


Orthoparamyxovirinae C Proteins Have a Common Origin and a Common Structural Organization

 , , , , ,
, , , , , 
Abstract
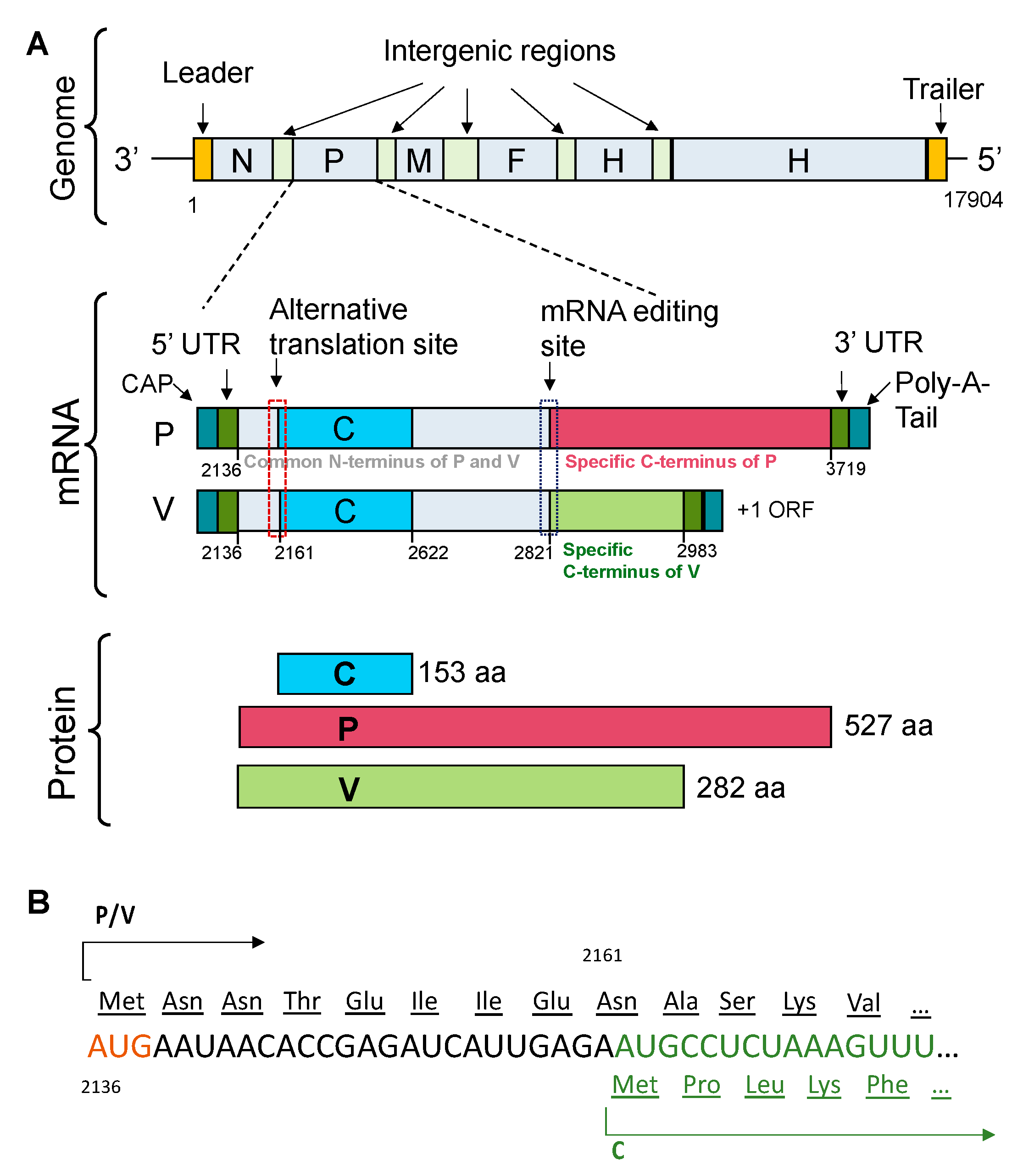
:1. Introduction
2. Materials and Methods
2.1. Disorder Metaprediction
2.2. Protein Expression and Purification
2.3. Size Exclusion Chromatography Coupled with Multiangle Laser Light Scattering Experiments [41]
2.4. Size Exclusion Chromatography Coupled with Small Angle X-ray Scattering Experiments [43]
2.5. X-ray Crystallography
2.6. AlphaFold Predictions
2.7. Cell Culture
2.8. Plasmids Constructs
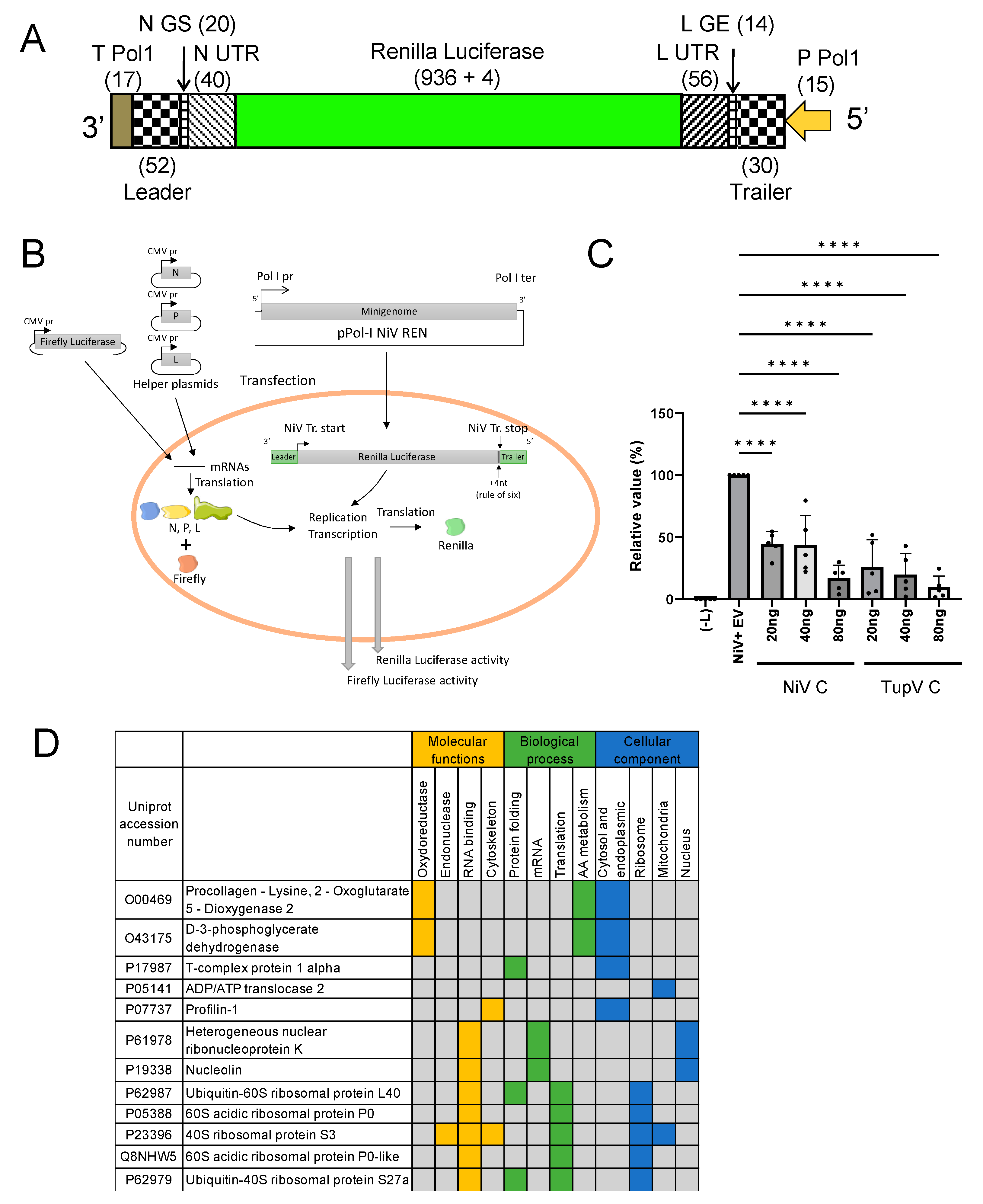
2.9. Minireplicon Assay
2.10. TupV C Protein Interactions with the Human Proteome
3. Results
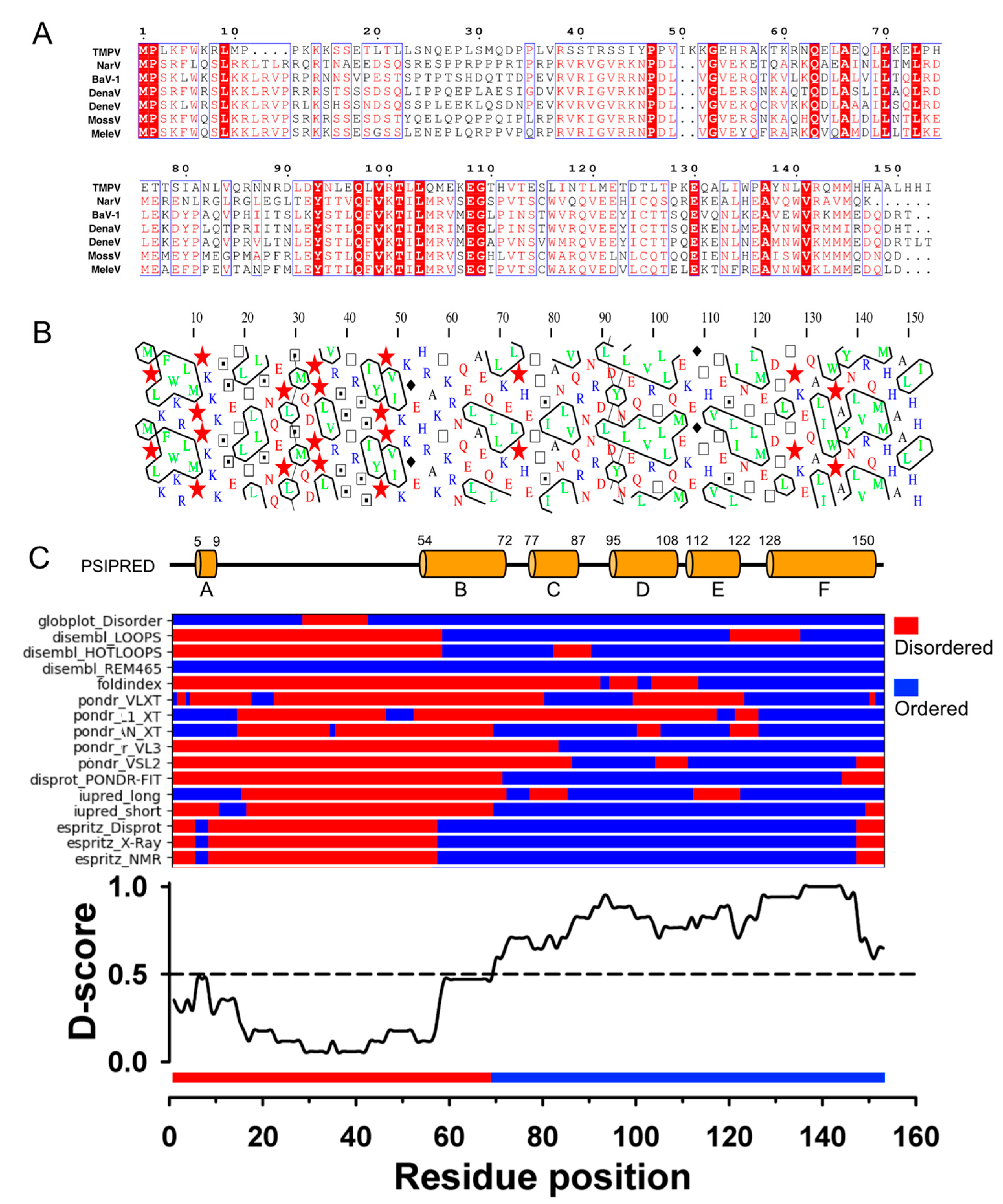
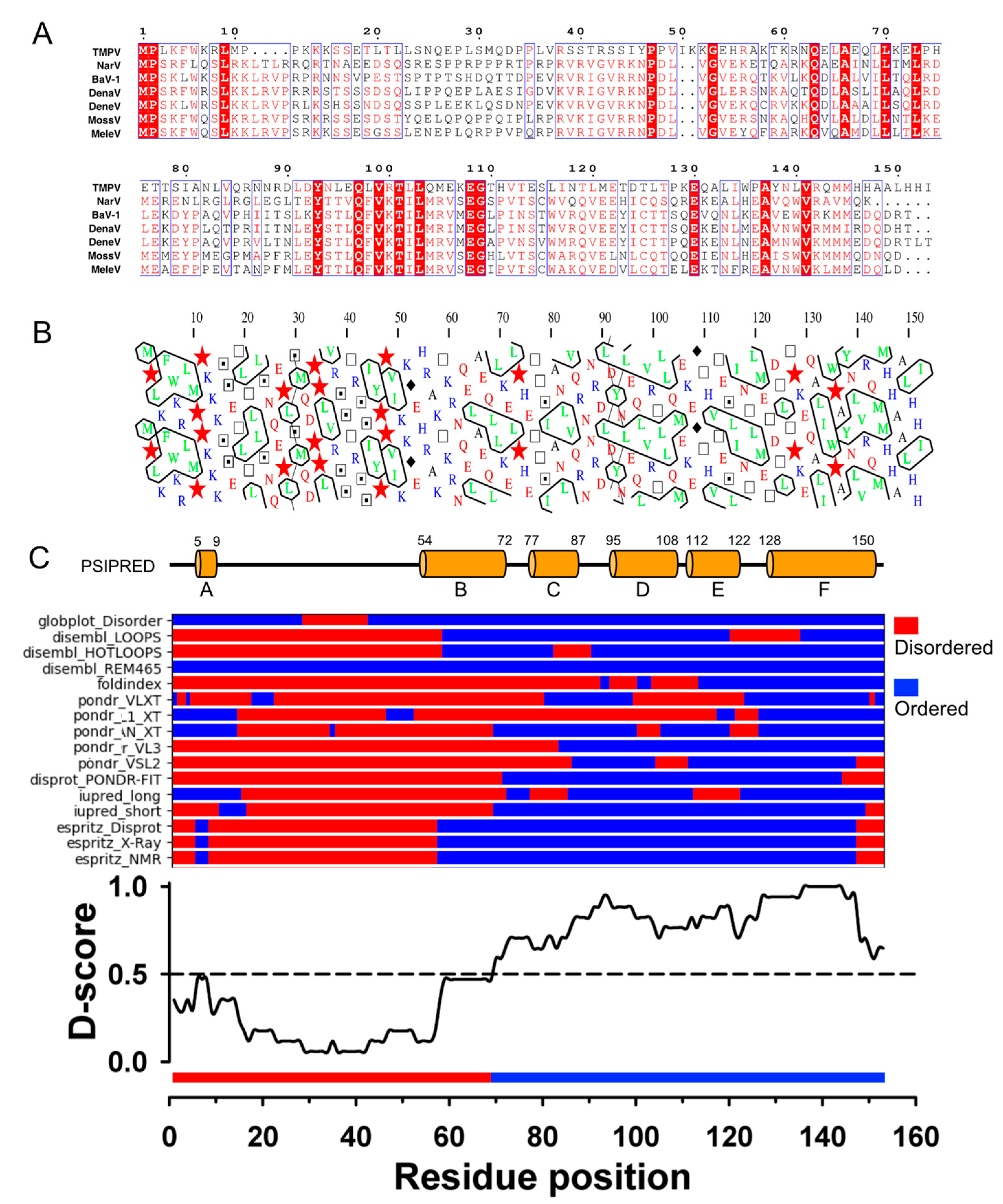
3.1. TupV C Protein Contains a Predicted Structured Region (aa 54−153)
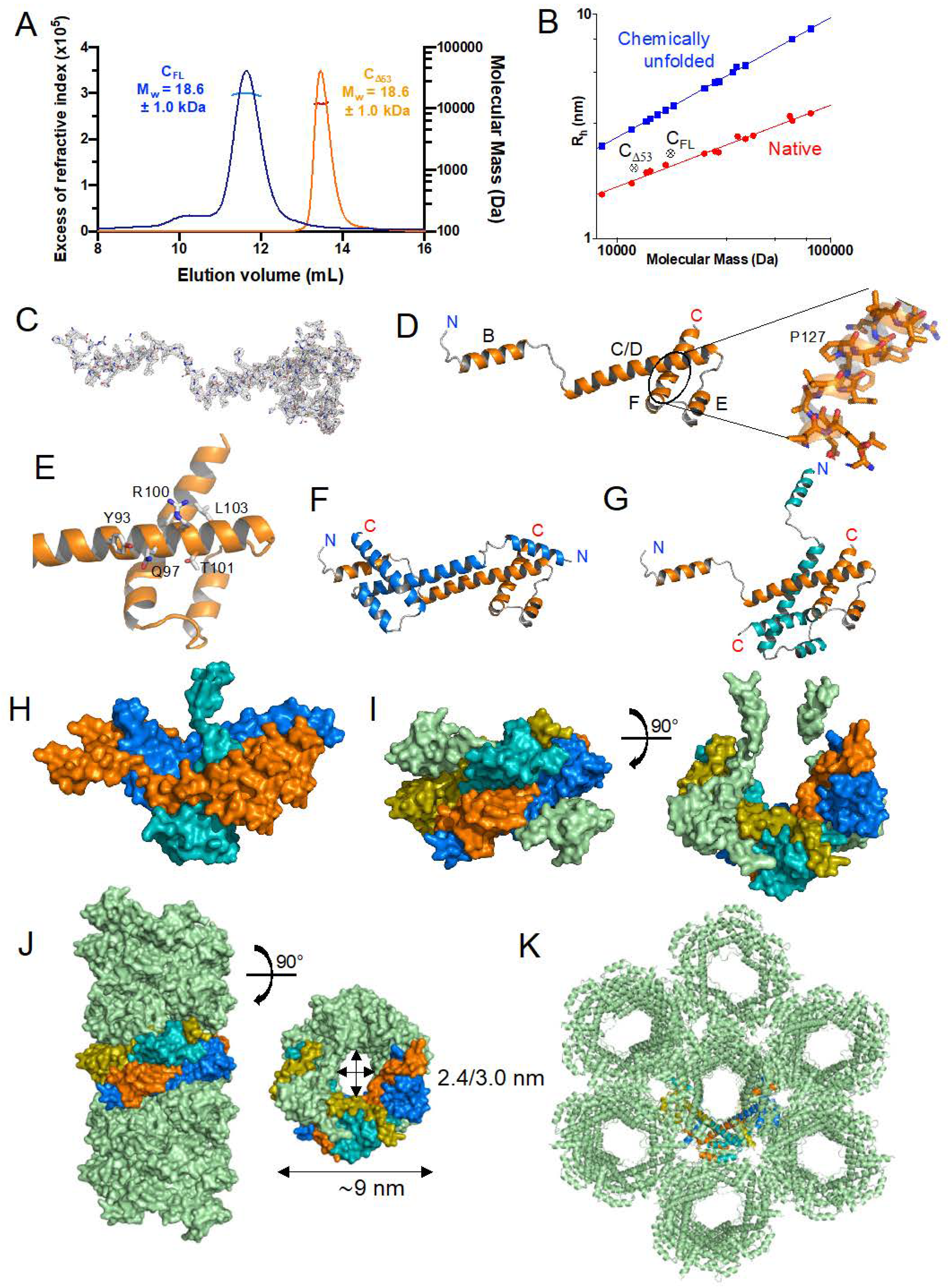
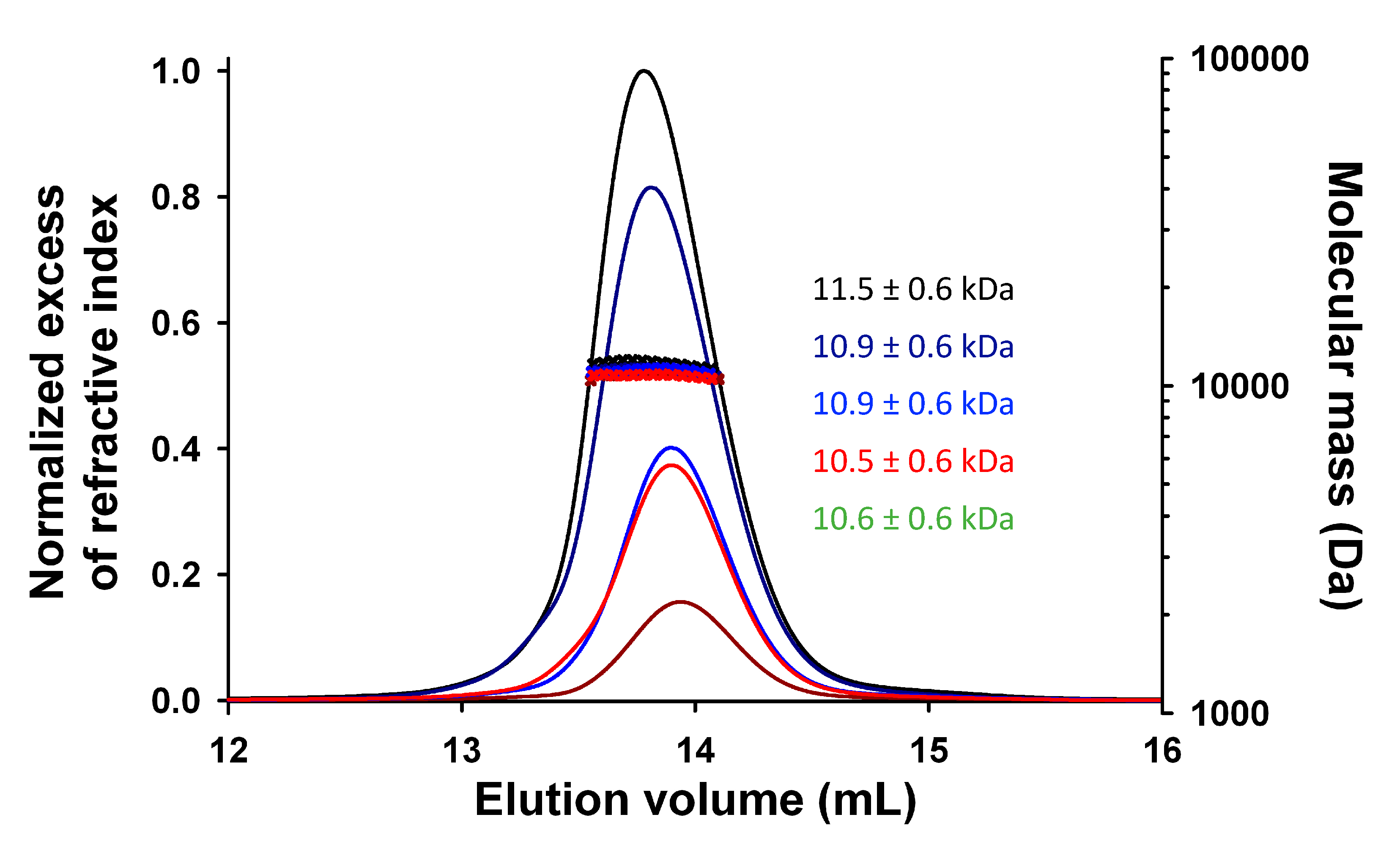
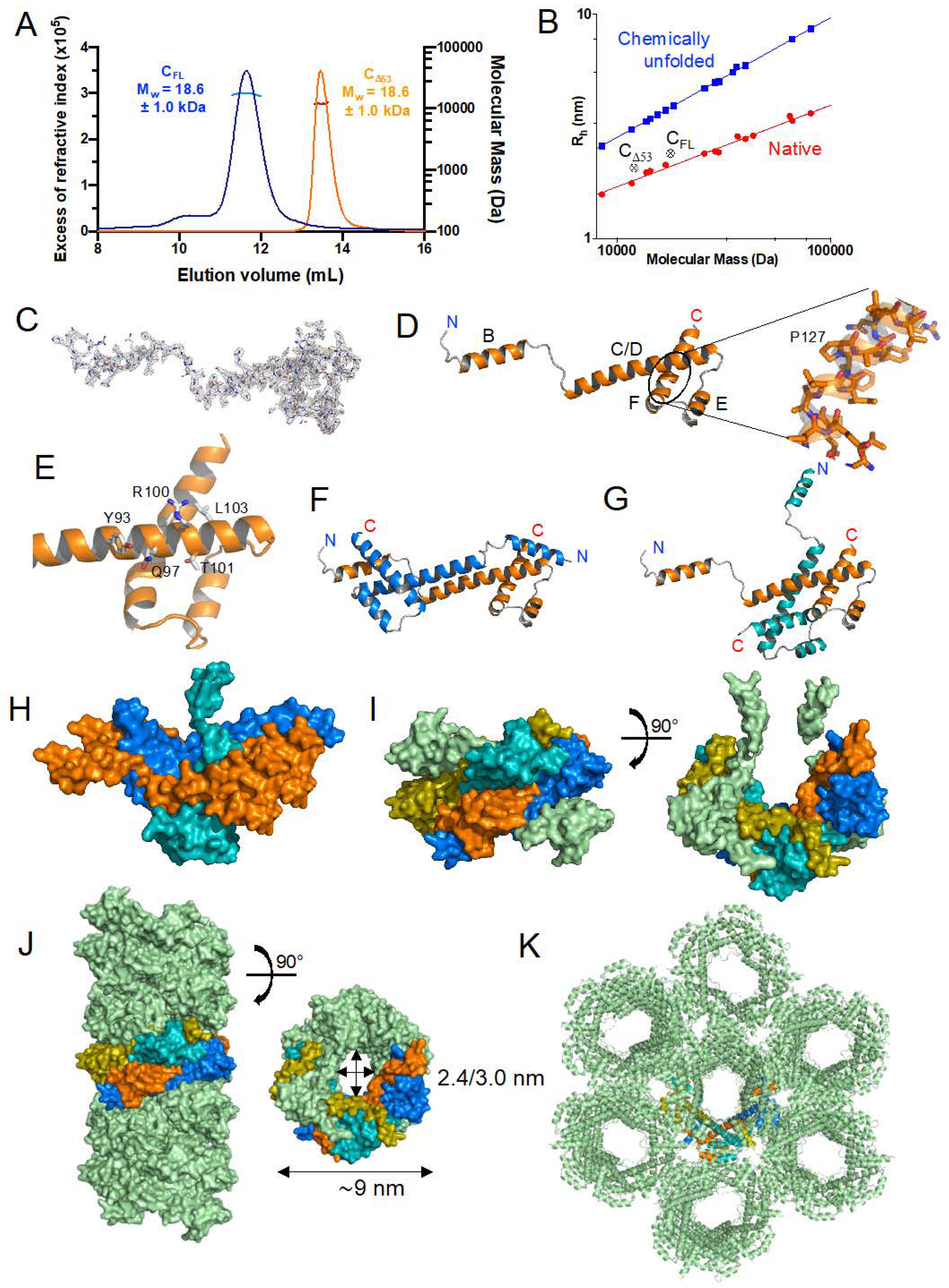
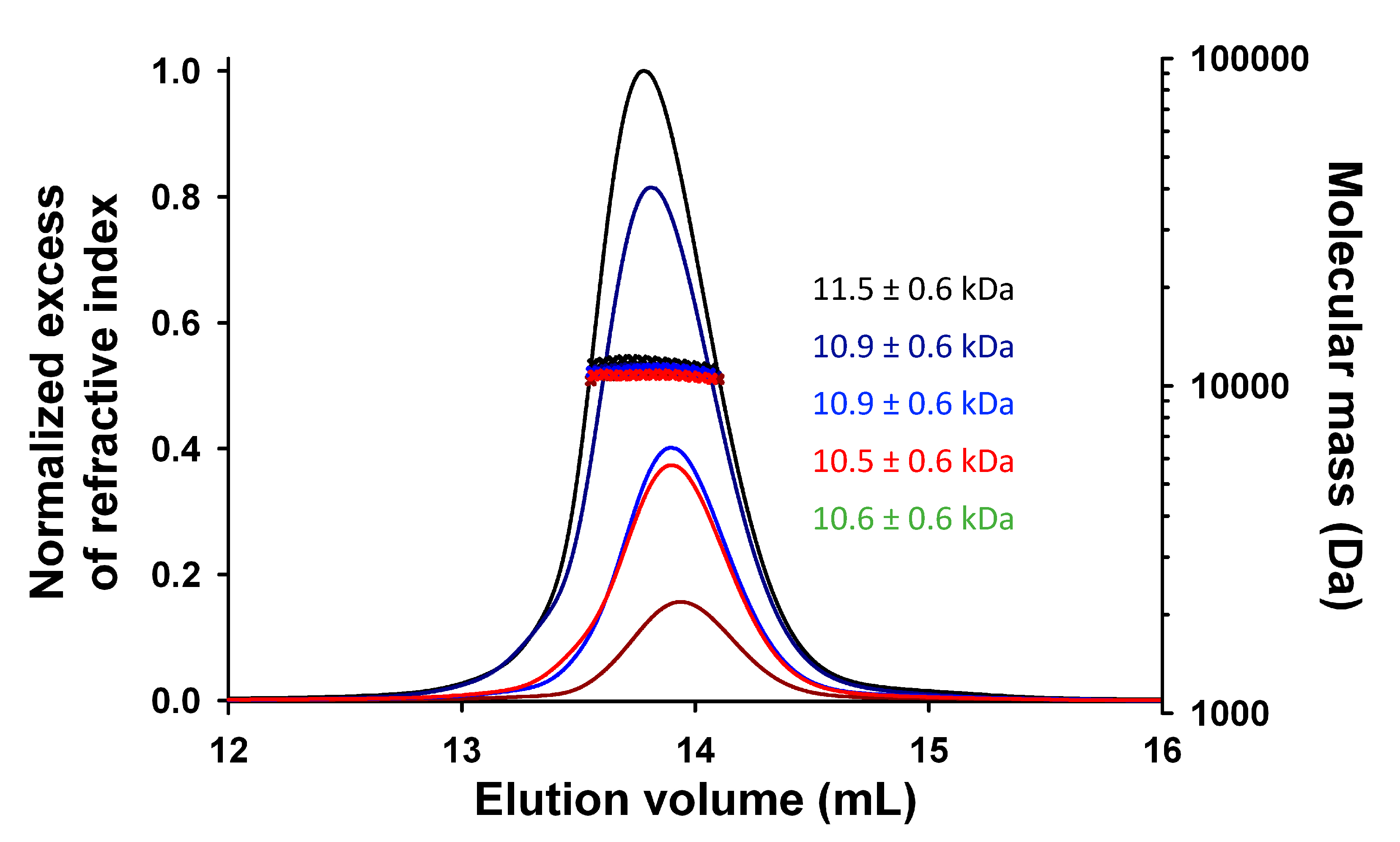
3.2. Expression, Purification, and Quality Control of N-Terminally Truncated and Full-Length TupV C Proteins
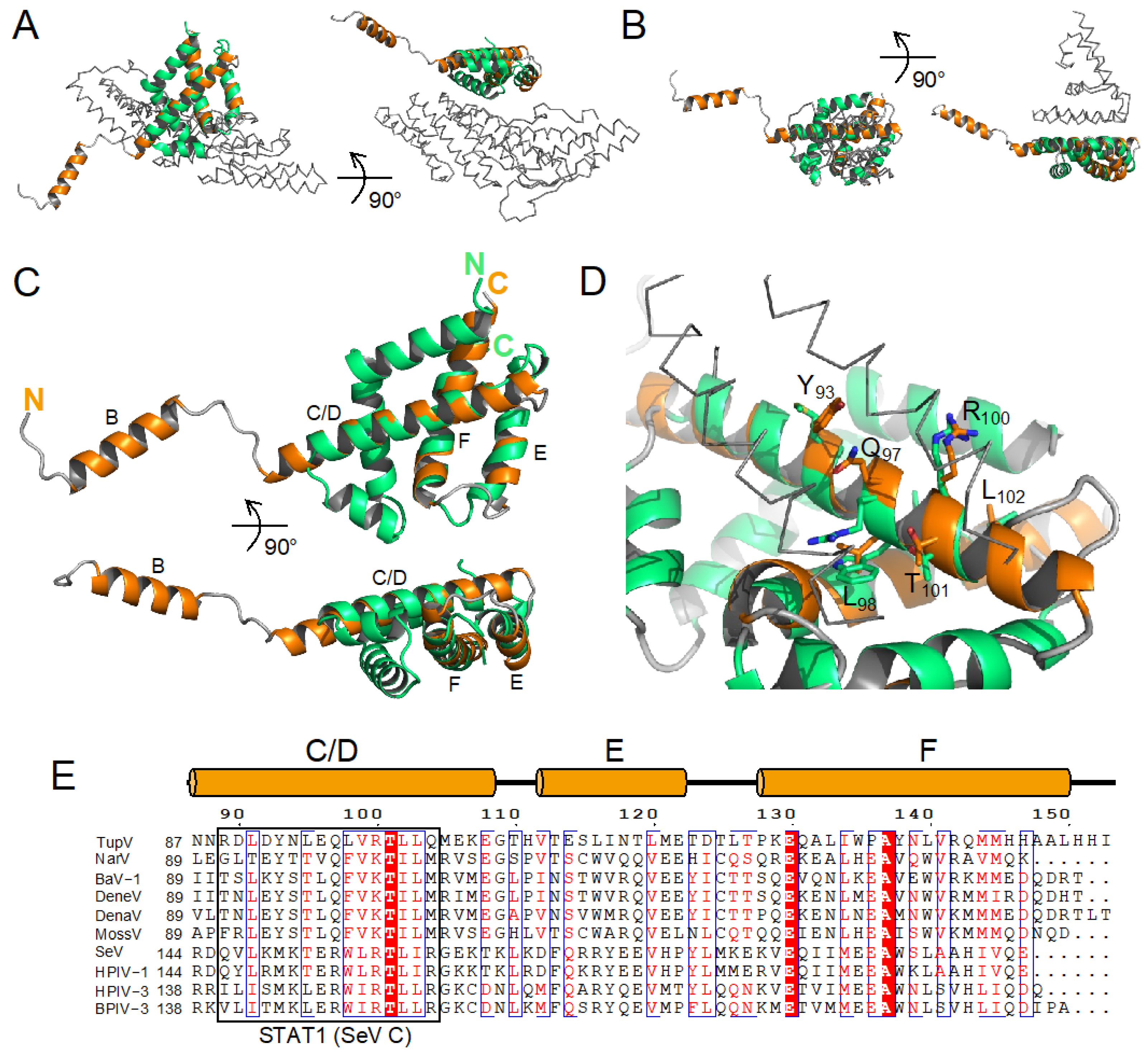
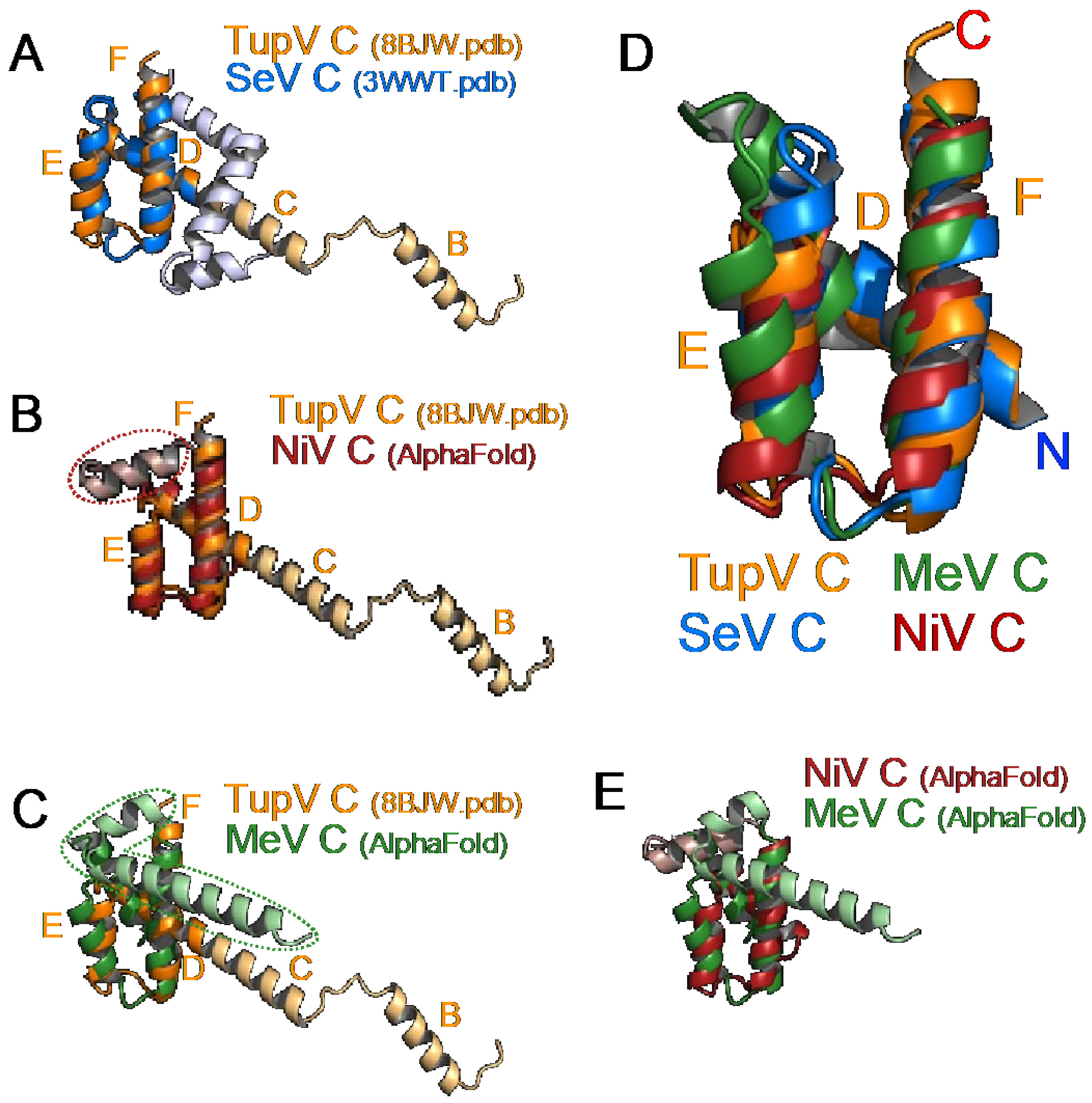
3.3. Crystal Structure of the Folded C-Terminal Domain Revealed Structural Similarity with the Sendai Virus C Protein
3.4. The C Proteins of NiV, MeV, and SeV Contain a Core of 3 Helices That Superpose to Helices D-E-F of TupV C
3.5. TupV CΔ53 Does Not Multimerize in Solution, Unlike in the Crystal
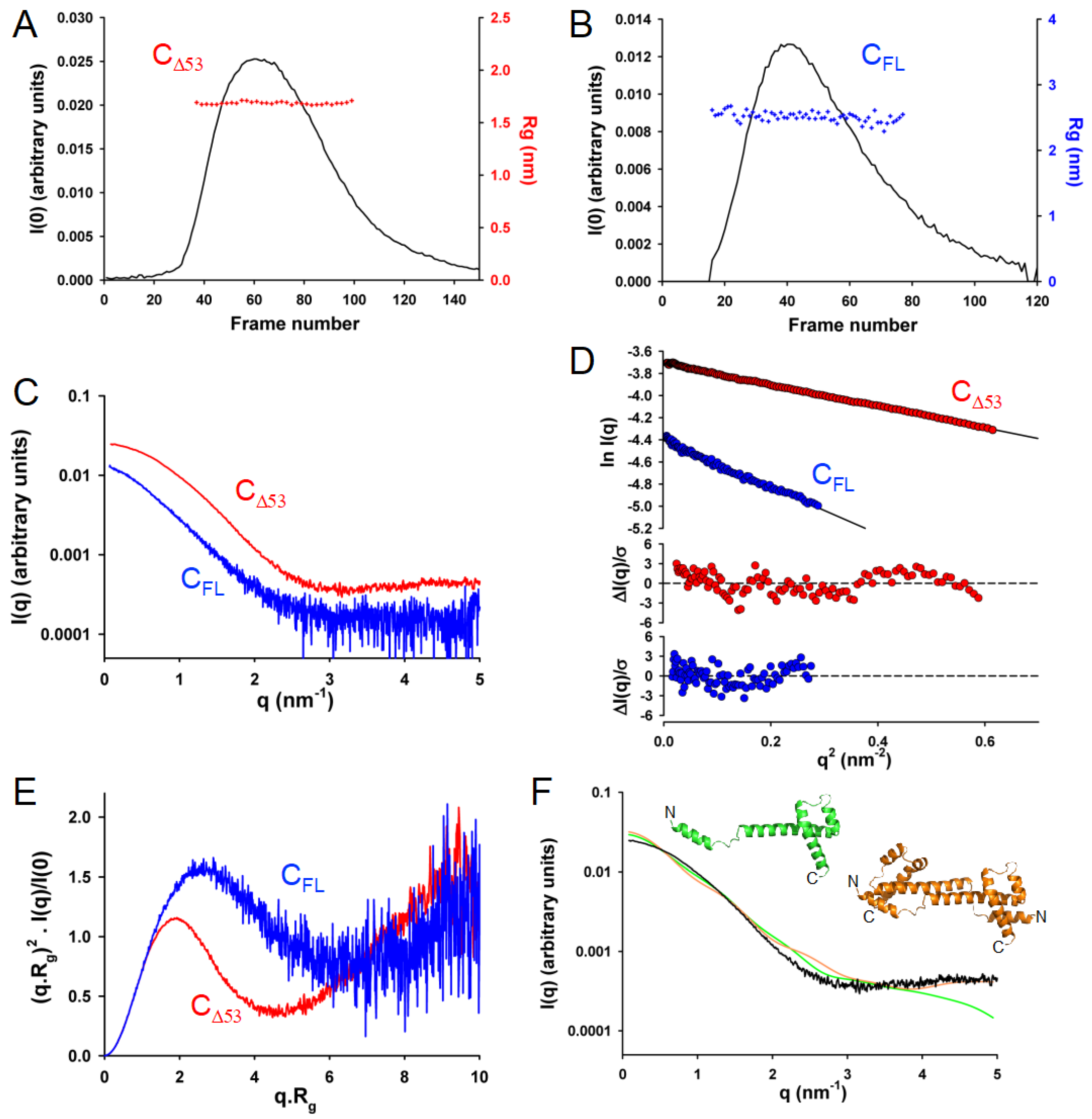
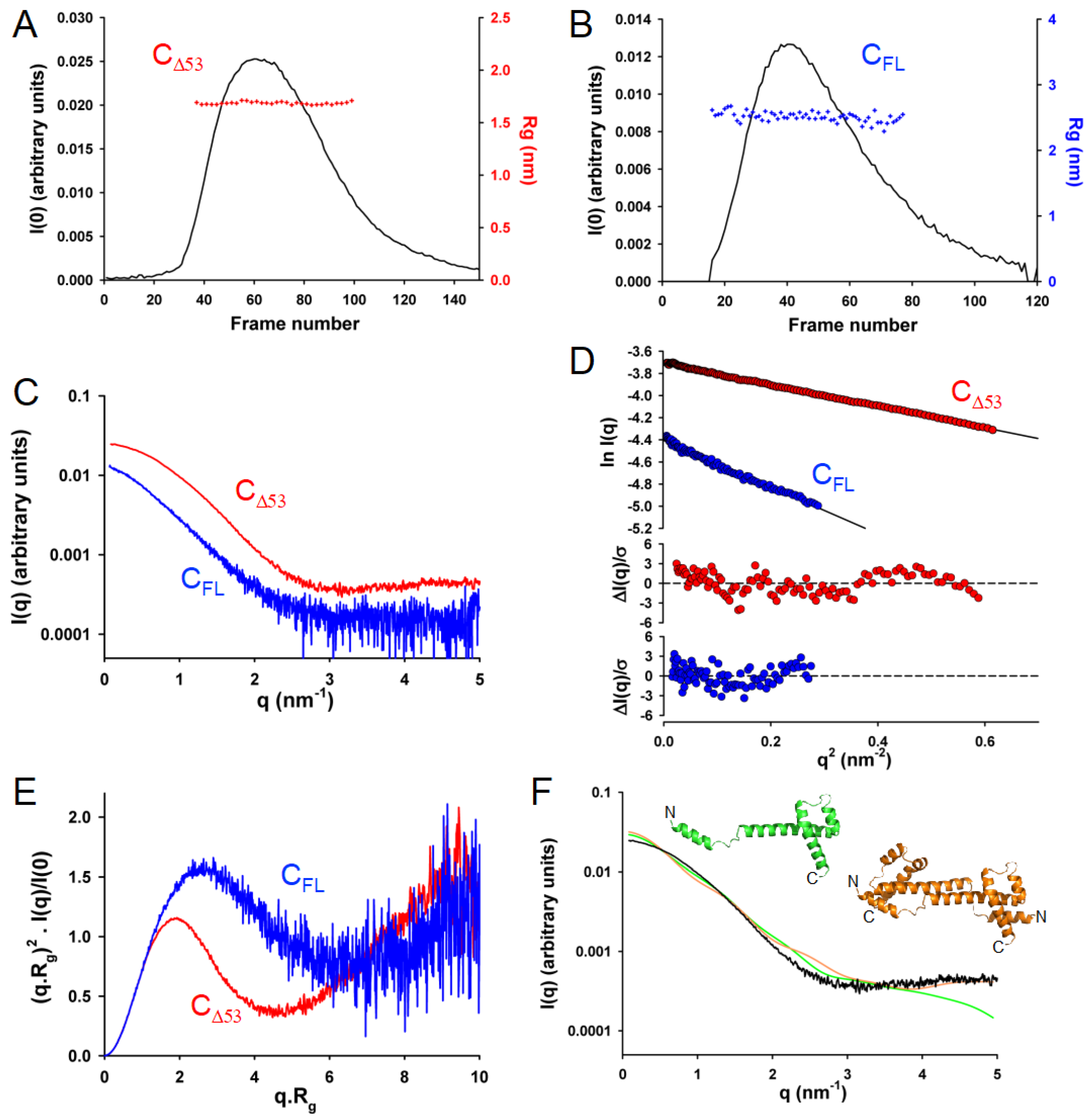
3.6. In Solution, TupV C Contains Both Disordered and Globular Regions
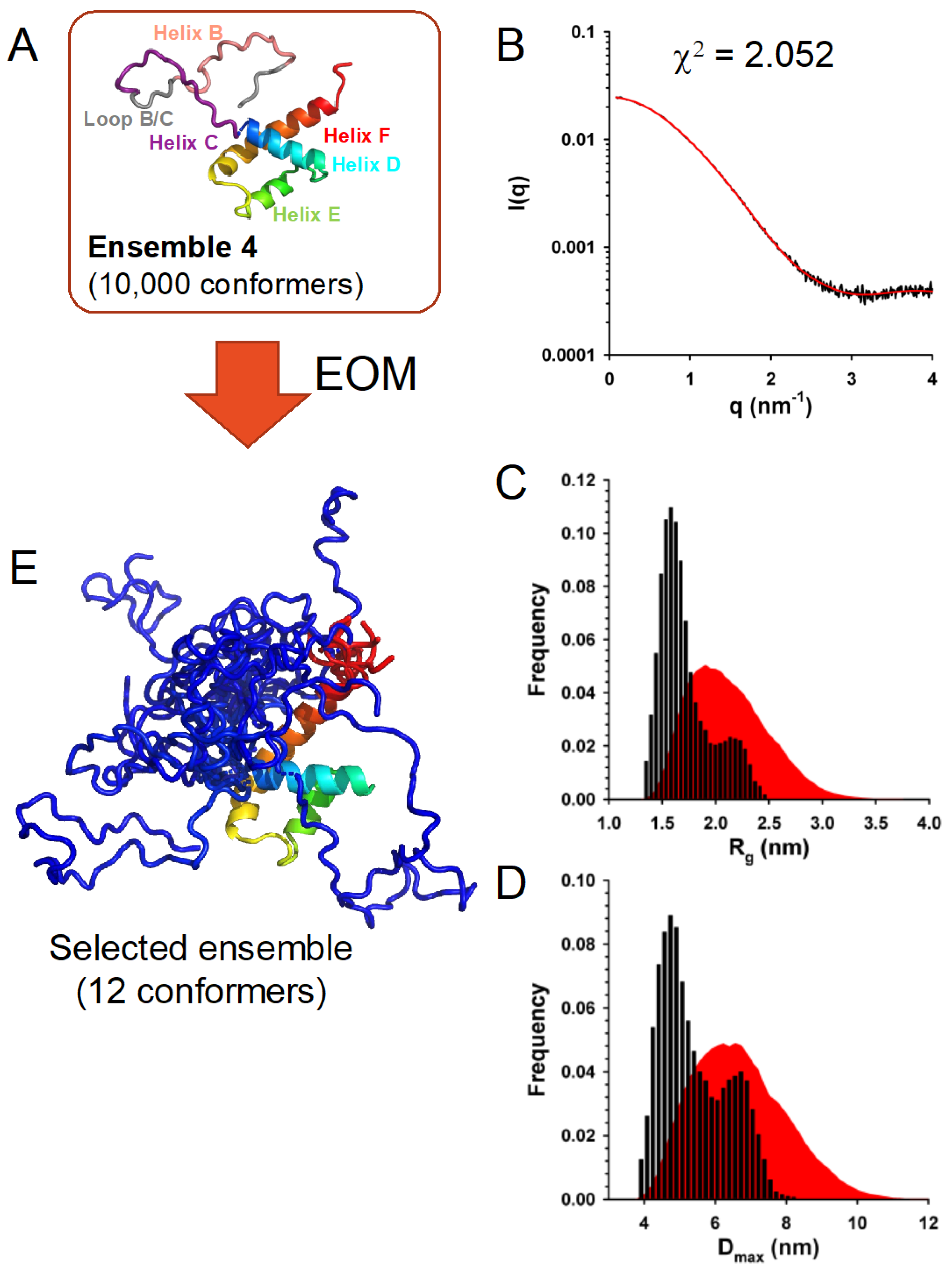
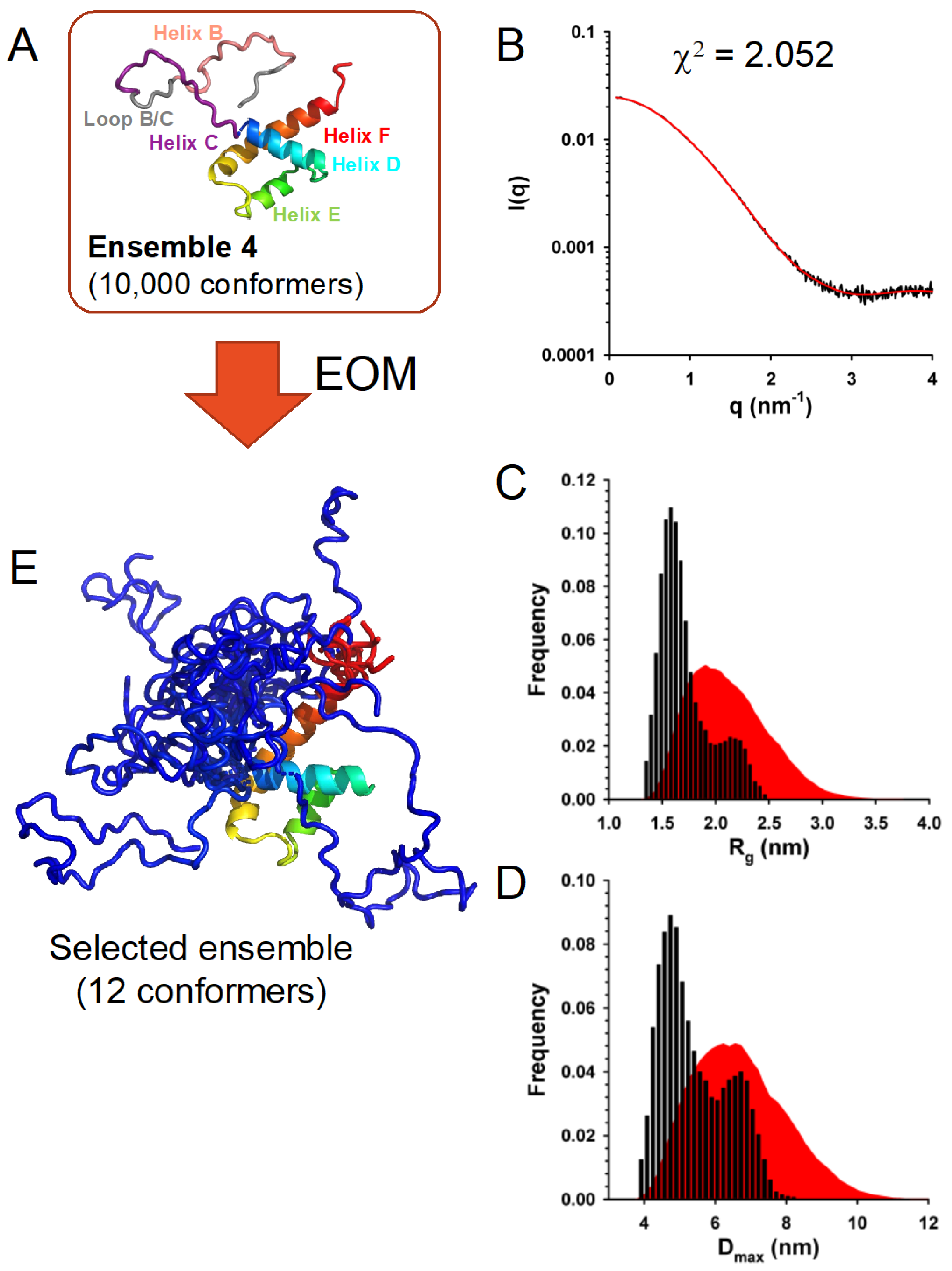
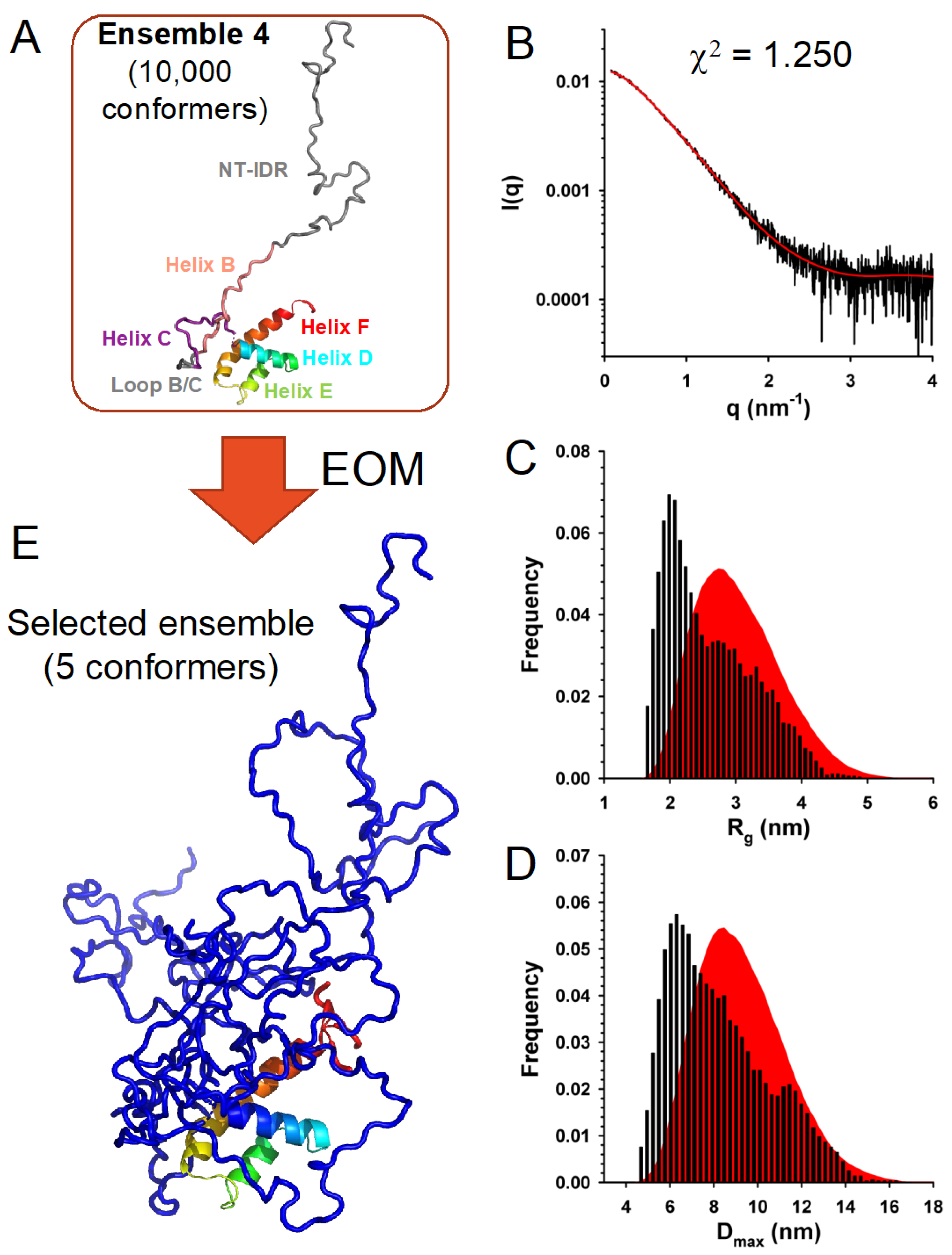
3.7. In Solution, TupV C Is Well Described by Ensembles of Conformers with a Disordered N-Terminal and Fluctuating Helices B and C
3.8. TupV C Protein Cross Inhibits Nipah Virus Minigenome Expression
3.9. TupV C Does Not Interact with Human STAT1 Nor with Known Interactors of Measles Virus and Nipah Virus C Proteins
4. Discussion
4.1. Structural Comparisons and Predictions
4.2. The C Protein Originated in the Ancestor of Orthoparamyxovirinae
- (a)
- The C coding region appeared in the common ancestor of Orthoparamyxovirinae and later diverged in sequence beyond recognition in respiroviruses and aquaparamyxoviruses. In this scenario, all C proteins are homologous.
- (b)
- The C coding region appeared independently in the ancestor of respiroviruses and aquaparamyxoviruses and in the rest of the Orthoparamyxovirinae. In this scenario, the C protein of respiroviruses and aquaparamyxoviruses would not be homologous to that of other Orthoparamyxovirinae.
- (a)
- All Orthoparamyxovirinae C proteins are encoded in the same frame with respect to P (i.e., the +1 frame), and overlap at exactly the same position within the P gene (the very 5′ end);
- (b)
- All Orthoparamyxovirinae C proteins have the same architecture (proven or predicted), composed of an N-terminal disordered region and C-terminal helical domain.
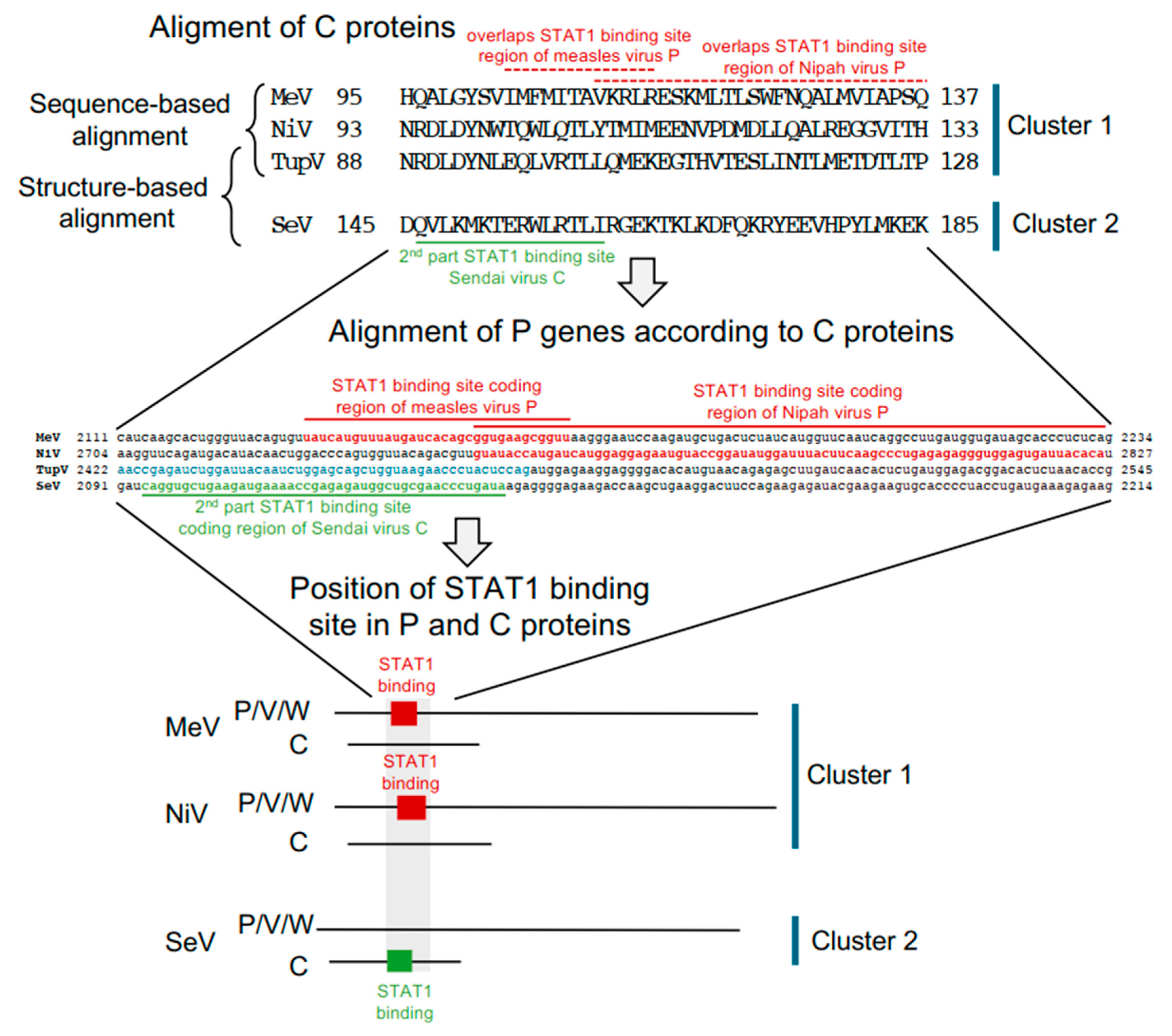
4.3. A STAT1-binding Site Is Encoded by Exactly the Same RNA Region of the P Gene but in Different Reading Frames (P or C Coding Region) in Orthoparamyxovirinae
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: The Viruses and Their Replication; Fields, B.N., Knipe, D.M., Howler, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 957–995. [Google Scholar]
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L. Hendra and Nipah Viruses: Different and Dangerous. Nat. Rev. Microbiol. 2006, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Vanmechelen, B.; Meurs, S.; Horemans, M.; Loosen, A.; Maes, T.J.; Laenen, L.; Vergote, V.; Koundouno, F.R.; Magassouba, N.; Konde, M.K.; et al. The Characterization of Multiple Novel Paramyxoviruses Highlights the Diverse Nature of the Subfamily Orthoparamyxovirinae. Virus Evol. 2022, 8, veac061. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Muller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Rasche, A.; Yordanov, S.; Seebens, A.; et al. Bats Host Major Mammalian Paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, J.H.; Wolf, Y.I.; Krupovic, M.; Zhang, Y.Z.; Maes, P.; Dolja, V.V.; Koonin, E.V. Classify Viruses—The Gain Is Worth the Pain. Nature 2019, 566, 318–320. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Amarasinghe, G.K.; Anthony, S.J.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. Taxonomic Update for Phylum Negarnaviricota (Riboviria: Orthornavirae), Including the Large Orders Bunyavirales and Mononegavirales. Arch. Virol. 2020, 165, 3023–3072. [Google Scholar] [CrossRef]
- Pringle, C.R. The Order Mononegavirales—Current Status. Arch. Virol. 1997, 142, 2321–2326. [Google Scholar]
- Douglas, J.; Drummond, A.J.; Kingston, R.L. Evolutionary History of Cotranscriptional Editing in the Paramyxoviral Phosphoprotein Gene. Virus Evol. 2021, 7, veab028. [Google Scholar] [CrossRef]
- Hausmann, S.; Garcin, D.; Delenda, C.; Kolakofsky, D. The Versatility of Paramyxovirus RNA Polymerase Stuttering. J. Virol. 1999, 73, 5568–5576. [Google Scholar] [CrossRef] [Green Version]
- Vidal, S.; Curran, J.; Kolakofsky, D. A Stuttering Model for Paramyxovirus P MRNA Editing. Embo J. 1990, 9, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Curran, J.; Kolakofsky, D. Editing of the Sendai Virus P/C MRNA by G Insertion Occurs during MRNA Synthesis via a Virus-Encoded Activity. J. Virol. 1990, 64, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.M.; Lamb, R.A.; Paterson, R.G. Two mRNAs That Differ by Two Nontemplated Nucleotides Encode the Amino Coterminal Proteins P and V of the Paramyxovirus SV5. Cell 1988, 54, 891–902. [Google Scholar] [CrossRef]
- Tidona, C.A.; Kurz, H.W.; Gelderblom, H.R.; Darai, G. Isolation and Molecular Characterization of a Novel Cytopathogenic Paramyxovirus from Tree Shrews. Virology 1999, 258, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, C.; Guillaume, V.; Volchkova, V.A.; Pohl, C.; Jacquot, F.; Looi, R.Y.; Wong, K.T.; Legras-Lachuer, C.; Volchkov, V.E.; Lachuer, J.; et al. Nonstructural Nipah Virus C Protein Regulates Both the Early Host Proinflammatory Response and Viral Virulence. J. Virol. 2012, 86, 10766–10775. [Google Scholar] [CrossRef] [Green Version]
- Siering, O.; Sawatsky, B.; Pfaller, C.K. C Protein Is Essential for Canine Distemper Virus Virulence and Pathogenicity in Ferrets. J. Virol. 2021, 95, e01840-20. [Google Scholar] [CrossRef]
- Pfaller, C.K.; Radeke, M.J.; Cattaneo, R.; Samuel, C.E. Measles Virus C Protein Impairs Production of Defective Copyback Double-Stranded Viral RNA and Activation of Protein Kinase R. J. Virol. 2014, 88, 456–468. [Google Scholar] [CrossRef] [Green Version]
- Pfaller, C.K.; Bloyet, L.-M.; Donohue, R.C.; Huff, A.L.; Bartemes, W.P.; Yousaf, I.; Urzua, E.; Clavière, M.; Zachary, M.; de Masson d’Autume, V.; et al. The C Protein Is Recruited to Measles Virus Ribonucleocapsids by the Phosphoprotein. J. Virol. 2019, 94, e01733-19. [Google Scholar] [CrossRef]
- Sleeman, K.; Bankamp, B.; Hummel, K.B.; Lo, M.K.; Bellini, W.J.; Rota, P.A. The C, V and W Proteins of Nipah Virus Inhibit Minigenome Replication. J. Gen. Virol. 2008, 89, 1300–1308. [Google Scholar] [CrossRef]
- Park, M.S.; Shaw, M.L.; Munoz-Jordan, J.; Cros, J.F.; Nakaya, T.; Bouvier, N.; Palese, P.; Garcia-Sastre, A.; Basler, C.F. Newcastle Disease Virus (NDV)-Based Assay Demonstrates Interferon-Antagonist Activity for the NDV V Protein and the Nipah Virus V, W, and C Proteins. J. Virol. 2003, 77, 1501–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrer, K.M.J.; Pfaller, C.K.; Conzelmann, K.-K. Measles Virus C Protein Interferes with Beta Interferon Transcription in the Nucleus. J. Virol. 2012, 86, 796–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, A.; Yun, T.; Vigant, F.; Pernet, O.; Won, S.T.; Dawes, B.E.; Bartkowski, W.; Freiberg, A.N.; Lee, B. Nipah Virus C Protein Recruits Tsg101 to Promote the Efficient Release of Virus in an ESCRT-Dependent Pathway. PLoS Pathog. 2016, 12, e1005659. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.K.; Søgaard, T.M.; Karlin, D.G. Evolution and Structural Organization of the C Proteins of Paramyxovirinae. PLoS ONE 2014, 9, e90003. [Google Scholar] [CrossRef]
- Fan, Y.; Huang, Z.Y.; Cao, C.C.; Chen, C.S.; Chen, Y.X.; Fan, D.D.; He, J.; Hou, H.L.; Hu, L.; Hu, X.T.; et al. Genome of the Chinese Tree Shrew. Nat. Commun. 2013, 4, 1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.; Liu, R.; Chen, C.S. Tree Shrew (Tupaia belangeri) as a Novel Laboratory Disease Animal Model. Zool. Res. 2017, 38, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerard, F.C.A.; Ribeiro, E.A.; Leyrat, C.; Ivanov, I.; Blondel, D.; Longhi, S.; Ruigrok, R.W.H.; Jamin, M. Modular Organization of Rabies Virus Phosphoprotein. J. Mol. Biol. 2009, 388, 978–996. [Google Scholar] [CrossRef] [PubMed]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A Simple Tool to Predict Whether a given Protein Sequence Is Intrinsically Unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence Complexity of Disordered Protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A Meta-Predictor of Intrinsically Disordered Amino Acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K. Predicting Intrinsic Disorder from Amino Acid Sequence. Proteins 2003, 53 (Suppl. 6), 566–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting Heterogeneous Sequence Properties Improves Prediction of Protein Disorder. Proteins 2005, 61 (Suppl. 7), 176–182. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. Structural Bioinformatics IUPred: Web Server for the Prediction of Intrinsically Unstructured Regions of Proteins Based on Estimated Energy Content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [Green Version]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein Disorder Prediction: Implications for Structural Proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring Protein Sequences for Globularity and Disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, I.; Martin, A.J.M.; di domenico, T.; Tosatto, S.C.E. Espritz: Accurate and Fast Prediction of Protein Disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callebaut, I.; Labesse, G.; Durand, P.; Poupon, A.; Canard, B.; Chomilier, J.; Henrissat, B.; Mornon, J.P. Deciphering Protein Sequence Information through Hydrophobic Cluster Analysis (HCA): Current Status and Perspectives. Cell. Mol. Life Sci. 1997, 53, 621–645. [Google Scholar] [CrossRef]
- Yabukarski, F.; Lawrence, P.; Tarbouriech, N.; Bourhis, J.M.; Delaforge, E.; Jensen, M.R.; Ruigrok, R.W.; Blackledge, M.; Volchkov, V.; Jamin, M. Structure of Nipah Virus Unassembled Nucleoprotein in Complex with Its Viral Chaperone. Nat. Struct. Mol. Biol. 2014, 21, 754–759. [Google Scholar] [CrossRef]
- Jensen, M.R.; Yabukarski, F.; Communie, G.; Condamine, E.; Mas, C.; Volchkova, V.; Tarbouriech, N.; Bourhis, J.M.; Volchkov, V.; Blackledge, M.; et al. Structural Description of the Nipah Virus Phosphoprotein and Its Interaction with STAT1. Biophys. J. 2020, 118, 2470–2488. [Google Scholar] [CrossRef]
- Ribeiro, E.A.; Favier, A.; Gerard, F.C.A.; Leyrat, C.; Brutscher, B.; Blondel, D.; Ruigrok, R.W.H.; Blackledge, M.; Jamin, M. Solution Structure of the C-Terminal Nucleoprotein-RNA Binding Domain of the Vesicular Stomatitis Virus Phosphoprotein. J. Mol. Biol. 2008, 382, 525–538. [Google Scholar] [CrossRef]
- Martinez, N.; Ribeiro, E.A., Jr.; Leyrat, C.; Tarbouriech, N.; Ruigrok, R.W.H.; Jamin, M.; Ribeiro, E.A.; Leyrat, C.; Tarbouriech, N.; Ruigrok, R.W.H.; et al. Structure of the C-Terminal Domain of Lettuce Necrotic Yellows Virus Phosphoprotein. J. Virol. 2013, 87, 9569–9578. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Wyatt, P.J. Submicrometer Particle Sizing by Multiangle Light Scattering Following Fractionation. J. Colloid Interface Sci. 1998, 197, 9–20. [Google Scholar] [CrossRef]
- Uversky, V.N. Use of Fast Protein Size-Exclusion Liquid Chromatography to Study the Unfolding of Proteins Which Denature through the Molten Globule. Biochemistry 1993, 32, 13288–13298. [Google Scholar] [CrossRef]
- Pérez, J.; Thureau, A.; Vachette, P. SEC-SAXS: Experimental Set-up and Software Developments Build up a Powerful Tool. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2022; Volume 677, pp. 221–249. [Google Scholar]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A Comprehensive Data Analysis Suite for Small-Angle Scattering from Macromolecular Solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural Characterization of Flexible Proteins Using Small-Angle X-Ray Scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Pape, T.; Schneider, T.R. HKL2MAP: A Graphical User Interface for Macromolecular Phasing with SHELX Programs. J. Appl. Cryst. 2004, 37, 843–844. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ciancanelli, M.J.; Volchkova, V.A.; Shaw, M.L.; Volchkov, V.E.; Basler, C.F. Nipah Virus Sequesters Inactive STAT1 in the Nucleus via a P Gene-Encoded Mechanism. J. Virol. 2009, 83, 7828–7841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakqori, G.; Kochs, G.; Haller, O.; Weber, F. Functional L Polymerase of La Crosse Virus Allows in Vivo Reconstitution of Recombinant Nucleocapsids. J. Gen. Virol. 2003, 84, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Halpin, K.; Bankamp, B.; Harcourt, B.H.; Bellini, W.J.; Rota, P.A. Nipah Virus Conforms to the Rule of Six in a Minigenome Replication Assay. J. Gen. Virol. 2004, 85, 701–707. [Google Scholar] [CrossRef]
- Beaty, S.M.; Park, A.; Won, S.T.; Hong, P.; Lyons, M.; Vigant, F.; Freiberg, A.N.; TenOever, B.R.; Duprex, W.P.; Lee, B. Efficient and Robust Paramyxoviridae Reverse Genetics Systems. mSphere 2017, 2, e00376-16. [Google Scholar] [CrossRef] [Green Version]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 Years On. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Oda, K.; Matoba, Y.; Irie, T.; Kawabata, R.; Fukushi, M.; Sugiyama, M.; Sakaguchi, T. Structural Basis of the Inhibition of STAT1 Activity by Sendai Virus C Protein. J. Virol. 2015, 89, 11487–11499. [Google Scholar] [CrossRef] [Green Version]
- Oda, K.; Matoba, Y.; Sugiyama, M.; Sakaguchi, T. Structural Insight into the Interaction of Sendai Virus C Protein with Alix To Stimulate Viral Budding. J. Virol. 2021, 95, JVI0081521. [Google Scholar] [CrossRef]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A Tool for Multiple Protein Sequence and Structure Alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]
- Svergun, D.I. Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J. Appl. Cryst. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Hansen, S. Bayesian Estimation of Hyperparameters for Indirect Fourier Transformation in Small-Angle Scattering. J. Appl. Cryst. 2000, 33, 1415–1421. [Google Scholar] [CrossRef]
- Martinez-Gil, L.; Vera-Velasco, N.M.; Mingarro, I. Exploring the Human-Nipah Virus Protein-Protein Interactions. J. Virol. 2017, 91, e01461-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meignié, A.; Combredet, C.; Santolini, M.; Kovács, I.A.; Douché, T.; Gianetto, Q.G.; Eun, H.; Matondo, M.; Jacob, Y.; Grailhe, R.; et al. Proteomic Analysis Uncovers Measles Virus Protein C Interaction with P65–IASPP Protein Complex. Mol. Cell. Proteom. 2021, 20, 100049. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Parisien, J.P.; Horvath, C.M. Nipah Virus V Protein Evades Alpha and Gamma Interferons by Preventing STAT1 and STAT2 Activation and Nuclear Accumulation. J. Virol. 2002, 76, 11476–11483. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.J.; Wang, L.-F.; Horvath, C.M. Hendra Virus V Protein Inhibits Interferon Signaling by Preventing STAT1 and STAT2 Nuclear Accumulation. J. Virol. 2003, 77, 11842–11845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaux, P.; von Messling, V.; Songsungthong, W.; Springfeld, C.; Cattaneo, R. Tyrosine 110 in the Measles Virus Phosphoprotein Is Required to Block STAT1 Phosphorylation. Virology 2007, 360, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, B.W.; Molloy, M.P.; Jaschke, P.R. Overlapping genes in natural and engineered genomes. Nat. Rev. Genet. 2022, 23, 154–168. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C∆53 Native | C∆53 Pt Derivative | |
|---|---|---|
| Data collection | ||
| Beamline | SOLEIL-PX1 | SOLEIL-PX1 |
| Wavelength (Å) | 1.07169 | 1.07169 |
| Space group | P3121 | P21 |
| a,b,c (Å) | 135.7, 135.7, 63.1 | 135.6, 135.6, 64 |
| a,b,g (°) | 90, 90, 120 | 90, 90, 120 |
| Resolution range (Å) 1 | 68.4–2.4 | 46.5–2.7 |
| Rmerge 1 | 0.085–0.661 | 0.078–2.46 |
| I/sI 1 | 18.9–4.4 | 16.3–0.9 |
| Completeness (%) 1 | 100–100 | 99.5–93.3 |
| Total reflections 1 | 481,705–43,842 | 25,1742–17,210 |
| Unique reflections 1 | 27,136–2824 | 18,927–1316 |
| Multiplicity 1 | 17.8–15.5 | |
| Anomalous completeness (%) 1 | n/a | 99.3–91.7 |
| Anomalous multiplicity 1 | n/a | 6.9–6.7 |
| DelAnom correlation 1 | n/a | 0.432–0.042 |
| Refinement | n/a | |
| Rwork/Rfree | 0.22 / 0.25 | |
| No. water | 49 | |
| R.m.s. deviations | ||
| Bond length (Å) | 0.0099 | |
| Bond angle (°) | 1.5851 | |
| Ramachadran statistic 2 | n/a | |
| Favoured (%) | 98.35 | |
| Allowed (%) | 1.47 | |
| Outlier (%) | 0.18 | |
| PDB code | 8BJW | n/a |
| CFL | CΔ53 | |
|---|---|---|
| Data collection parameters | ||
| Instrument | SOLEIL-SWING | |
| Energy (keV) | 12.000 | |
| Detector | EigerX-4M | |
| Detector distance (m) | 2 | |
| Exposure (s per image) | 1 | |
| q range (Å−1) | 0.008–0.50 | |
| Column | S75inc 5/150 GL | |
| Flow rate (mL.min−1) | 0.3 | |
| Sample concentrations (mg.mL−1) | ||
| Injection volume (μL) | 50 | 50 |
| Temperature (K) | 293 | 293 |
| Guinier Analysis | ||
| I(0) (A.U.) | 1.2·10−2 ± 0.03·10−2 | 2.4·10−2 ± 0.01·10−2 |
| Rg (nm) | 2.5 ± 0.1 | 1.7 ± 0.1 |
| qrgmin | 0.2951 | 0.2702 |
| qrgmax | 1.3070 | 1.3027 |
| P(R) analysis 1 | ||
| Dmax (nm) | 10.5 | 6.4 |
| Molecular weight 2 | ||
| Theoretical Mw (kDa) 3 | 17,807 | 11,897 |
| Measured Mw (kDa) | 18.6 ± 0.8 | 12.3 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, A.; Chan Mine, E.; Gaifas, L.; Leyrat, C.; Volchkova, V.A.; Baudin, F.; Martinez-Gil, L.; Volchkov, V.E.; Karlin, D.G.; Bourhis, J.-M.; et al. Orthoparamyxovirinae C Proteins Have a Common Origin and a Common Structural Organization. Biomolecules 2023, 13, 455. https://doi.org/10.3390/biom13030455
Roy A, Chan Mine E, Gaifas L, Leyrat C, Volchkova VA, Baudin F, Martinez-Gil L, Volchkov VE, Karlin DG, Bourhis J-M, et al. Orthoparamyxovirinae C Proteins Have a Common Origin and a Common Structural Organization. Biomolecules. 2023; 13(3):455. https://doi.org/10.3390/biom13030455
Chicago/Turabian StyleRoy, Ada, Emeric Chan Mine, Lorenzo Gaifas, Cédric Leyrat, Valentina A. Volchkova, Florence Baudin, Luis Martinez-Gil, Viktor E. Volchkov, David G. Karlin, Jean-Marie Bourhis, and et al. 2023. "Orthoparamyxovirinae C Proteins Have a Common Origin and a Common Structural Organization" Biomolecules 13, no. 3: 455. https://doi.org/10.3390/biom13030455