anglicheau, D.; Tharaux, P.L. MicroRNAs in kidney injury and disease. Nat. Rev. Nephrol. 2022, 18, 643–662. [Google Scholar] [CrossRef]
- A Gebeshuber, C.; Kornauth, C.; Dong, L.; Sierig, R.; Seibler, J.; Reiss, M.; Tauber, S.; Bilban, M.; Wang, S.; Kain, R.; et al. Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat. Med. 2013, 19, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Ichii, O.; Horino, T. MicroRNAs associated with the development of kidney diseases in humans and animals. J. Toxicol. Pathol. 2018, 31, 23–34. [Google Scholar] [CrossRef]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef]
- Simpson, R.J.; Lim, J.W.; Moritz, R.L.; Mathivanan, S. Exosomes: Proteomic insights and diagnostic potential. Expert Rev. Proteom. 2009, 6, 267–283. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Pan, Y.; Hu, J.; Ma, J.; Qi, X.; Zhou, H.; Miao, X.; Zhang, W.; Jia, L. MiR-193a-3p and miR-224 mediate renal cell carcinoma progression by targeting al-pha-2,3-sialyltransferase IV and the phosphatidylinositol 3 kinase/Akt pathway. Mol. Carcinog. 2018, 57, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Mamoori, A.; Gopalan, V.; Lam, A.K.Y. Role of miR-193a in Cancer: Complexity and Factors Control the Pattern of its Expression. Curr. Cancer Drug Targets 2018, 18, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Grossi, I.; Salvi, A.; Abeni, E.; Marchina, E.; De Petro, G. Biological Function of MicroRNA193a-3p in Health and Disease. Int. J. Genom. Proteom. 2017, 2017, 5913195. [Google Scholar] [CrossRef] [PubMed]
- Kitiyakara, C.; Eggers, P.; Kopp, J.B. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am. J. Kidney Dis. 2004, 44, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Ronco, P.; Beck, L.; Debiec, H.; Fervenza, F.C.; Hou, F.F.; Jha, V.; Sethi, S.; Tong, A.; Vivarelli, M.; Wetzels, J. Membranous nephropathy. Nat. Rev. Dis. Primers. 2021, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.J.; Jarad, G.; Cunningham, J.; Goldberg, S.; Schermer, B.; Harfe, B.D.; McManus, M.T.; Benzing, T.; Miner, J.H. Podocyte-specific deletion of Dicer alters cytoskeletal dynamics and causes glo-merular disease. J. Am. Soc. Nephrol. 2008, 19, 2150–2158. [Google Scholar] [CrossRef]
- Shi, S.; Yu, L.; Chiu, C.; Sun, Y.; Chen, J.; Khitrov, G.; Merkenschlager, M.; Holzman, L.B.; Zhang, W.; Mundel, P.; et al. Podocyte-Selective Deletion of Dicer Induces Proteinuria and Glomerulosclerosis. J. Am. Soc. Nephrol. 2008, 19, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zheng, C.; Fan, Y.; Zeng, C.; Chen, Z.; Qin, W.; Zhang, C.; Zhang, W.; Wang, X.; Zhu, X.; et al. Downregulation of microRNA-30 facilitates podocyte injury and is prevented by glucocorti-coids. J. Am. Soc. Nephrol. 2014, 25, 92–104. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, C.; Chen, H.; Li, L.; Tu, Y.; Liu, C.; Shi, S.; Zen, K.; Liu, Z. Evaluation of MicroRNAs miR-196a, miR-30a-5P, and miR-490 as Biomarkers of Disease Activity among Patients with FSGS. Clin. J. Am. Soc. Nephrol. 2014, 9, 1545–1552. [Google Scholar] [CrossRef]
- Krebs, C.F.; Kapffer, S.; Paust, H.-J.; Schmidt, T.; Bennstein, S.B.; Peters, A.; Stege, G.; Brix, S.R.; Meyer-Schwesinger, C.; Müller, R.-U.; et al. MicroRNA-155 Drives TH17 Immune Response and Tissue Injury in Experimental Crescentic GN. J. Am. Soc. Nephrol. 2013, 24, 1955–1965. [Google Scholar] [CrossRef]
- Shaffi, S.K.; Galas, D.; Etheridge, A.; Argyropoulos, C. Role of MicroRNAs in Renal Parenchymal Diseases-A New Dimension. Int. J. Mol. Sci. 2018, 19, 1797. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, X.; Nie, F.; Liu, T.; Yu, X.; Wang, H.; Li, Q.; Peng, R.; Mao, Z.; Zhou, Q.; et al. miR-135 family members mediate podocyte injury through the activation of Wnt/β-catenin signaling. Int. J. Mol. Med. 2015, 36, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Stolz, D.B.; Kiss, L.P.; Monga, S.P.; Holzman, L.B.; Liu, Y. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J. Am. Soc. Nephrol. 2009, 20, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Müller-Deile, J.; Dannenberg, J.; Schroder, P.; Lin, M.H.; Miner, J.H.; Chen, R.; Bräsen, J.-H.; Thum, T.; Nyström, J.; Staggs, L.B.; et al. Podocytes regulate the glomerular basement membrane protein nephron-ectin by means of miR-378a-3p in glomerular diseases. Kidney Int. 2017, 92, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, X.; Fu, X.; Yang, Y.; Chen, J.; Lin, W. MicroRNA-26a: An Emerging Regulator of Renal Biology and Disease. Kidney Blood Press Res. 2019, 44, 287–297. [Google Scholar] [CrossRef]
- Gao, D.; Yu, P.; Jing, S.; Yan, C.; Ding, D.; Qiao, Y.; Wu, G. miR-193a as a potential mediator of WT-1/synaptopodin in the renoprotective effect of Losartan on diabetic kidney. Can. J. Physiol. Pharmacol. 2022, 100, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ren, Y.; Li, J. Application of miR-193a/WT1/PODXL axis to estimate risk and prognosis of idiopathic membranous nephropathy. Ren. Fail. 2019, 41, 704–717. [Google Scholar] [CrossRef]
- Dieter, C.; Assmann, T.S.; Costa, A.R.; Canani, L.H.; de Souza, B.M.; Bauer, A.C.; Crispim, D. MiR-30e-5p and MiR-15a-5p Expressions in Plasma and Urine of Type 1 Diabetic Patients with Diabetic Kidney Disease. Front. Genet. 2019, 10, 563. [Google Scholar] [CrossRef]
- Donderski, R.; Szczepanek, J.; Naruszewicz, N.; Naruszewicz, R.; Tretyn, A.; Skoczylas-Makowska, N.; Tyloch, J.; Odrowąż-Sypniewska, G.; Manitius, J. Analysis of profibrogenic microRNAs (miRNAs) expression in urine and serum of chronic kidney disease (CKD) stage 1–4 patients and their relationship with proteinuria and kidney function. Int. Urol. Nephrol. 2021, 54, 937–947. [Google Scholar] [CrossRef]
- Lai, J.Y.; Luo, J.; O’connor, C.; Jing, X.; Nair, V.; Ju, W.; Randolph, A.; Ben-Dov, I.Z.; Matar, R.N.; Briskin, D.; et al. MicroRNA-21 in Glomerular Injury. J. Am. Soc. Nephrol. 2015, 26, 805–816. [Google Scholar] [CrossRef]
- He, F.; Peng, F.; Xia, X.; Zhao, C.; Luo, Q.; Guan, W.; Li, Z.; Yu, X.; Huang, F. MiR-135a promotes renal fibrosis in diabetic nephropathy by regulating TRPC1. Diabetologia 2014, 57, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Gambardella, J.; Sardu, C.; Lombardi, A.; Santulli, G. Functional Role of miR-155 in the Pathogenesis of Diabetes Mellitus and Its Complications. Noncoding RNA 2021, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Venkatesh, I.; Villanueva, V.; Wei, H.; Geraghty, T.; Rajagopalan, A.; Helmuth, R.W.; Altintas, M.M.; Faridi, H.M.; Gupta, V. Podocyte-specific deletion of miR-146a increases podocyte injury and diabetic kidney disease. Front. Med. 2022, 9, 897188. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Y.; Li, L.; Su, B.; Yang, L.; Fan, W.; Yin, Q.; Chen, L.; Cui, T.; Zhang, J.; et al. Involvement of inflammation-related miR-155 and miR-146a in diabetic nephropathy: Implica-tions for glomerular endothelial injury. BMC Nephrol. 2014, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kwan, B.C.H.; Lai, F.M.M.; Chow, K.M.; Li, P.K.T.; Szeto, C.C. Urinary miR-21, miR-29, and miR-93: Novel biomarkers of fibrosis. Am. J. Nephrol. 2012, 36, 412–418. [Google Scholar] [CrossRef]
- Ma, H.; Li, X.; Yu, S.; Hu, Y.; Yin, M.; Zhu, F.; Xu, L.; Wang, T.; Wang, H.; Li, H.; et al. Deletion of the miR-25/93/106b cluster induces glomerular deposition of immune complexes and renal fibrosis in mice. J. Cell. Mol. Med. 2021, 25, 7922–7934. [Google Scholar] [CrossRef]
- Zheng, Z.; Guan, M.; Jia, Y.; Wang, D.; Pang, R.; Lv, F.; Xiao, Z.; Wang, L.; Zhang, H.; Xue, Y. The coordinated roles of miR-26a and miR-30c in regulating TGFβ1-induced epitheli-al-to-mesenchymal transition in diabetic nephropathy. Sci. Rep. 2016, 6, 37492. [Google Scholar] [CrossRef]
- Liu, D.; Chen, R.; Ni, H.; Liu, H. miR-181a Improved Renal Inflammation by Targeting TNF-α in a Diabetic Nephropathy Animal Model. Nephron 2022, 146, 637–646. [Google Scholar] [CrossRef]
- Du, G.; Xiao, M.; Zhang, X.; Wen, M.; Pang, C.; Jiang, S.; Sang, S.; Xie, Y. Alpinia oxyphylla Miq. extract changes miRNA expression profiles in db-/db- mouse kidney. Biol. Res. 2017, 50, 9. [Google Scholar] [CrossRef]
- Yang, S.; Fei, X.; Lu, Y.; Xu, B.; Ma, Y.; Wan, H. miRNA-214 suppresses oxidative stress in diabetic nephropathy via the ROS/Akt/mTOR signaling pathway and uncoupling protein 2. Exp. Ther. Med. 2019, 17, 3530–3538. [Google Scholar] [CrossRef]
- Li, Z.; Yin, H.; Hao, S.; Wang, L.; Gao, J.; Tan, X.; Yang, Z. miR-200 family promotes podocyte differentiation through repression of RSAD2. Sci. Rep. 2016, 6, 27105. [Google Scholar] [CrossRef] [PubMed]
- Müller-Deile, J.; Sopel, N.; Ohs, A.; Rose, V.; Gröner, M.; Wrede, C.; Christoph, W.; Jan, H.; Christoph, D.; Kerstin, A.; et al. Glomerular Endothelial Cell-Derived microRNA-192 Regulates Nephronectin Ex-pression in Idiopathic Membranous Glomerulonephritis. J. Am. Soc. Nephrol. 2021, 32, 2777–2794. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Xiong, X.; Tang, B.; Ji, T.; Li, X.; Qu, X.; Li, W. hsa-miR-199b-3p Prevents the Epithelial-Mesenchymal Transition and Dysfunction of the Renal Tubule by Regulating E-cadherin through Targeting KDM6A in Diabetic Nephropathy. Oxid. Med. Cell. Longev. 2021, 2021, 8814163. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, D.; Wang, F.; Liu, J.; Huang, B.; Baker, M.A.; Yin, J.; Wu, R.; Liu, X.; Regner, K.R.; et al. Endogenous miR-204 Protects the Kidney against Chronic Injury in Hypertension and Diabetes. J. Am. Soc. Nephrol. 2020, 31, 1539–1554. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.-W.; Jiang, C.-M.; Wan, C.; Zhang, M.; Zhang, Q.-Y.; Zhao, M.; Yang, B.; Zhu, D.-L.; Han, X. Upregulation of MiR-126 Delays the Senescence of Human Glomerular Mesangial Cells Induced by High Glucose via Telomere-p53-p21-Rb Signaling Pathway. Curr. Med. Sci. 2018, 38, 758–764. [Google Scholar] [CrossRef]
- Chen, N.X.; Kiattisunthorn, K.; O’Neill, K.D.; Chen, X.; Moorthi, R.N.; Gattone, V.H.; Allen, M.R.; Moe, S.M. Decreased MicroRNA Is Involved in the Vascular Remodeling Abnormalities in Chronic Kidney Disease (CKD). PLoS ONE 2013, 8, e64558. [Google Scholar] [CrossRef]
- Ge, X.; Xi, L.; Wang, Q.; Li, H.; Xia, L.; Cang, Z.; Peng, W.; Huang, S. Circular RNA Circ_0000064 promotes the proliferation and fibrosis of mesangial cells via miR-143 in diabetic nephropathy. Gene 2020, 758, 144952. [Google Scholar] [CrossRef]
- Xiao, S.; Yang, Y.; Liu, Y.T.; Zhu, J. Liraglutide Regulates the Kidney and Liver in Diabetic Nephropathy Rats through the miR-34a/SIRT1 Pathway. J. Diabetes Res. 2021, 2021, 8873956. [Google Scholar] [CrossRef]
- Jha, A.; Saha, S.; Ayasolla, K.; Vashistha, H.; Malhotra, A.; Skorecki, K.; Singhal, P.C. MiR193a Modulation and Podocyte Phenotype. Cells 2020, 9, 1004. [Google Scholar] [CrossRef]
- Shankland, S.J.; Smeets, B.; Pippin, J.W.; Moeller, M.J. The emergence of the glomerular parietal epithelial cell. Nat. Rev. Nephrol. 2014, 10, 158–173. [Google Scholar] [CrossRef]
- Kietzmann, L.; Guhr, S.S.; Meyer, T.N.; Ni, L.; Sachs, M.; Panzer, U.; Stahl, R.A.; Saleem, M.A.; Kerjaschki, D.; Gebeshuber, C.A.; et al. MicroRNA-193a Regulates the Transdifferentiation of Human Parietal Epithelial Cells toward a Podocyte Phenotype. J. Am. Soc. Nephrol. 2015, 26, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Bharati, J.; Chander, P.N.; Singhal, P.C. Parietal Epithelial Cell Behavior and Its Modulation by microRNA-193a. Biomolecules 2023, 13, 266. [Google Scholar] [CrossRef] [PubMed]
- Eng, D.G.; Sunseri, M.W.; Kaverina, N.V.; Roeder, S.S.; Pippin, J.W.; Shankland, S.J. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015, 88, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Paliwal, N.; Ayasolla, K.; Vashistha, H.; Jha, A.; Chandel, N.; Chowdhary, S.; Saleem, M.A.; Malhotra, A.; Chander, P.N.; et al. Disruption of APOL1-miR193a Axis Induces Disorganization of Podocyte Actin Cytoskeleton. Sci. Rep. 2019, 9, 3582. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Vashistha, H.; Lan, X.; Chandel, N.; Ayasolla, K.; Shoshtari, S.S.M.; Aslam, R.; Paliwal, N.; Abbruscato, F.; Mikulak, J.; et al. Role of Apolipoprotein L1 in Human Parietal Epithelial Cell Transition. Am. J. Pathol. 2018, 188, 2508–2528. [Google Scholar] [CrossRef]
- Jessee, J.; Kopp, J.B. APOL1-miR-193 Axis as a Bifunctional Regulator of the Glomerular Parietal Epithelium: Maintaining Parietal Cell Phenotype versus Promoting Podocyte Differentiation. Am. J. Pathol. 2018, 188, 2461–2463. [Google Scholar] [CrossRef]
- Mishra, A.; Ayasolla, K.; Kumar, V.; Lan, X.; Vashistha, H.; Aslam, R.; Hussain, A.; Chowdhary, S.; Shoshtari, S.M.; Paliwal, N.; et al. Modulation of apolipoprotein L1-microRNA-193a axis prevents podocyte dediffer-entiation in high-glucose milieu. Am. J. Physiol.-Ren. Physiol. 2018, 314, F832–F843. [Google Scholar] [CrossRef]
- Singh, T.; Ayasolla, K.; Rai, P.; Chandel, N.; Haque, S.; Lederman, R.; Husain, M.; Vethantham, V.; Chawla, A.; Vashistha, H.; et al. AT1R blockade in adverse milieus: Role of SMRT and corepressor complexes. Am. J. Physiol. Physiol. 2015, 309, F189–F203. [Google Scholar] [CrossRef]
- Fogo, A.B. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat. Rev. Nephrol. 2015, 11, 76–87. [Google Scholar] [CrossRef]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 Genetic Variants in Focal Segmental Glomerulosclerosis and HIV-Associated Nephropathy. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef]
- Lan, X.; Jhaveri, A.; Cheng, K.; Wen, H.; Saleem, M.A.; Mathieson, P.W.; Mikulak, J.; Aviram, S.; Malhotra, A.; Skorecki, K.; et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal mem-brane permeability. Am. J. Physiol.-Ren. Physiol. 2014, 307, F326–F336. [Google Scholar] [CrossRef]
- Beckerman, P.; Bi-Karchin, J.; Park, A.S.D.; Qiu, C.; Dummer, P.D.; Soomro, I.; Boustany-Kari, C.M.; Pullen, S.S.; Miner, J.H.; A Hu, C.-A.; et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017, 23, 429–438. [Google Scholar] [CrossRef] [PubMed]
- De Vriese, A.S.; Sethi, S.; Nath, K.A.; Glassock, R.J.; Fervenza, F.C. Differentiating Primary, Genetic, and Secondary FSGS in Adults: A Clinicopathologic Approach. J. Am. Soc. Nephrol. 2018, 29, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Wang, J.; Zhang, L.; Sun, W.; Xu, X.; Zhang, K. Plasma miR-193a-3p can be a potential biomarker for the diagnosis of diabetic nephropathy. Ann. Clin. Biochem. 2021, 58, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Wang, Z.; Zhou, J.; Zhang, Y. Higher Urine Exosomal miR-193a Is Associated with a Higher Probability of Primary Focal Segmental Glomerulosclerosis and an Increased Risk of Poor Prognosis Among Children with Nephrotic Syndrome. Front. Cell Dev. Biol. 2021, 9, 727370. [Google Scholar] [CrossRef] [PubMed]
- Bukosza, E.N.; Kratochwill, K.; Kornauth, C.; Schachner, H.; Aufricht, C.; Gebeshuber, C.A. Podocyte RNA sequencing reveals Wnt- and ECM-associated genes as central in FSGS. PLoS ONE 2020, 15, e0231898. [Google Scholar] [CrossRef] [PubMed]
- Kerjaschki, D. 2015 Homer W. Smith Award: The Podocyte from Periphery to Center Stage. J. Am. Soc. Nephrol. 2016, 27, 3266–3270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, W.; Chen, H.-M.; Liu, C.; Wu, J.; Shi, S.; Liu, Z.-H. Plasma MicroRNA-186 and Proteinuria in Focal Segmental Glomerulosclerosis. Am. J. Kidney Dis. 2015, 65, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Umanath, K.; Lewis, J.B. Update on Diabetic Nephropathy: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 71, 884–895. [Google Scholar] [CrossRef]
- Barutta, F.; Bellini, S.; Gruden, G. Mechanisms of podocyte injury and implications for diabetic nephropathy. Clin. Sci. 2022, 136, 493–520. [Google Scholar] [CrossRef]
- Chandel, N.; Ayasolla, K.; Wen, H.; Lan, X.; Haque, S.; Saleem, M.A.; Malhotra, A.; Singhal, P.C. Vitamin D receptor deficit induces activation of renin angiotensin system via SIRT1 modulation in podocytes. Exp. Mol. Pathol. 2017, 102, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Bai, J.; Cui, S.; Fu, B.; Yin, Z.; Cai, G.; Chen, X. Renal progenitor cells modulated by angiotensin II receptor blocker (ARB) medication and differentiation towards podocytes in anti-thy1.1 nephritis. Ann. Transl. Med. 2020, 8, 355. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S. New “Antigens” in Membranous Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.; Shen, L.; Deng, Y. Improvement of membranous nephropathy by inhibition of miR-193a to affect podocytosis via targeting WT1. J. Cell Biochem. 2019, 120, 3438–3446. [Google Scholar] [CrossRef] [PubMed]
- Trevisani, F.; Ghidini, M.; Larcher, A.; Lampis, A.; Lote, H.; Manunta, P.; Alibrandi, M.T.S.; Zagato, L.; Citterio, L.; Dell’Antonio, G.; et al. MicroRNA 193b-3p as a predictive biomarker of chronic kidney disease in patients undergoing radical nephrectomy for renal cell carcinoma. Br. J. Cancer 2016, 115, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Precazzini, F.; Detassis, S.; Imperatori, A.S.; Denti, M.A.; Campomenosi, P. Measurements Methods for the Development of MicroRNA-Based Tests for Cancer Diagnosis. Int. J. Mol. Sci. 2021, 22, 1176. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Arce, L.; Natarajan, R. MicroRNAs and their role in progressive kidney diseases. Clin. J. Am. Soc. Nephrol. 2009, 4, 1255–1266. [Google Scholar] [CrossRef]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti–microRNA-21 oligonucleotides prevent Alport nephropathy pro-gression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Y.; Zhou, J.; Zhang, Y. Urinary Exosomal miR-193a Can Be a Potential Biomarker for the Diagnosis of Primary Focal Segmental Glomerulosclerosis in Children. Biomed. Res. Int. 2017, 2017, 7298160. [Google Scholar] [CrossRef]
- Liu, F.; Chen, J.; Luo, C.; Meng, X. Pathogenic Role of MicroRNA Dysregulation in Podocytopathies. Front. Physiol. 2022, 13, 948094. [Google Scholar] [CrossRef]
- Selvaskandan, H.; Pawluczyk, I.; Barratt, J. Clinical application of microRNAs in glomerular diseases. Nephrol. Dial. Transplant. 2023, 38, 1375–1384. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNA | Mechanism of Action |
|---|---|
| miR-193a (FSGS, DN, MN) [3,26,27] | Targets Wilm’s tumor protein 1, disrupts podocytes. |
| miR-30 family (FSGS, DN) [19,28] | Modulates fibrosis and inflammation. |
| miR-29 family (FSGS, DN, MN) [29] | Regulates extracellular matrix. |
| miR-21 (FSGS, DN, MN) [30] | Promotes fibrosis and inflammation. |
| miR-135a (FSGS, DN) [31] | Inhibits transient receptor potential cation channel 1, alters calcium signaling. |
| miR-155 (FSGS, DN, MN) [32] | Contributes to podocyte injury. |
| miR-146a (FSGS, DN, MN) [33,34] | Regulates inflammation and fibrosis. |
| miR-93 (FSGS, DN, MN) [35,36] | Modulates podocyte dysfunction. |
| miR-25 (FSGS, DN) [36] | Impairs podocyte function and regulates pathways involved in DN progression. |
| miR-26a (FSGS, DN) [37] | Alters podocyte signaling pathways. |
| miR-181a (FSGS, DN) [38] | Modulates inflammatory response, targets tumor necrosis factor-alpha. |
| miR-378 (FSGS, DN) [39] | Modulates transforming growth factor-β1 signaling pathway. |
| miR-214 (FSGS, DN) [40] | Regulates fibrosis and inflammation. |
| miR-200 family (FSGS, DN, MN) [41] | Affects epithelial-to-mesenchymal transition. |
| miR-192 (FSGS, DN, MN) [42] | Modulates podocyte function. |
| miR-199a-3p (FSGS, DN) [43] | Affects podocyte integrity and protects tubular epithelial cells from high-glucose injury. |
| miR-204 (FSGS, MN, DN) [44] | Regulates podocyte injury. |
| miR-126 (FSGS, DN, MN) [45] | Regulates angiogenesis and inflammation. |
| miR-125b (DN, MN) [46] | Regulates inflammation and podocyte injury. |
| miR-143 (FSGS, DN) [47] | Affects podocyte structure and function. |
| miR-34a (MN, DN) [48] | Regulates podocyte apoptosis and fibrosis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bharati, J.; Kumar, M.; Kumar, N.; Malhotra, A.; Singhal, P.C. MicroRNA193a: An Emerging Mediator of Glomerular Diseases. Biomolecules 2023, 13, 1743. https://doi.org/10.3390/biom13121743
Bharati J, Kumar M, Kumar N, Malhotra A, Singhal PC. MicroRNA193a: An Emerging Mediator of Glomerular Diseases. Biomolecules. 2023; 13(12):1743. https://doi.org/10.3390/biom13121743
Chicago/Turabian StyleBharati, Joyita, Megan Kumar, Neil Kumar, Ashwani Malhotra, and Pravin C. Singhal. 2023. "MicroRNA193a: An Emerging Mediator of Glomerular Diseases" Biomolecules 13, no. 12: 1743. https://doi.org/10.3390/biom13121743
APA StyleBharati, J., Kumar, M., Kumar, N., Malhotra, A., & Singhal, P. C. (2023). MicroRNA193a: An Emerging Mediator of Glomerular Diseases. Biomolecules, 13(12), 1743. https://doi.org/10.3390/biom13121743